
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceR-VAPOL-phosphoric acid based 1H and 13C-NMR for sensing of chiral amines and acids†
Durga Prasada,
Santosh Mogurampellyb and
Sachin R. Chaudhari *a
*a
aDepartment of Spice and Flavour Science, CSIR-Central Food Technological Research Institute, Mysore, Karnataka 570020, India. E-mail: sachinnmr@cftri.res.in
bDepartment of Physics, Indian Institute of Technology Jodhpur, Karwar, Rajasthan 342037, India
First published on 13th January 2020
Abstract
Enantiomers have significant importance in pharmaceuticals, biology and modern chemistry and therefore distinguishing and quantifying the enantiomeric forms is of utmost importance. Herein, we propose diphenyl-3,3′-biphenanthryl-4,4′-diyl phosphate (R-VAPOL-PA) as a promising chiral solvating agent to discriminate amines and acids of poly-functional groups such as chiral amines, amino alcohols and hydroxy acids. The methodological approach involves using the nature of hydrogen bonds and ion pairs as a mode of weak interactions to form diastereomers where the probe is associated with enantiomers. The resulting diastereomer difference in the NMR spectrum enables the chiral discrimination with a complete baseline peak separation and an accurate enantiomeric excess (ee) analysis. We also carried out density functional theory (DFT) calculations to understand the complex formation to explain enantiodiscrimination by analysing the formation and stability of different chiral complexes. The binding energy differences between enantiomeric forms revealed by DFT calculations are qualitatively in agreement with the diastereomer difference in the NMR spectrum and unequivocally establishes the suggested experimental protocol of R-VAPOL-PA-based enantiomeric discrimination.
Introduction
Chiral molecules and particularly enantiomers have attracted huge attention due to their indispensable role in technological applications including pharmaceuticals, biology and modern chemistry.1,2 Quantifying the nature of governing molecular interactions and structural characterization is essential for such applications and requires efficient characterization tools.3–5 The development of combinatorial chemistry and asymmetric synthesis made it possible to generate libraries of enantiomers. Spectroscopic and chromatographic approaches are generally utilized to determine the enantiomeric excess (ee), enantiodiscrimination and assignment of absolute configuration.6–14 Specifically, analytical tools such as chiral HPLC, GC, CD, mass spectrometry and NMR methods have been widely considered for enantiomer discrimination.6–14 However, visual recognition between enantiomers which can be one of the simple approaches and fast measurement of enantiomeric excess can be achieved by 1H-NMR spectroscopy.13,15 The NMR aided enantiodiscrimination involves diasterospecific interactions brought by enantiopure host molecules called chiral auxiliaries.13,15One of the simplest and practical approaches in enantiodiscrimination is to use a chiral solvating agent (CSA) where weak interactions play a dominant role in diastereomer formation.16 This can be achieved simply by mixing chiral solvating agent (host) and chiral analyte (guest) followed by NMR analysis compared to use chiral derivatizing agent.13,17–19 A number of chiral solvating agents are available in the literature such as macrocyclics,20–22 metal complexes,23 hydrogen bonding and ion-pair agents;20,24–26 some of which are synthetic and natural.27–34 Recently, we have also made a significant contribution in enantiodiscrimination and proposed various chiral solvating agents to the existing list of chiral auxiliary.16,35–37 Unfortunately, the use of single host chiral auxiliary is limited to a particular monofunctional group and therefore the enantiodiscrimination of polyfunctional group13 requires various CSA depending on the sample of interest. Undeniably, the resolution of chiral amines and acids is poor with most of traditional CSAs due to the formation of soluble and insoluble salt between amine and acids. Besides, even a small amount of chemical shift difference leads to improper baseline corrections and exhibit insufficient peak separation, and therefore limits the applicability of CSA. Therefore, there is a need for better probing strategies involving, for instance achieving the resolution by a single probe and apply in a more generalized and practical way.36
Motivated by the above issues, herein, we propose the C2-symmetric vaulted biaryl chiral ligand derivative (R)-2,2′-diphenyl-3,3′-biphenanthryl-4,4′-diyl phosphate called as R-VAPOL-PA (I) (Fig. 1) as a protean chiral solvating agent. Due to the presence of acid group, it can act as a proton donor in promoting the formation of hydrogen bond and participate in weak ion-pair interactions.38,39 The nature of hydrogen bond formation and ion pair interactions associated with R-VAPOL-PA makes it an advanced CSA for discrimination of amines and acids of functional groups. We demonstrate the use of R-VAPOL-PA by considering chiral amines which can form salts. Thus, the mode of weak molecular interactions can be successfully employed for the chiral discrimination of enantiomers bearing amine groups such as chiral amines and amino alcohols. We also explored using the R-VAPOL-PA agent for discriminating enantiomers containing a carboxylic acids and its derivative such as the hydroxy acids and acids etc. Unfortunately, we find that the interactions were not sufficiently strong enough to clearly establish the difference in the NMR spectrum which hampers the ee analysis. However, considering a systematic application of weak interactions data documented in crystal engineering,40 it is possible to rationalize the types of weak molecular interactions required to form stable diastereomers. Inspired by this, we introduce an additional component along with R-VAPOL-PA, which acts as a linker to promote hydrogen bonding, and ion-pair formation. Because of the induced interaction mechanism, the host and guest molecules form a stable complex and allow us to visualize the discrimination. In the R-VAPOL-PA-based enantiodiscrimination framework of polyfunctional groups such as chiral acids, hydroxy acids, amines and amino alcohols, we propose the R-VAPOL-PA as the primary chiral solvating agent with and without 4-dimethylaminopyridine (DMAP) as a mediator.
