
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel method to identify group-specific non-catalytic pockets of human kinome for drug design†
Huiwen Wanga,
Zeyu Guana,
Jiadi Qiua,
Ya Jiaa,
Chen Zengab and
Yunjie Zhao *a
*a
aDepartment of Physics, Institute of Biophysics, Central China Normal University, Wuhan 430079, China. E-mail: yjzhaowh@mail.ccnu.edu.cn
bDepartment of Physics, The George Washington University, Washington DC 20052, USA
First published on 10th January 2020
Abstract
Kinase proteins have been intensively investigated as drug targets for decades because of their crucial involvement in many biological pathways. Most kinase drugs target the catalytic ATP pocket, which is highly conserved across the kinome, and as such often leads to potential side effects. It is thus highly desirable to develop non-ATP-competitive drugs that inhibit kinase activity via allosteric interactions. However, to elucidate the complex allosteric mechanism, it is essential to build a novel method to characterize a comprehensive non-catalytic pocket for the structurally well-covered human kinome. In this work, we developed a hybrid approach of sequence, structure and network analysis on 168 representative kinases to identify group-specific non-catalytic pockets. The geometric analysis was performed to cluster these pockets and to identify group-specific non-catalytic pockets based on their shape and location characteristics. Subsequent sequence evolutionary analysis reveals the crucial residues of each pocket that will likely interact with inhibitors binding to the pocket. These residues thus serve as potential biomarkers of each pocket for inhibitor design. Moreover, the residue–residue interaction network analysis was performed to elucidate the complex allosteric mechanism of these non-catalytic pockets. The final list of 14 group-specific non-catalytic pockets and their characterized structural, sequence and network features can be an enabling dataset for drug design effort at the human kinome level. The developed hybrid approach is able to identify group-specific non-catalytic pockets and will benefit the research related to human kinome drug design.
1. Introduction
Human kinase proteins are one class of the most important regulators for cellular pathways, which are associated with biological processes such as cell-cycle regulation, metabolism, differentiation and apoptosis.1,2 Aberrant kinase activity may cause a large diversity of diseases, such as cancer, psoriasis, alopecia areata, and chronic neurodegenerative disease.3 Thus, the identification of potentially druggable pockets on human kinase proteins would be essential to new kinase drug development.4,5Kinase proteins have been intensively investigated as drug targets for decades.6–8 Currently, 518 kinases encoded in the human genome9,10 are classified into eight groups (CK1, STE, CAMK, AGC, CMGC, TK, TKL, and RGC). Structurally, most of the kinase proteins share a similar topology that consists of N-terminal and C-terminal lobes.11 The N-terminal lobe, containing five β strands and at least one α C-helix, is highly conserved in the human kinome, while the C-terminal lobe, which is composed of α helix, activation loop, and substrate binding groove, shows more sequence variation.
At present, kinase inhibitors are broadly classified into two classes depending on whether an inhibitor is ATP-competitive or allosteric.12 The former is further refined as type I or II for targeting active or inactive ATP pocket, respectively, while the latter as type III or IV for targeting non-catalytic pockets near or far away from the ATP pocket, respectively. The vast majority of kinase drugs currently in clinical use are ATP-competitive inhibitors. While the pockets targeted by some allosteric inhibitors remain unknown, a few are known to target certain allosteric pockets near the ATP pocket.12,13 Volkamer et al.9 systematically analyzed the geometric characteristics of the ATP pocket in the human kinome. It is found that the geometric characteristics of the ATP pocket are highly conserved. Thus, the ATP-competitive drugs may cause undesirable side effects such as hand-foot skin reaction, hypertension and acute renal failure.14–16 Therefore, the highly selective allosteric inhibitors (type III and IV) with minimal side effects are widely needed. There are some case studies reported for allosteric inhibitors. For example, the inhibitor trametinib targets the non-catalytic pockets for MEK and BRAF kinase proteins.17 Cobimetinib is another MEK allosteric inhibitor currently in clinical trials as an anticancer agent.18 Chen et al.19 developed some peptide inhibitors binding to the allosteric pocket. The experiments show that these peptides can break the CDK2/Cyclin interface and decrease the kinase activity. Hu et al.20 identified some novel allosteric inhibitors that interrupted the interaction between CDK2 and Cyclin A3. Wylie et al.21 developed the allosteric inhibitor ABL001 experimentally. This inhibitor binds to the allosteric pocket of the ABL1 kinase. This binding leads to the C-terminal helix formation and decreases the kinase activity. However, both the number of known allosteric inhibitors and kinase targets were very limited.
It is easier to develop or screen for new drugs to target known pockets.22 Barnash et al.23 believed that target-oriented drug development not only complements a disease-focused approach but also reduces the risk of side effects. Several studies have attempted to identify specific non-catalytic pockets for drug design. For example, Chen et al.24 identified one non-catalytic pocket on CDK2 and developed corresponding peptides to inhibit CDK2 activity. Ma et al.4 proposed some potentially non-catalytic pockets in six human kinase proteins by correlation analysis between allosteric sites and catalytic sites. Given that the kinome is now well covered structurally, a comprehensive analysis of all potentially allosteric pockets may shed light on generic mechanisms of allosteric kinase inhibition.
In this article, we performed cluster analysis to identify the group-specific non-catalytic (GSNC) pockets by location distance and shape distance. The group-specific non-catalytic (GSNC) pockets are highly conserved only in one or several groups but share little shape similarity in other kinase groups. A total of 29 GSNC pockets were identified in seven groups. Then, we further clustered these GSNC pockets into 14 pockets at the kinome level. Some of the 14 pockets are shared by one or several groups. The inhibitors targeting these pockets will have minimal side effects for the diseases that involve in one or a few groups only. Moreover, we performed sequence conservation and network analysis to explain the allosteric mechanism of these GSNC pockets. The developed hybrid approach is able to identify group-specific non-catalytic pockets and will benefit the research related to human kinome drug design.
2. Results
2.1. Structural coverage of human kinases
We have built a kinase structure dataset (struKin dataset) from the human kinome. The RGC was removed due to no crystal structures in this group. Therefore, there are seven groups (AGC, CK1, STE, CAMK, CMGC, TK, TKL) containing 168 kinase structures in our dataset (struKin dataset) (see the Method section for details). The kinase family is well covered by crystal structures (Fig. 1B).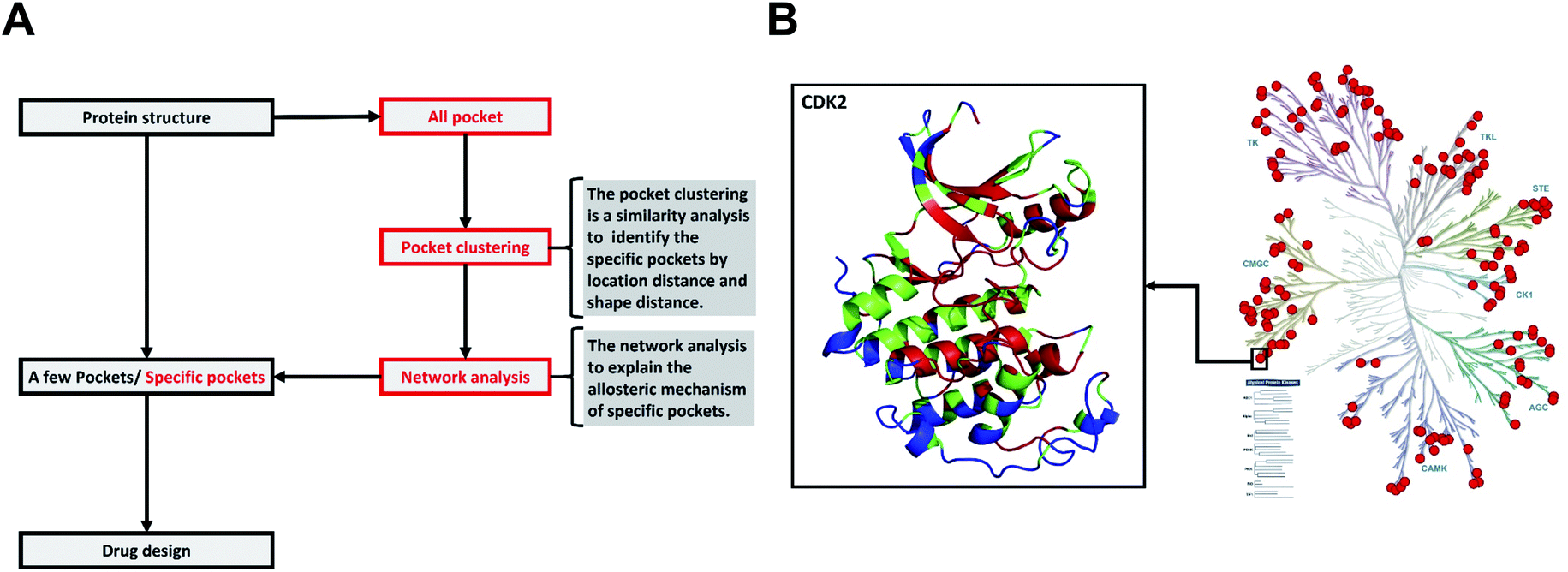 | ||
| Fig. 1 The difference between the traditional method and our method, and the distribution of 168 human kinases in struKin dataset on the kinome tree. (A) The difference between the traditional method and our method. The traditional method is to design and screen drugs directly by analyzing pockets of one or several proteins. We first obtained all pockets of a class of proteins and clustered these pockets with pocket structure features to identify specific pockets. Then the network analysis was performed to explain the allosteric mechanism of specific pockets. (B) The distribution of 168 human kinases in struKin dataset on kinome tree. The red dots represent each structure. For example, one red dot represents the CDK2 structure (PDB ID: 4ACM41). The CDK2 structure is colored in conservation scores with red (conserved residue), green (average residues), and blue (variable residues), respectively. The ATP pocket is highly conserved while T-loop, C-terminal helix, and the area below the C-helix are variable. | ||
Structurally, the N-terminal conformations including ATP pocket are highly similar in the entire human kinome. However, the C-terminal conformations are different. In addition, we also performed sequence evolutionary analysis to infer the crucial residues and projected the evolutionary scores of each residue onto the tertiary structures. The highly conserved ATP pocket (colored in red) indicates that the ATP pocket maintains the structure for biological function while some non-catalytic pockets are variable (Fig. 1B).
2.2. Signatures of the ATP pocket
To obtain the structural values, we extracted all ATP pockets from struKin dataset using DoGSiteScorer.25,26 Fig. S1† shows the volume, depth, and surface area values of all ATP pockets for 168 kinase structures. The average volume value is 626 (±206) Å3. The mid-value volumes of ATP pockets in each group are spread similarly including slightly larger (STE groups, 614 Å3), similar (CAMK groups, 600 Å3) and slightly smaller pockets (TKL groups, 593 Å3; CMGC groups, 591 Å3; TK groups, 567 Å3; AGC groups 565 Å3; CK1 groups, 542 Å3). The results agree with the previous findings reported by Volkamer et al.9 The average depth and surface area values are 17.34 (±3.58) Å and 718.89 (±239.96) Å2, respectively.In terms of sequence features, we performed an evolutionary analysis of all ATP pockets using ConSurf.27,28 The continuous conservation scores are divided into a discrete scale of 9 grades with grade 1 indicating the most variable positions and grade 9 the most conserved positions. Table S1† shows the ATP pockets have a mean value of 7.34 (±0.16). The high conservation scores and small standard deviation values suggest that ATP pockets are highly conserved. For example, there are 42 residues in the ATP pocket of MAP2K2 kinase (PDB ID: 1S9I29). We re-numbered column positions of the 42 residues from the 168 kinase sequences alignment. Fig. S2A† shows the sequence variations for ATP binding sites by analyzing 15 available kinase/ATP complex structures in struKin dataset. We divided the binding sites into hydrogen bonds and hydrophobic interactions (see Datasets S1, S2, and S3 for details†). The hydrogen bonding residues are mainly located at four positions. Two charged residues are located at positions 11(K) and 22(D/E). Another two residues are located at positions 34 and 37 (mostly N/D) form ATP interactions via magnesium or manganese ions (Fig. S3†). The hydrophobic interaction residues are mainly located at eight positions. The residues in position 1 are mainly L or I, positions 2/4 are G, position 8 is V, position 9 is A, position 21 are M, F or T, position 23 are F, L or Y, and position 35 are L or M, respectively. These residues, which interact with ATP to form hydrogen bonds and hydrophobic interactions, are mainly uniformly distributed in the three phosphate groups and adenosine regions of ATP respectively (shown in Fig. S2B†).