 | ||
| Fig. 1 2D representation of the structure of R-VAPOL-PA (I), mode of interactions between R-VAPOL-PA and chiral amines (II) (for example molecule 1) and ternary ion-pair complex with chiral acids and third component 4-dimethylaminopyridine (III). | ||
Except for the chiral acids, we are able to achieve proper baseline corrections for all investigated chiral amines and mandelic acids and their derivatives. The R-VAPOL-PA-based CSA proposed in these studies also aids in the measurement of enantiomeric excess, (ee). We have also performed DFT calculations to shed light on the nature of interactions and binding energies of different enantiomers and to support the NMR results.
Results and discussions
The R-VAPOL-PA (I) chiral complexes have been effectively used in enantioselective reaction.41–43 Since the R-VAPOL-PA bears the acid functional group, it can act as a proton donor and can promote ion transfer in case of amine and amino-alcohols. Based on the interacting nature of R-VAPOL-PA, we hypothesize that chiral amines are good candidates for testing the applicability of R-VAPOL-PA as a CAS for enantiodiscrimination. To rationalize this, we have recorded the 1H-NMR spectrum of the racemic mixture of alphamethylbenzylamine (hereafter referred to as molecule 1, Fig. 3) with and without R-VAPOL-PA in the solvent CDCl3, and the respective spectra are given in Fig. 2a. Methyl proton and alpha proton of molecule 1 in the absence of R-VAPOL-PA show a single peak at 1.4 and 4.1 ppm, respectively (Fig. 2a lower trace), and as expected, the enantiomers not resolved in the achiral environment. However, the addition of one equivalent of R-VAPOL-PA chiral agent is seen to split the single peak into two distinguishable peaks around 0.75 and 3.1 ppm for methyl proton and alpha proton respectively (Fig. 2a upper trace). Moreover, the R-VAPOL-PA introduces a chemical shift difference (ΔδR/S) of 0.07 ppm for methyl and 0.19 ppm for alpha proton. The above effects arise due to the formation of either R, R or R, S diastereomers. The above interpretation was further confirmed by analysing the spectrum of one of the enantiomers of molecule 1, specifically for instance by considering the molecule 1 with R-VAPOL-PA ligand. The 1H-NMR shows only peak with either R enantiomer form Fig. 2b, however for the racemic mixture separate peak of each enantiomers were observed thereby confirming that R-VAPOL-PA can be successfully used as a chiral solvating agent for discrimination of chiral amines. | ||
| Fig. 2 (a) 1H-NMR spectrum of racemic molecule 1 in CDCl3 with and without 1 equivalent of R-VAPOL-PA showing only alpha proton and methyl proton respectively (b) 1H-NMR stack plot of R and R/S molecule 1 with 1 equivalent of R-VAPOL-PA along with the chemical structure in the schematic shows the chemical shift difference in ppm (ΔδR/S) observed at the respective proton sites. (c) Effect of concentration of CSA (R-VAPOL-PA) on the chemical shift difference of molecule 1 in the solvent CDCl3. (d) 1H-NMR spectrum of R/S molecule 1 showing discriminate aromatic protons with R-VAPOL-PA as CSA. All the spectra were recorded at 500 MHz magnetic field. The black circles indicate the peaks corresponding to both isomers. | ||
We have also varied the concentration of CSA as it is known to affect the chemical shift difference (ΔδR/S). The ΔδR/S were measured for different equivalents of R-VAPOL-PA (0.5 to 3 equivalents) for the molecule 1 on α-H proton peak in 1H-NMR and resultant comparative spectra are shown in Fig. 2c. As results are shown in Fig. 2c, experimental data suggest that 0.5 to 1 equivalent of CSA is sufficient for successful enantiodiscrimination. An interesting feature of the proposed CSA approach is that the discrimination achieved for protons not only at the chiral centre but also protons in the vicinity of the chiral centre. This is clearly seen from Fig. 2d where discrimination is achieved on aliphatic as well as aromatic protons of molecule 1. In order to explore the wide utility of R-VAPOL-PA-based approach, we considered testing the approach on various chiral amines and amino alcohols (Fig. 3). The use of 1H-NMR is typically limited because of the multiplicity pattern and cramped in chemical shift dispersions, thereby