2.3. Group-specific non-catalytic (GSNC) pockets in different kinase groups
We collected the information related to kinase-disease associations from KinMap website.30 The KinMap website is an online tool that facilitates interactive navigation through kinase knowledge by linking biochemical, structural, and disease association data to the human kinome tree. And the kinase-disease associations are from the Center for Therapeutic Target Validation (CTTV) platform.30 To explain the kinase-related side effects, we selected four kinds of diseases: cancer, lymphoblastic, brain disease, and endometriosis. Cancer and lymphoblastic leukemia involve in most kinase groups (Fig. S4A and B†). However, brain disease and endometriosis only involve in one or a few groups (Fig. S4C and D†). Thus, identifying group-specific non-catalytic pockets for one or several groups but not for the entire kinome would be helpful to reduce potential drug's side effects in treatment.The group-specific non-catalytic (GSNC) pockets were identified by pocket location and shape similarity. We first aligned kinase structures to a reference structure for each group. Then, we detected all pockets using DoGSiteScorer.25,26 The group-specific non-catalytic pockets (GSNC pockets) were clustered by the following criterion.
(a) Location distance (LD) of 8 Å between geometric centers of two pockets was used to quantify the position similarity of two pockets.
(b) Shape distance (SD) of 2.5 was used to measure the shape similarity of two pockets. The volume, depth, and surface values in SD are able to help screen the drug size, length, and interaction groups, respectively.
(c) The coverage rate of the similar pocket for each group is greater than 80%.31
Thus, a total of 29 typical GSNC pockets and 49 non-GSNC pockets (the coverage rates of the similar pockets for a given group are not greater than 80%.) were identified as listed in Table 1. And the 29 typical GSNC pockets were visualized as shown in Fig. 2.
| Group | Kinase | PDB ID | GSNC pockets | Non-GSNC pockets |
|---|---|---|---|---|
| CMGC | CLK1 | 1Z57 (ref. 56) | p2, p3, p4, p6, p7 | p1, p5, p8 |
| AGC | AKT1 | 4GV1 (ref. 59) | p1, p3, p4, p5, p8 | p2, p6, p7, p9, p10, p11, p12, p13, p14 |
| TKL | PIPK2 | 5J7B60 | p1, p3, p5 | p2, p4, p6, p7, p8, p9, p10, p11 |
| TK | JAK1 | 3EYG58 | p1, p2, p3, p6, p8 | p4, p5, p7, p9 |
| CAMK | CaMK1α | 4FG8 (ref. 61) | p1, p2, p3 | p4, p5, p6, p7, p8, p9, p10, p11 |
| STE | MST3 | 3A7I62 | p5, p9 | p1, p2, p3, p4, p6, p7, p8, p10, p11, p12, p13 |
| CK1 | CK1α | 5FQD63 | p1, p2, p3, p6, p7, p12 | p4, p5, p8, p9, p10, p11 |
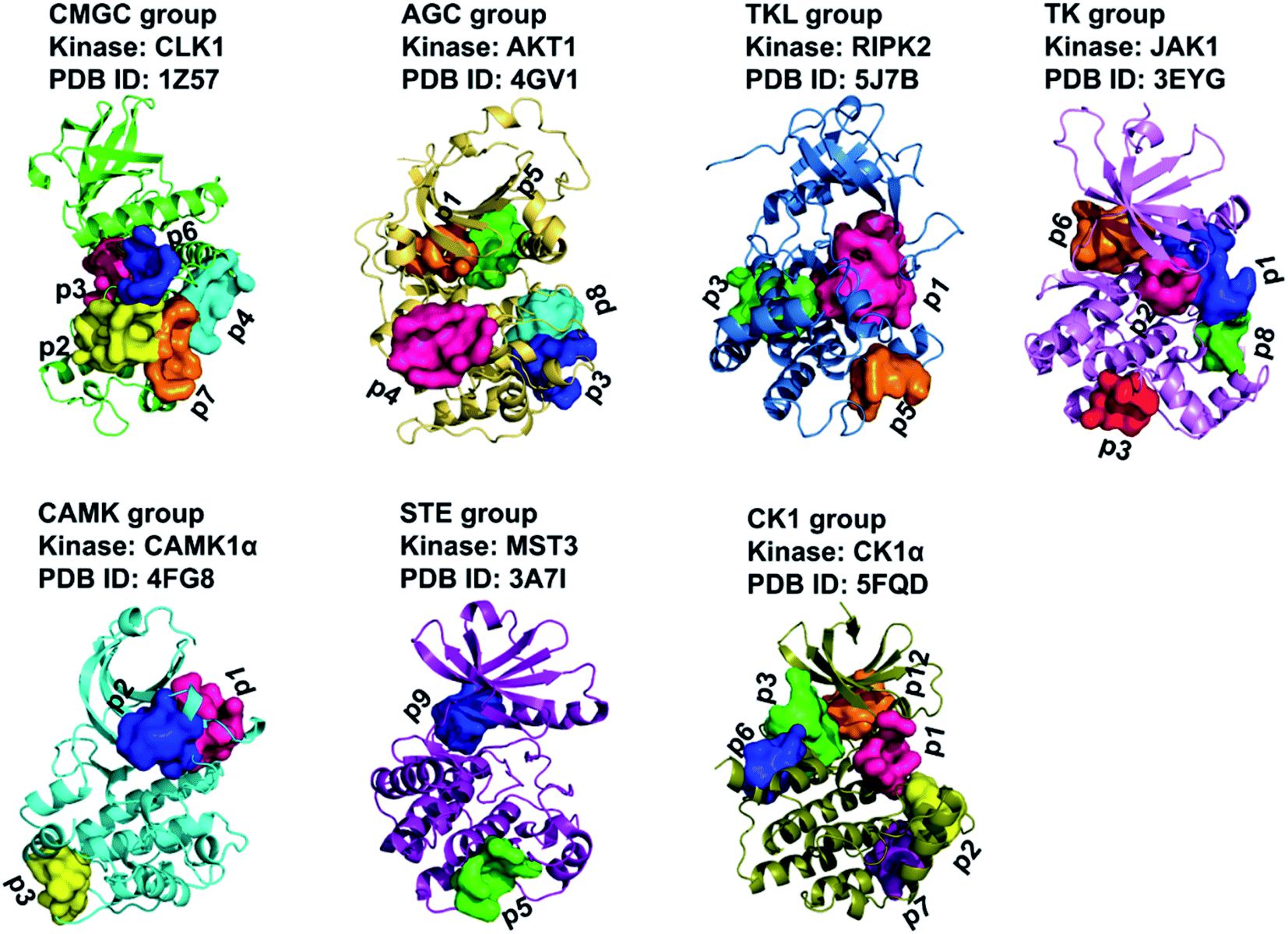 | ||
| Fig. 2 A total of 29 group-specific non-catalytic (GSNC) pockets in seven kinase groups. The kinase structures and identified GSNC pockets are shown as cartoon and surface, respectively. | ||
2.4. Typical shape characteristics of GSNC pockets
To obtain the shape features for GSNC pockets, we analyzed the volume, depth, and surface area values. Volume is the most important characterization in QSAR calculation for drug design.32 As in the lock and key model,33,34 a drug will not bind to a pocket if the drug cannot physically fit within the size of the pocket. The result shows that the average volume value of 29 GSNC pockets is 205 (±124) Å3 (Fig. 3A). Depth of pocket plays an important role in drug design. If the depth of the drug is greater or less than that of the pocket, the drug cannot be firmly integrated with the pocket. The result shows that the average depth value of 29 GSNC pockets is about 10.0 (±3.9) Å (Fig. 3B). When the size of the drug matches the volume and depth of the pocket, surface area value is able to help define the interaction types for drug design. The average surface area value of 29 GSNC pockets is about 368 (±185) Å2 (Fig. 3C).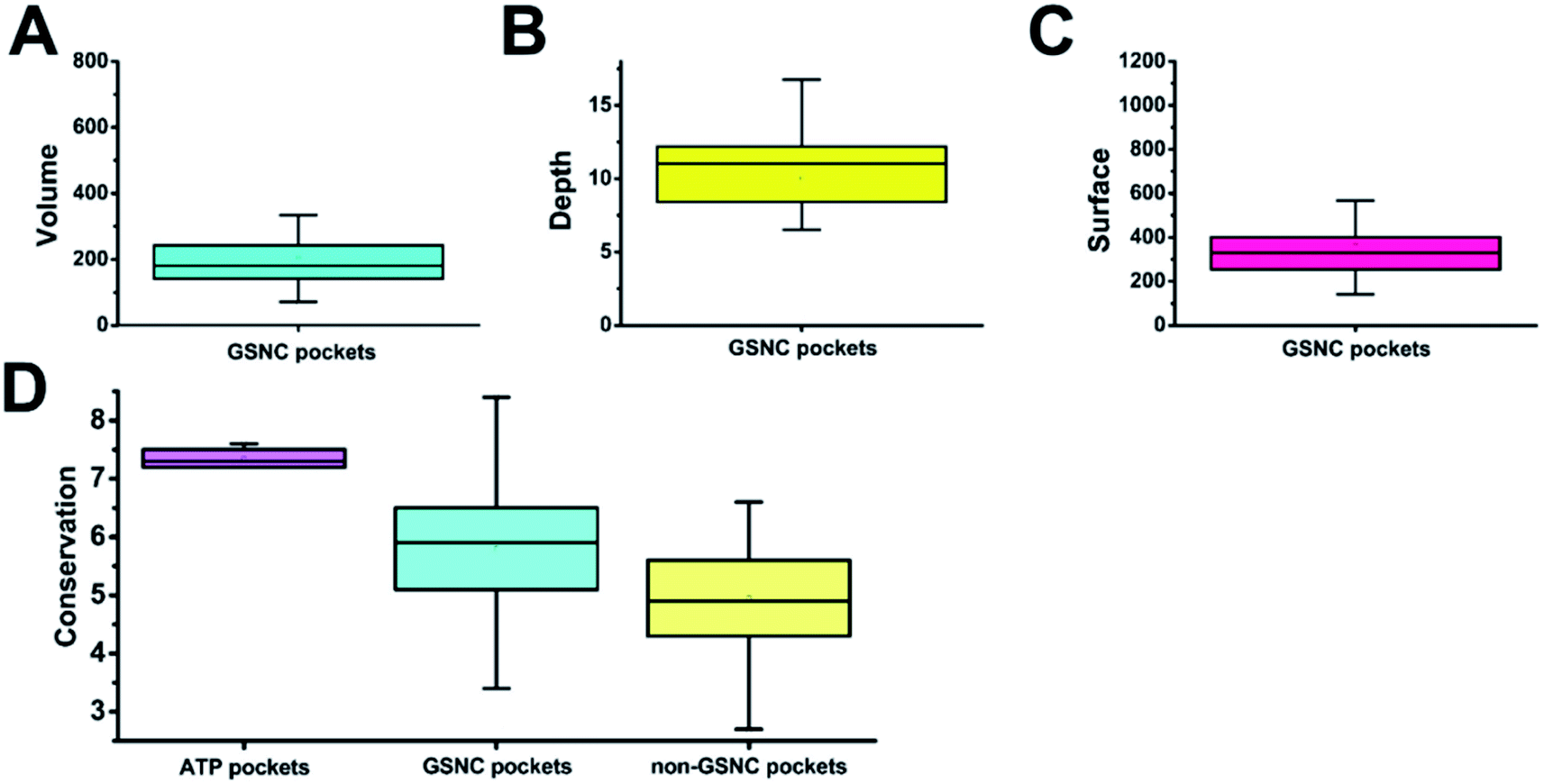 | ||
| Fig. 3 The geometric structure characteristics of 29 group-specific non-catalytic (GSNC) pockets and conservation analysis for all pockets. The average of volume (A), depth (B), and surface area (C) values of 29 GSNC pockets are 205 (±124) Å3, 10.0 (±3.9) Å and 368 (±185) Å2, respectively. (D) The average conservation scores of ATP pockets, GSNC pockets, and non-GSNC pockets are 7.34 ± 0.16, 5.79 ± 1.24 and 4.95 ± 1.13, respectively. | ||
Unlike the large ATP-binding pockets with volume around 600 Å3, the identified GSNC pockets are smaller with volume around 200 Å3. Previous research reported that the small non-catalytic pockets may act as allosteric sites for kinase inhibition. For example, Ma et al.4 identified 13 non-catalytic pockets in 6 human kinases (CDK2, CK2, Chk1, MAP14, MAP8, and c-Abl). The average volume, surface area and depth values of these 13 non-catalytic pockets are 263.27 (±110.58) Å3, 414.64 (±148.25) Å2 and 10.23 (±4.75) Å (Table S2†). Another experiment performed by Comess et al. demonstrated a small non-catalytic pocket (volume = 140.67 Å3, surface area = 182.60 Å2, depth = 8.59 Å) in JNK1α1 is able to bind an inhibitor (PDB ID: 3O2M35). This pocket is located at the same position as p2 of CLK1 kinase in the CMGC group. In addition, some allosteric inhibitors (such as PDB ID: 4M12 (ref. 36)) were developed to target a non-catalytic pocket of ITK kinase. This pocket is located at the same position as p6 (volume = 158.85 Å3, surface area = 334.14 Å2, depth = 11.79 Å) of JAK1 in TK group. Taken together, these results show that the identified GSNC pockets may act as allosteric sites for inhibitor binding.