the resonances of enantiomers can be overlapped. In such a situation, the use of heteronuclei such as 13C instead of 1H-NMR can be advantageous due to large chemical shift dispersions.44–47 For example, the 1H-NMR spectrum of 1-aminoindane (molecule 4), the aliphatic region for all the protons shows multiplicity pattern due to nearby protons making it difficult to extract the chemical shift difference and hampers the analysis of ee (Fig. S1†). However, the 13C-{1H}-NMR gives singlet at each site reduces imbricate in resonances from each diastereomers. These results can be confirmed by 13C-{1H}-NMR spectrum given in Fig. 4 and besides, the 13C-spectrum of molecule 4 gives large chemical shift difference up to 0.34 ppm for some sites, which straightforwardly allows for extraction and quantifications. The obtained large chemical shift difference makes the R-VAPOL-PA a promising CSA candidate for enantiodiscrimination compared to traditional CSAs reported in the literature.20,44,48 Additionally, 0.5 to 1 equivalent is sufficient to achieve a higher resolution, whereas widely utilized Pirkle salt required 4 equivalents and causes the problem of spectral overwhelming.44 We have also explored the utility of 13C-{1H} spectrum for other chiral amines and amino alcohols and the corresponding results are summarized in Fig. 3. The results indicate that in all cases, the R-VAPOL-PA clearly discriminates the enantiomers containing amines and amino alcohol functional groups (molecule 1 to 5). In some cases, we have achieved a large chemical shift difference up to 0.36 and 0.20 ppm for 13C and 1H, respectively which is much larger compared to earlier reported CSA (Table 1).20,44,48 The discriminate ability of BINOL based phosphoric acid49 and R-VAPOL-PA were also compared. This can judge by the comparisons of chemical shift difference obtained of diastereomers form under similar condition. For that, we have selected molecule 1 as a probe. 1H and 13C-NMR were recorded of the 1:BINAP and 1:R-VAPOL-PA complex. The extracted chemical shift difference is compared in the Table 1 (below trace). From the Table 1 it indicates that in case of BINAP, the methyl group (CH3) yields large ΔδR/S compared to R-VAPOL-PA. On other hand, situation is opposite in case alpha proton. The possible explanation could be effect of degree of deshielding or shielding of naphthyl ring in case of BINAP and phenanthryl ring of R-VAPOL-PA. These shielding and deshielding effects often enhance the magnitude of enantio-differentiation in the NMR spectra of analytes. However, overall comparisons with spectral overlapping in aromatic resonances (7–9 ppm region in 1H), higher ΔδR/S in case of 13C, R-VAPOL-PA can be better alternative and choice over the BINAP.
 | ||
| Fig. 3 Measured ΔδR/S values of discriminated protons and carbons for polyfunctional chiral molecules. ΔδR/S displayed in light and bold fonts corresponds to 1H and 13C-NMR, respectively. | ||
 | ||
| Fig. 4 13C{1H}-NMR spectra of racemic 1-aminoindane (molecule 4, 1 equivalent) in CDCl3 with 1 equivalent of R-VAPOL-PA added. The red closed circles indicate the peaks corresponding to each isomer and the chemical structure in the schematic shows the chemical shift difference in ppm (ΔδR/S) at the respective carbon sites. | ||
| CSA | 1H (ppm) | 13C (ppm) |
|---|---|---|
| a R-BINOL = (R)-(+)-1,1′-Bi(2-naphthol).b R-PA = Pirkle's alcohol.c R-BINAP = (R)-(−)-1,1′-Binaphthyl-2,2′-diyl hydrogenphosphate.d For assignment of the protons refer structure in the Fig. 4 (1-aminoindane) and Fig. 2 (alphamethylbenzylamine). | ||
| Analyte: 1-aminoindane (4) (ΔδR/S) | ||
| R-BINOLa | 0.030 (1), 0.030 (3), 0.009 (3′), 0.012 (2), 0.056 (2′) | 0 (no discrimination observed) |
| R-PAb | 0.015 (1), 0.100 (3), 0.045 (3′), 0.07 (2), 0.076 (2′) | 0.047 (1), 0.074 (2), 0.033 (3) |
| R-VAPOL-PA | 0.078 (1), 0.040 (3), 0.070 (3′), 0.306 (2), 0.142 (2′) | 0.125 (1), 0.356 (2), 0.028 (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Analyte: alphamethylbenzylamine(1) (ΔδR/S) | ||
| R-BINAPc | CH3 – 0.24 (1), CH – 0.05 (2) | CH3 – 0.26 (1), CH – 0.21 (2) |
| R-VAPOL-PA | CH3 – 0.08 (1), CH – 0.14 (2) | CH3 – 0.25 (1), CH – 0.26 (2) |