2.5. Sequence evolutionary analysis of GSNC pockets
Capra et al.37 showed that tertiary structural information combined with sequence evolutionary characteristics are able to predict ligand-binding sites. Therefore, we also analyzed the sequence variations of the GSNC pockets.First, we compared the sequence conservation scores of identified GSNC pockets and non-GSNC pockets on the protein surface (Fig. 3D and Table S1†). The identified GSNC pockets (average conservation score = 5.79 ± 1.24) are less conserved than the ATP pockets, but more conserved than non-GSNC pockets (average conservation score = 4.95 ± 1.13). These results suggest that there may be some more conserved residues within each GSNC pocket that can serve as the biomarkers for the GSNC pockets.
Then, we identified the crucial residues for different GSNC pockets using WebLogo.38,39 For example, p7 (TL) pocket from CLK1 kinase in CMGC group is able to accommodate a new class of inhibitors distinct from the traditional ATP-competitive inhibitors. We performed sequence variation analysis of this pocket using all kinase sequences (Fig. 4A) and CMGC group sequences (Fig. 4B). Residue Tyr180 (position 4) is highly conserved in CMGC group but shows variation in the entire kinome. This result indicates that Tyr180 may be a crucial residue for p7 (TL) pocket of CLK1 kinase in CMGC group. This observation agrees with previous experiment.24 In addition, Yang et al.40 demonstrated that the Glu–Arg pair serves as a center hub of connectivity between these two structurally conserved elements in EPKs. Mutations of either residue would disrupt communication between the two segments as well as the rest of the protein, leading to altered catalytic activity and enzyme regulation. Residue Pro271 (position 12) in CDK2 (PDB ID: 4ACM41) shields the Glu–Arg ion pair from solvent, which suggests that Pro271 (position 12) may also be a crucial residue served as biomarker for the p7 (TL) pocket of CLK1 kinase in CMGC group.
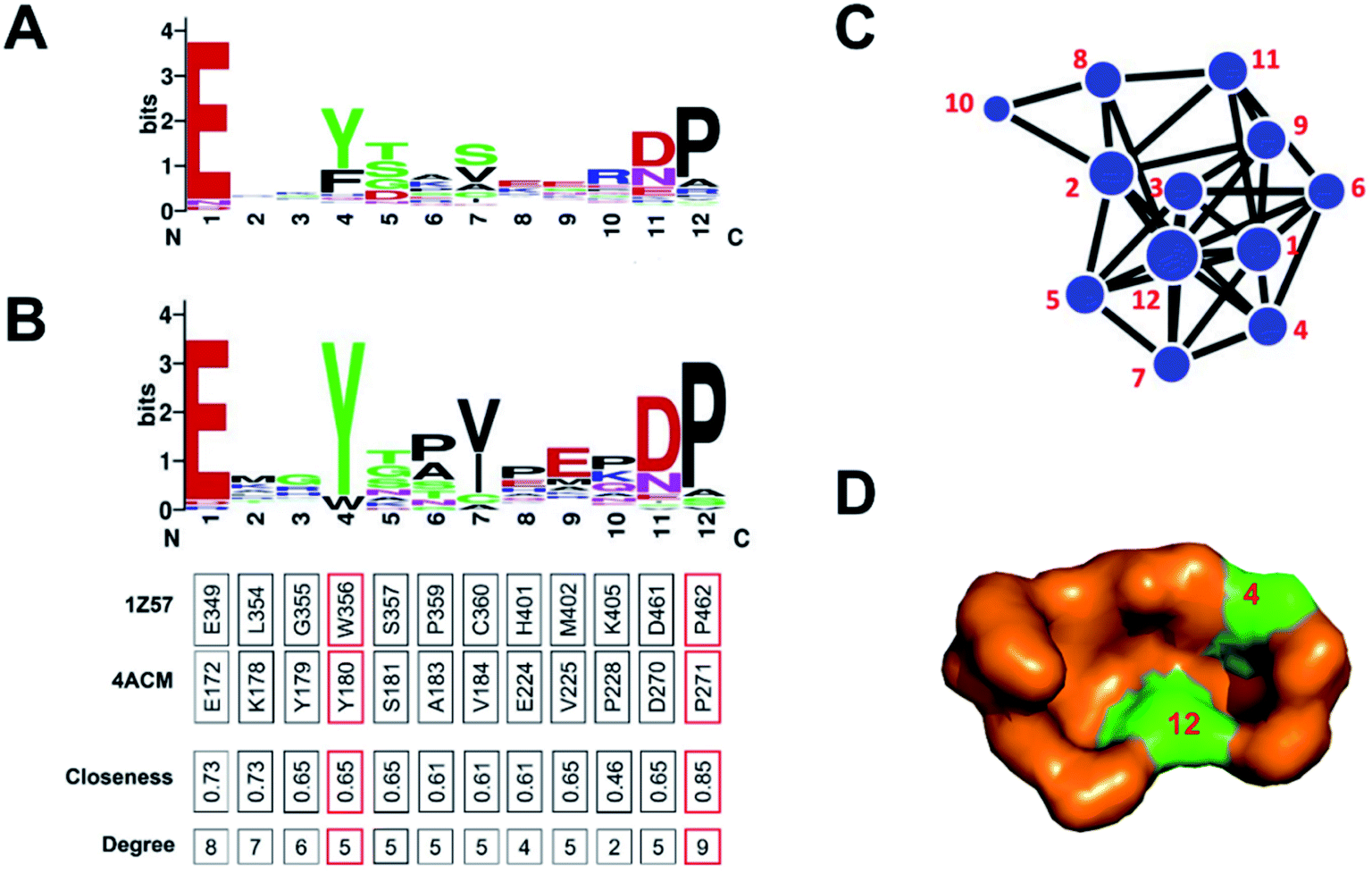 | ||
| Fig. 4 Sequence variation analysis of p7 (TL) pocket from CLK1 kinase (PDB ID: 1Z57 (ref. 56)) in the CMGC group. The differences between (A) kinome level and (B) CMGC group level shows specificity. The network representation (C) and surface model (D) of p7 pocket indicate that Tyr180 (position 4) and Pro271 (position 12) may be the crucial residues for p7 (TL) pocket from CLK1 kinase in the CMGC group. This observation agrees with previous experiments.24,40 | ||
2.6. Network analysis of GSNC pockets and experimental verification
In a connected network, the closeness of a node is defined as the inverse of the sum of its shortest distances to all other nodes (see to the section Methods). Previous researches suggested that closeness analysis is able to identify critical residues for binding.42,43 Indeed, benchmark tests showed that the closeness values successfully identified 70% of the protein binding sites.43 Thus, we performed a closeness analysis to infer binding pockets. The pocket closeness is defined by the average closeness of all residues in the pocket. The identified closeness value of GSNC pockets (0.35 ± 0.02) is smaller than ATP pockets (0.37 ± 0.01) but larger than non-GSNC pockets (0.33 ± 0.03) (Table 2 and Fig. 5A). These results suggest that there may be a small portion of residues in the GSNC pockets that are highly conserved. These crucial residues can thus serve as biomarkers of GSNC pockets.| Group | Reference kinase | PDB ID | ATP pocket | GSNC pocket's ranking | Non-GSNC pocket's ranking |
|---|---|---|---|---|---|
| CMGC | CLK1 | 1Z57 | p0 (0.350) | p3(0.371) > p4(0.339) = p6(0.339) > p2(0.321) > p7(0.320) | p1(0.333) > p8(0.290) > p5(0.272) |
| AGC | AKT1 | 4GV1 | p0(0.360) | p1(0.371) > p5(0.367) > p4(0.350) > p8(0.331) > p3(0.313) | p12(0.363) > p13(0.360) > p7(0.347) > p10(0.310) > p6(0.308) > p14(0.307) > p11(0.299) > p9(0.296) > p2(0.285) |
| TKL | PIPK2 | 5J7B | p0(0.362) | p1(0.368) > p3(0.356) > p5(0.325) | p8(0.351) > p11(0.350) > p7(0.346) p4(0.343) > p10(0.335) > p9(0.325) p2(0.322) > p6(0.291) |
| TK | JAK1 | 3EYG | p0(0.374) | p2(0.408) > p1(0.370) > p6(0.369) > p8(0.356) > p3(0.324) | p4(0.343) > p7(0.319) > p9(0.315) > p5(0.275) |
| CAMK | CAMK1α | 4FG8 | p0(0.372) | p2(0.357) > p1(0.346) > p3(0.325) | p5(0.375) > p7(0.371) > p6(0.367) > p10(0.362) > p4(0.356) > p8(0.336) > p9(0.328) > p11(0.298) |
| STE | MST3 | 3A7I | p0(0.375) | p9(0.375) > p5(0.326) | p12(0.395) > p8(0.371) > p4(0.367) > p7(0.347) > p1(0.332) > p13(0.331) > p3(0.328) > p10(0.324) > p6(0.323) > p11(0.315) > p2(0.301) |
| CK1 | CK1α | 5FQD | p0(0.364) | p1(0.386) > p7(0.356) > p3(0.351) > p2(0.347) > p6(0.344) > p12(0.339) | p10(0.391) > p11(0.382) > p4(0.353) > p8(0.349) > p5(0.323) > p9(0.318) |
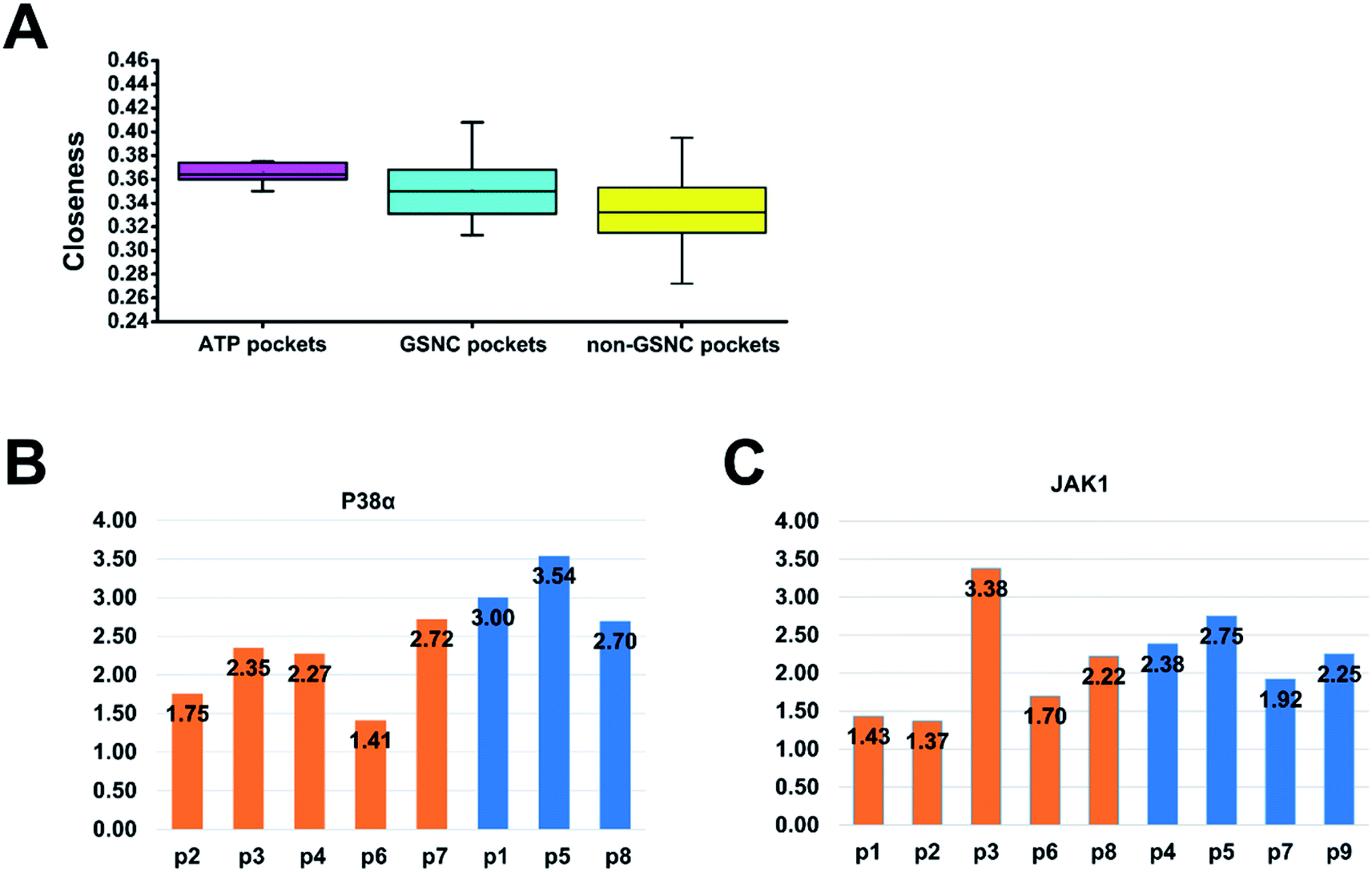 | ||
| Fig. 5 The network (closeness and shortest paths) analysis of pockets. (A) The pocket closeness is defined by the average closeness of all residues in the pocket. The results show that the average closeness values of ATP pockets, group-specific non-catalytic (GSNC) pockets, and non-GSNC pockets are 0.37 ± 0.01, 0.35 ± 0.02 and 0.33 ± 0.03 respectively. (B and C) The average shortest paths of the GSNC pockets (orange) and the non-GSNC pockets (blue) to Asp–Phe–Gly (DFG) residues. A large conformational change for DFG residues at the N terminus of the activation segment determines whether the kinase is active or inactive.45 The shortest path of one pocket to a residue is the average shortest path of all residues in the pocket to the residue. (B) For p38α kinases, the results indicate that the average shortest path of GSNC pockets (2.10 ± 0.52) is smaller than those of non-GSNC pockets (3.08 ± 0.43), and the shortest paths of 5 GSNC pockets to DFG residues are ranked as the following: p6 < p2 < p4 < p3 < p7. (C) For JAK1 kinase, the results indicate that the average shortest path of GSNC pockets to DFG residues (2.02 ± 0.83) is less than those of non-GSNC pockets (2.33 ± 0.34), and the average shortest path of 5 GSNC pockets to DFG residues are ranked as the following: p2 < p1 < p6 < p8 < p3. | ||