After the successful application of R-VAPOL-PA as chiral solvating agent for chiral discrimination of various amine and amino alcohols, we have tested it on a different class of functional groups such as hydroxyl acids and acids. Our results suggest that the R-VAPOL-PA as CSA is insufficient for the discrimination of acids. For example, the discrimination of mandelic acid (molecule 6, Fig. 3) with –COOH as the functional group was not achieved with R-VAPOL-PA alone. Explicitly, in the presence of R-VAPOL-PA, the 1H-NMR spectrum of molecule 6 shows very small chemical changes in the spectrum with chemical shift difference of 0.01 ppm compared to a larger shift of 0.20 ppm observed for molecule 1. We speculate that the acid group present in R-VAPOL-PA does not interact strongly with the acid functional group of the guest enantiomer under investigation and undermines the formation of stable diastereomer. To check the veracity of the above interpretation, we performed the NMR experiments of R-VAPOL-PA for different other functional groups such as acid, hydroxy acids. In all the above cases, we could not achieve a proper baseline correction indicating the limited applicability of R-VAPOL-PA for enantiodiscrimination for acids and hydroxy acids. To understand the stability/lifetime of diastereomers form R-VAPOL-PA with amines and acids, we have compared the chemical shift (δ-1H) of molecule 1 and molecule 6 inbound and unbound states. The obtained chemical shift difference in 1H-NMR of molecule 1 for CH3 proton peak and for molecule 6 (α-H proton peak) between unbound and bound state was 0.90 and 0.19 ppm respectively. The large chemical shift difference (0.90 ppm) observed for molecule 1 gives an idea about stability of the association and thus indicates that the molecule 1 forms a more stable complex and interacts strongly with R-VAPOL-PA compared to molecule 6.
To support these studies and to understand the role of the acid functional group of R-VAPOL-PA in the enantiodiscrimination, we calculated the binding energies of molecule 1 and 6 with R-VAPOL-PA complexes using the DFT calculations. The interatomic forces were derived within the framework of DFT using the Perdew, Burke and Ernzerhof (PBE)-GGA approximation for the exchange–correlation energy functional.50 The interaction between valence electrons and ionic cores was represented by the Troullier–Martin norm-conserving pseudopotentials.51 The van der Waals dispersion corrections were treated using the Tkatchenko–Scheffler parameters.52 Additional details of the DFT calculations were provided in the ESI.† As shown in Fig. 5, the binding energies calculated for R-VAPOL-PA-1 and R-VAPOL-PA-6 complex were −108.44 and −28.53 kcal mol−1, respectively implying that R-VAPOL-PA interacts strongly with molecule 1 compared to molecule 6. The above observations conclusively demonstrate that the acid group present on the R-VAPOL-PA inhibits the strong interaction with the host enantiomer molecules. Therefore, the chiral discrimination of acid group-containing enantiomers becomes difficult with R-VAPOL-PA CSA and an advanced CSAs are required for proper enantiodiscrimination.
 | ||
| Fig. 5 The DFT optimized geometries of (a) R-VAPOL-PA-1, (b) R-VAPOL-PA-6 and (c) R-VAPOL-PA-6-DMAP complexes. The calculated binding energies (in kcal mol−1) and OH distance (in Å) in hydrogen bonds obtained for the respective complexes were also mentioned. | ||
To increase the interaction of host and guest (acid and its derivatives) molecules and to form stable diastereomers, we considered the possibility of adding a third compound which can possibly act as a linker between host and guest. Towards the above objective, we chose to introduce 4-dimethylaminopyridine (DMAP) third agent which contains nitrogen and thus basic in nature as a linker agent. The –POOH group on R-VAPOL-PA can donate a proton to nitrogen of DMAP and form ion-pair as shown in Fig. 1(III). The acid moiety then can form a hydrogen bond with one of the oxygen of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–OH groups. The resulting ternary component is shown in Fig. 1(III). The results obtained from the 1H-NMR spectrum of molecule 6 with DMPA along with R-VAPOL-PA shown in Fig. 6a now enables us to observe enhanced discrimination which, otherwise, was not possible by R-VAPOL-PA alone.
P–OH groups. The resulting ternary component is shown in Fig. 1(III). The results obtained from the 1H-NMR spectrum of molecule 6 with DMPA along with R-VAPOL-PA shown in Fig. 6a now enables us to observe enhanced discrimination which, otherwise, was not possible by R-VAPOL-PA alone.
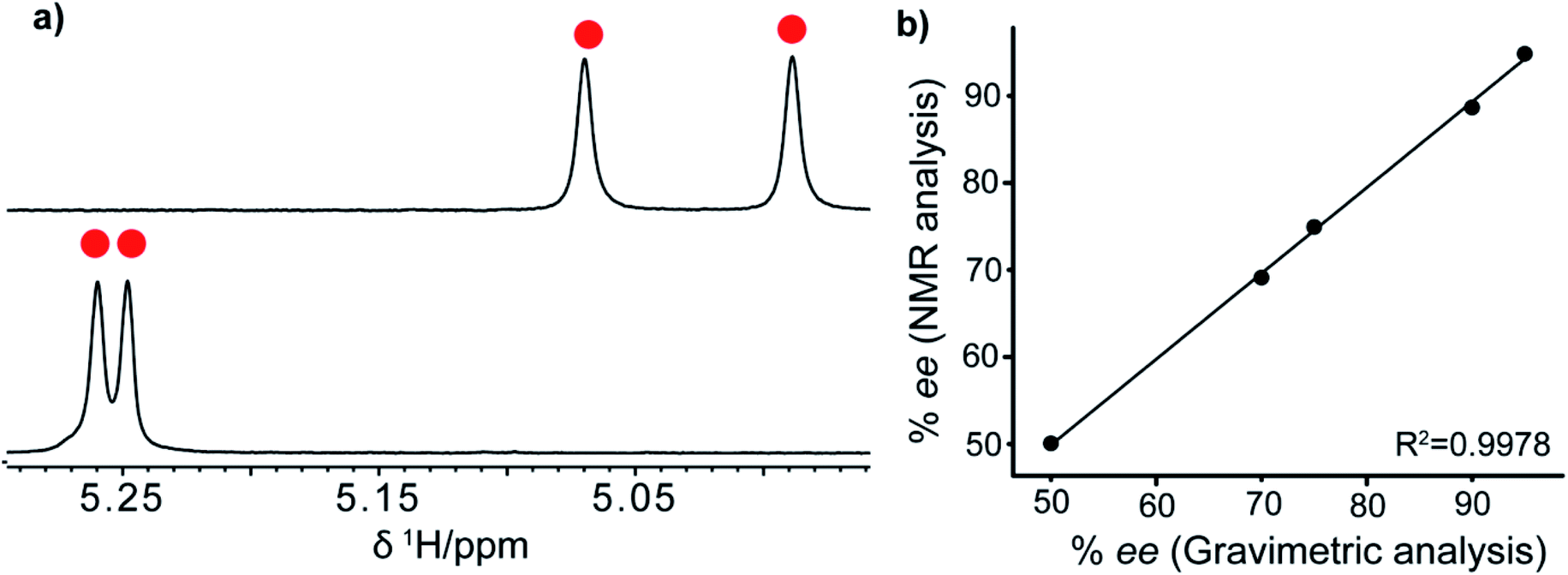 | ||
| Fig. 6 (a) 1H-NMR spectrum of mandelic acid (molecule 6): R-VAPOL-PA with (upper spectrum) and without DMAP (lower spectrum) showing only alpha proton and (b) plot between NMR and gravimetric analyses in determining the ee for molecule 1 (see Table 2 for details). | ||
Further, we calculated the binding energy for the stable diastereomer of ternary ion-pair-III complex using the first principle density functional theory (DFT) calculations. The obtained binding energy, −123.83 kcal mol−1 is comparable to that of the R-VAPOL-PA-1 complex (−108.44 kcal mol−1) which provides further evidence for the formation of stable diastereomers observed with NMR experiments especially with the comparisons of the chemical shift difference between bound and unbound state as discussed earlier. The integration of ion-pair and hydrogen bonding interactions result in the stable diastereomeric complex. The results lead to separate signal for each diastereomers and yields a very good resolution in NMR spectrum.
Thus, by rationalising the weak interactions by means of additional linker solvent, we can tune the sensing property of some of given chiral ligand. Further, we have explored the different concentration of R-VAPOL-PA and DMAP, however, 2 equivalents was found to better and higher concentration does not show significant changes in enantiodiscrimination. The optimized R-VAPOL-PA-based protocol has been applied to other functional groups such as acids and we achieved the enantiodiscrimination for all investigated systems. These results are consolidated in Fig. 3.