We hypothesize that the closeness analysis is able to qualitatively identify the non-catalytic pockets, and more suitable for drug design. To do this, we constructed the protein network from the kinase crystal structures, and then computed the closeness values of all surface residues (without ATP pockets) and classified the pockets into three categories: (1) most likely drug binding pockets (high closeness values), (2) likely drug binding pockets (intermediate closeness values), and (3) unlikely drug binding pockets (small closeness values). Fig. 6 shows the top 10 high closeness residues on kinase surface for most likely drug binding pockets identification with experiment validations. The residues colored in red, purple and green are located at inhibitor binding GSNC pockets with experimental validation, GSNC pockets without inhibitor, and surface residues, respectively. For example, for the p38α kinase, nine high closeness residues are located at four GSNC pockets (p2, p3, p4 and p6 from CLK1 kinase in the CMGC group) as shown in Fig. 6A. Five of the nine high closeness residues are located at p2 pocket from CLK1 kinase in CMGC group that are able to bind a molecule (molecule name: 46A; Kd = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM; PDB ID: 3O2M35). Fig. 6B shows another example in the TK group. Seven high closeness residues are located at four GSNC pockets (p1, p2, p6 and p8 from JAK1 kinase of TK group). Two out of the seven high closeness residues are located at p2 pocket from JAK1 kinase in the TK group which are able to bind molecule (molecule name: 0O7; IC50 = 4200 nM; PDB ID: 4EBV44). Two of the seven high closeness residues are able to bind a molecule (molecule name: 1YZ; Kd = 900 nM; PDB ID: 4M12 (ref. 36)) located at p6 pocket from JAK1 kinase in TK group. The results suggest that the closeness analysis is able to qualitatively identify the useful GSNC pockets for drug design. Other GSNC pockets with high closeness residues are potential binding pockets.
000 nM; PDB ID: 3O2M35). Fig. 6B shows another example in the TK group. Seven high closeness residues are located at four GSNC pockets (p1, p2, p6 and p8 from JAK1 kinase of TK group). Two out of the seven high closeness residues are located at p2 pocket from JAK1 kinase in the TK group which are able to bind molecule (molecule name: 0O7; IC50 = 4200 nM; PDB ID: 4EBV44). Two of the seven high closeness residues are able to bind a molecule (molecule name: 1YZ; Kd = 900 nM; PDB ID: 4M12 (ref. 36)) located at p6 pocket from JAK1 kinase in TK group. The results suggest that the closeness analysis is able to qualitatively identify the useful GSNC pockets for drug design. Other GSNC pockets with high closeness residues are potential binding pockets.