After establishing the sensing property of the R-VAPOL-PA-based CSA, we also measured the enantiomeric excess, by preparing different ratios of R and S enantiomer utilizing molecule 1 as a probe. The yielded and obtained results from NMR are compared in Fig. 6b and the data presented therein confirms that they agree with each other. The details of analysis, percent of enantiomeric excess and obtained integral values are provided in Table 2. Together, the results presented above suggest that the R-VAPOL-PA-based CSA is a promising approach for the measurement of enantiomeric excess within the allowed error in NMR experiments.
| (R) Enantiomer | (S) Enantiomer | ||||
|---|---|---|---|---|---|
| Gravimetric (ee) | Experimental (NMR) (ee) | Integral (abs) | Gravimetric | Experimental (NMR) (ee) | Integral (abs) |
| 50.00 | 50.1 | 1339481586 |
50.00 | 49.9 | 1335540873 |
| 70.00 | 69.1 | 5834674654 |
30.00 | 30.9 | 2607902814 |
| 75.00 | 74.9 | 3757999390 |
25.00 | 25.1 | 1257315684 |
| 87.00 | 88.7 | 5621687032 |
13.00 | 11.3 | 717636699 |
| 95.00 | 94.8 | 8656606700 |
5.00 | 5.2 | 472201958 |
In the extreme case when the 1H-NMR spectrum gets complicated due to the overlapping of resonance of diastereomers, one can use advanced NMR methods such as pure shift NMR, J-resolved, ω1-COSY, RES-TOCSY which offer enhancement in resolution in an overlap peak.36,53–56 In the case of chiral acid obtained small chemical shift difference between the discriminated resonance's thereby poor resolutions. In such a situation, we have demonstrated the 2D-1H-J-resolved experiment, as these experiments were fruitful to achieve the resolution. The 2D-1H-J-resolved spectrum for the sample 9 shown in Fig. 7, the separated peaks are highlighted with dotted lines. The obtained J-resolved spectrum shows resolution, where conventional 1D 1H-NMR fails to achieve it. We have also explored the discrimination ability using 13C-NMR for chiral acid hydroxy acids. However, results are not so encouraging as poor resolution was obtained in the case chiral acids, whereas mandelic acid and it is derivative able to show discrimination in 13C-NMR.
 | ||
| Fig. 7 2D 1H J-resolved spectrum NMR spectra of racemic 9 (1 equivalent) in CDCl3 with 2 equivalents of R-VAPOL-PA and DMAP were added. | ||
Conclusion
In summary, the present study demonstrated the applicability of R-VAPOL-PA as an efficient chiral solvating agent for discriminating a variety of polyfunctional group enantiomers. In the case of chiral amines, we showed that the R-VAPOL-PA is itself sufficient to discriminate the enantiomeric forms without ambiguity. However, for a range of other functional groups such as acid and its derivatives, mediator third agent such as DMAP is recommended for achieving a stable diastereoisomer complex to obtain an enhanced resolution. The proposed R-VAPOL-PA-based protocol is demonstrated to be sufficient for measuring the enantiomeric excess (ee) within the limits of typical errors associated with NMR measurements.Understanding the nature of weak molecular interactions in the analysis of enantiomers by R-VAPOL-PA as well as R-VAPOL-PA-DMAP is a powerful approach. Moreover, ion pair and hydrogen bonding interactions are complementary to form solid diastereomer where the ligand is attached to the probe. The discrimination was successfully probed by 1H and 13C probe nuclei with proper baseline corrections for chiral amines and amino alcohol. Along with multiple probe sites available for discriminations, selective protons of interest can be used to rationalize the enantiomer discrimination and ee. However, for the case of chiral acids where discrimination is not possible with 1H-NMR as the only method, an advance NMR approach is suggested.
The R-VAPOL-PA-based results are compared with those of widely utilized traditional CSAs such as R-BINOL and Pirkle's alcohol and found that the proposed R-VAPOL-PA-based CSA shows better performance. We also carried out DFT based calculations to characterize the weak interactions and found a good agreement with the experimental observations. The calculated binding energies give further insight into the strength of the governing molecular interactions occurring in chiral complexes. Since the use of traditional CSAs for chiral discrimination require additional multistep synthesis and owing to the difficulties associated with its applicability, the R-VAPOL-PA solvent-based approach proposed in this study has numerous advantages including the higher resolution, efficiency, practical applicability and commercial availability of the solvent.
Experimental section
Chemicals
All the chemicals used in the presented work were purchased from Sigma Aldrich.Solution-state NMR
Solution-state NMR experiments were performed on 500 MHz Bruker AVANCE spectrometers, equipped with a double resonance probe. The sample temperature was regulated to 298 ± 2 K. All the spectra were a reference to TMS as an internal standard.Preparation of samples
All the samples were prepared directly in the NMR sample tube and then used directly for NMR measurements. For the enantiomeric excess measurement (quantitative measurement) bulk solution of R and S of molecule 1 were prepared in solvent CDCl3 and then mixed in appropriate proportion to prepare different ratio of R and S enantiomers.1H spectra
The 1H spectra were acquired using a single pulse. Relaxation times (T1) were measured with a standard inversion-recovery sequence for a sample of molecule 1 for an accurate analysis of ee and 5T1 used in the 1H-NMR analysis. For all other experiments sample relaxation delay of 5 s and acquisition time, 3 s was used.13C{1H} spectra
The 13C-{1H} spectra were acquired using an ‘inverse gated decoupling experiment’. For all the experiments sample pulse angle of 90°, relaxation delay of 3 s and acquisition time 1.2 s were used.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank Dr Raghava Rao K. S. M. S., Director, and Dr Madhava Naidu M., HOD, SFS, CSIR-CFTRI, for extending the facilities and support this work. This project has received funding from Science and Engineering Research Board under start-up research grant with the grant agreement no. SRG/2019/000039.References
- (a) U. R. Prabhu, S. R. Chaudhari and N. Suryaprakash, Chem. Phys. Lett., 2010, 500, 334 CrossRef CAS; (b) H. A. Naqi, T. J. Woodman, S. M. Husbands and I. S. Blagbrough, Anal. Methods, 2019, 11, 3090 RSC.
- M. Ardej-Jakubisiak and R. Kawęcki, Tetrahedron: Asymmetry, 2008, 19, 2645 CrossRef CAS.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biol. Sci., 2006, 2, 85 CAS.
- E. Sanganyado, Z. Lu, Q. Fu, D. Schlenk and J. Gan, Water Res., 2017, 124, 527 CrossRef CAS PubMed.
- L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS PubMed.
- D. Parker, Chem. Rev., 1991, 91, 1441 CrossRef CAS.
- (a) J. K. Rugutt, H. H. Yarabe, S. A. Shamsi, D. R. Billodeaux, F. R. Fronczek and I. M. Warner, Anal. Chem., 2000, 72, 3887 CrossRef CAS PubMed; (b) F. Fowler, B. Voyer, M. Marino, J. Finzel, M. Veltri, N. M. Wachter and L. Huang, Anal. Methods, 2015, 7, 7907 RSC.
- R. Vespalec and P. Boček, Chem. Rev., 2000, 100, 3715 CrossRef CAS PubMed.
- Z.-P. Yao, T. S. M. Wan, K.-P. Kwong and C.-T. Che, Anal. Chem., 2000, 72, 5383 CrossRef CAS PubMed.
- L. Pu, Chem. Rev., 2004, 104, 1687 CrossRef CAS PubMed.
- T. J. Ward and D.-M. Hamburg, Anal. Chem., 2004, 76, 4635 CrossRef CAS PubMed.
- L. Wu, E. C. Meurer and R. G. Cooks, Anal. Chem., 2004, 76, 663 CrossRef CAS PubMed.
- T. J. Wenzel and C. D. Chisholm, Prog. Nucl. Magn. Reson. Spectrosc., 2011, 59, 1 CrossRef CAS PubMed.
- X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918 CrossRef CAS PubMed.
- S. R. Chaudhari and N. Suryaprakash, J. Indian Inst. Sci., 2014, 94, 484 Search PubMed.
- S. R. Chaudhari and N. Suryaprakash, New J. Chem., 2013, 37, 4025 RSC.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- H. Taji, M. Watanabe, N. Harada, H. Naoki and Y. Ueda, Org. Lett., 2002, 4, 2699 CrossRef CAS PubMed.
- S. Porto, J. Duran, J. M. Seco, E. Quinoa and R. Riguera, Org. Lett., 2003, 5, 2979 CrossRef CAS PubMed.
- L. Yang, T. Wenzel, R. T. Williamson, M. Christensen, W. Schafer and C. J. Welch, ACS Cent. Sci., 2016, 2, 332 CrossRef CAS PubMed.
- T. J. Wenzel and J. E. Thurston, J. Org. Chem., 2000, 65, 1243 CrossRef CAS PubMed.
- A. E. Lovely and T. J. Wenzel, Org. Lett., 2006, 8, 2823 CrossRef CAS PubMed.
- C. Zonta, A. Kolarovic, M. Mba, M. Pontini, E. P. Kundig and G. Licini, Chirality, 2011, 23, 796 CrossRef CAS PubMed.
- H. Huang, G. Bian, H. Zong, Y. Wang, S. Yang, H. Yue, L. Song and H. Fan, Org. Lett., 2016, 18, 2524 CrossRef CAS PubMed.
- S. Ito, M. Okuno and M. Asami, Org. Biomol. Chem., 2018, 16, 213 RSC.
- M. S. Seo, S. Jang and H. Kim, Chem. Commun., 2018, 54, 6804 RSC.
- S. A. Vignon, J. Wong, H. R. Tseng and J. F. Stoddart, Org. Lett., 2004, 6, 1095 CrossRef CAS PubMed.
- D. K. Bwambok, S. K. Challa, M. Lowry and I. M. Warner, Anal. Chem., 2010, 82, 5028 CrossRef CAS PubMed.
- D. Kumari, P. Bandyopadhyay and N. Suryaprakash, J. Org. Chem., 2013, 78, 2373 CrossRef CAS PubMed.
- S. Yu and L. Pu, Tetrahedron, 2015, 71, 745 CrossRef CAS.
- J. Yi, G. Du, Y. Yang, Y. Li, Y. Li and F. Guo, Tetrahedron: Asymmetry, 2016, 27, 1153 CrossRef CAS.
- G. Du, Y. Li, S. Ma, R. Wang, B. Li, F. Guo, W. Zhu and Y. Li, J. Nat. Prod., 2015, 78, 2968 CrossRef CAS PubMed.
- J. Redondo, A. Capdevila and I. Latorre, Chirality, 2009, 22, 472 Search PubMed.
- F. Yuste, R. Sánchez-Obregón, E. Díaz and M. A. García-Carrillo, Tetrahedron: Asymmetry, 2014, 25, 224 CrossRef CAS.
- S. R. Chaudhari and N. Suryaprakash, Org. Biomol. Chem., 2012, 10, 6410 RSC.
- A. Lakshmipriya, S. R. Chaudhari and N. Suryaprakash, Chem. Commun., 2015, 51, 13492 RSC.
- S. K. Mishra, S. R. Chaudhari and N. Suryaprakash, Org. Biomol. Chem., 2014, 12, 495 RSC.
- A. Rahman and X. Lin, Org. Biomol. Chem., 2018, 16, 4753 RSC.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- S. Tothadi, A. Mukherjee and G. R. Desiraju, Chem. Commun., 2011, 47, 12080 RSC.
- J. C. Antilla and W. D. Wulff, J. Am. Chem. Soc., 1999, 121, 5099 CrossRef CAS.
- G. B. Rowland, H. Zhang, E. B. Rowland, S. Chennamadhavuni, Y. Wang and J. C. Antilla, J. Am. Chem. Soc., 2005, 127, 15696 CrossRef CAS PubMed.
- Y. Liang, E. B. Rowland, G. B. Rowland, J. A. Perman and J. C. Antilla, Chem. Commun., 2007, 4477 RSC.
- M. Pérez-Trujillo, E. Monteagudo and T. Parella, Anal. Chem., 2013, 85, 10887 CrossRef PubMed.
- P. H. Menezes, S. M. C. Gonçalves, F. Hallwass, R. O. Silva, L. W. Bieber and A. M. Simas, Org. Lett., 2003, 5, 1601 CrossRef CAS PubMed.
- D. A. Tickell, M. F. Mahon, S. D. Bull and T. D. James, Org. Lett., 2013, 15, 860 CrossRef CAS PubMed.
- M. S. Silva, Molecules, 2017, 22, 274 CrossRef PubMed.
- J. Redondo, A. Capdevila and I. Latorre, Chirality, 2010, 22, 472 CAS.
- I. Pal, S. R. Chaudhari and N. Suryaprakash, New J. Chem., 2014, 38, 4908 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 1993 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- N. Lokesh, S. R. Chaudhari and N. Suryaprakash, Org. Biomol. Chem., 2014, 12, 993 RSC.
- N. Nath, P. Bordoloi, B. Barman, B. Baishya and S. R. Chaudhari, Magn. Reson. Chem., 2018, 56, 876 CrossRef CAS PubMed.
- S. R. Chaudhari and N. Suryaprakash, Chem. Phys. Lett., 2013, 555, 286 CrossRef CAS.
- M. Pérez-Trujillo, L. Castañar, E. Monteagudo, L. T. Kuhn, P. Nolis, A. Virgili, R. T. Williamson and T. Parella, Chem. Commun., 2014, 50, 10214 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional supporting data, figures, experimental and computational details. See DOI: 10.1039/c9ra07803g |
| This journal is © The Royal Society of Chemistry 2020 |