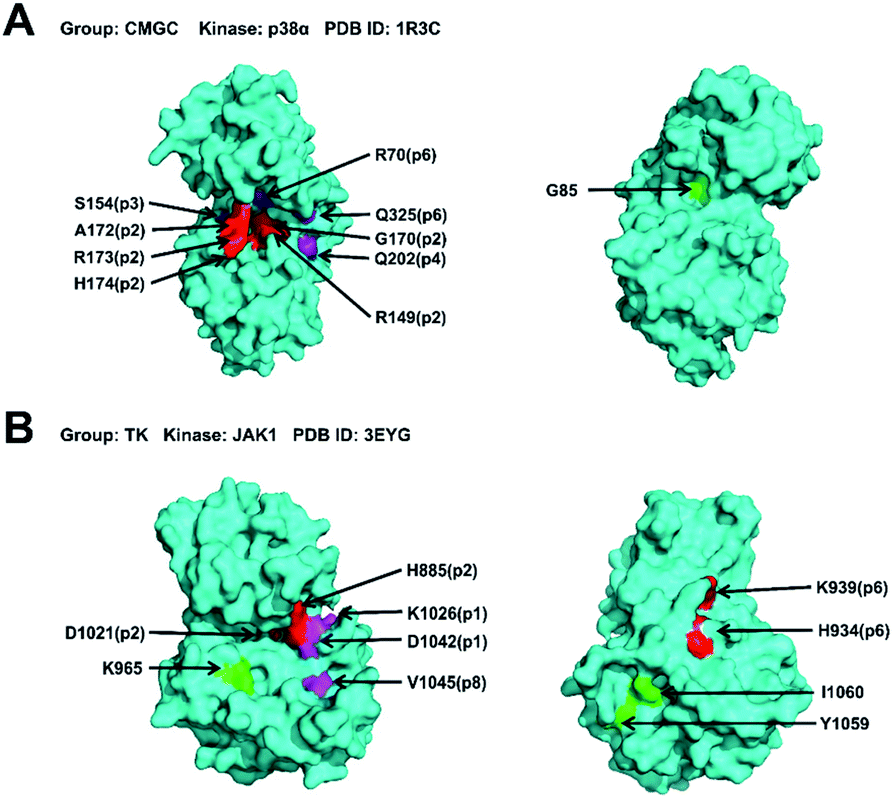 | ||
| Fig. 6 The top 10 high closeness residues on kinase surface from most likely drug binding pockets. The residues colored in red, purple and green are located at inhibitor binding GSNC pockets with experimental validation, GSNC pockets without inhibitor, and surface residues, respectively. (A) For the p38α kinase (PDB ID: 1R3C57), the nine high closeness residues are located at four GSNC pockets (p2, p3, p4 and p6 from CLK1 kinase in CMGC group). Five out of the nine high closeness residues are located at p2 pocket from CLK1 kinase in CMGC group which are able to bind molecule (molecule name: 46A; Kd = 16000 nM; PDB ID: 3O2M35). (B) Another example in the TK group. Seven high closeness residues are located at the four GSNC pockets (p1, p2, p6, and p8 from JAK1 kinase (PDB ID: 3EYG58)) of the TK group. Two out of the seven high closeness residues are located at p2 pocket from JAK1 kinase in the TK group which are able to bind molecule (molecule name: 0O7; IC50 = 4200 nM; PDB ID: 4EBV44). And two out of the seven high closeness residues are located at p6 pocket from JAK1 kinase in the TK group which are able to bind molecule (molecule name: 1YZ; Kd = 900 nM; PDB ID: 4M12 (ref. 36)). The results suggest that the closeness analysis is able to qualitatively identify the useful non-catalytic pockets for drug design. Other non-catalytic pockets with high closeness residues are potential druggable binding pockets. | ||
2.7. Quantitative analysis of the druggable pocket
The druggability calculation predicts the pocket druggability by analyzing the pocket topology characteristics (volume, surface, depth) and protein structure similarity.25,26 The druggability results of ATP, GSNC, and non-GSNC pockets are 0.59 ± 0.15, 0.34 ± 0.16, and 0.34 ± 0.18, respectively (Table S3†). The results show that the druggability model can distinguish the ATP and non-ATP pockets but cannot distinguish the GSNC and non-GSNC pockets. To further detail analyze, we collected all the available small molecules binding to allosteric sites (Table S4†). None of the highest druggability pockets have available small molecules. The druggability rankings are 11th out of 14 pockets in the AGC group, 3rd and 9th out of 9 pockets in the TK group, 2nd and 3rd out of 8 pockets in the CMGC group. Therefore, it is difficult to predict the very druggable pocket using druggability calculation.The closeness calculation predicts the druggable pocket by average the closeness values of all residues to the corresponding pocket. The previous conclusion shows that the closeness of GSNC pockets (0.35 ± 0.02) is smaller than ATP pockets (0.37 ± 0.01) but larger than non-GSNC pockets (0.33 ± 0.03) (Table 2 and Fig. 5A). The closeness rankings are 4th out of 14 pockets in the AGC group, 1st and 3rd out of 9 pockets in the TK group, 5th and 6th out of 8 pockets in the CMGC group. The performance of closeness calculation is better than druggability calculation. These two druggable pocket prediction strategies will provide guidance for people working in the field.
3. Discussion
The kinome is now well covered by tertiary structures, making it possible to identify potential allosteric binding pockets to reduce the side effect. However, a systematic study of non-catalytic pockets at the kinome scale has not been performed. There are only some case studies on the detection of allosteric binding pockets. In this article, we systematically analyzed the pockets of the entire human kinome by clustering pockets.A large conformational change for Asp–Phe–Gly (DFG) residues at the N terminus of the activation segment determines whether the kinase is active or inactive.45 Thus, we want to calculate the correlations between all non-catalytic pockets and the DFG residues. Based on the protein network, we calculated the average shortest paths between all non-catalytic pockets and DFG residues to quantify their correlation for p38α and JAK1 kinases. The shortest path of one pocket to a residue is the average shortest path of all residues in the pocket to the residue. The shorter the average path of the pocket to DFG residues, the stronger the ability of the pocket to regulate DFG residues. For p38α kinases (Fig. 5B), the average shortest path of GSNC pockets to DFG residues (2.10 ± 0.52) is less than that of non-GSNC pockets (3.08 ± 0.43). And the average shortest path of p2 pocket, to which a molecule binds (molecule name: 64A; Kd = 16000 nM; PDB ID: 3O2M35), to DFG residues is 1.75. The shortest paths of 5 GSNC pockets to DFG residues are ranked as follows p6 < p2 < p4 < p3 < p7. The results indicate that GSNC pockets are more likely to regulate DFG residues than non-GSNC pockets. For JAK1 kinase (Fig. 5C), the average shortest path of GSNC pockets to DFG residues (2.02 ± 0.83) is less than that of non-GSNC pockets (2.33 ± 0.34). And the average shortest path of p2 pocket, to which a molecule binds (molecule name: 0O7; IC50 = 4200 nM; PDB ID: 4EBV44), to DFG residues is 1.37. The average shortest paths of 5 GSNC pockets to DFG residues are ranked as follows p2 < p1 < p6 < p8 < p3. Again, the results indicate that GSNC pockets are more likely to regulate DFG residues than non-GSNC pockets.
We further clustered the 29 GSNC pockets at the kinome level using cutoffs of LD = 8 Å and SD = 2.5. Finally, a total of 14 GSNC pockets (GSNCp1 to GSNCp14 pockets) were identified in the entire human kinome as shown in Table S5† and Fig. 7. GSNCp1 pocket is shared by four groups (CMGC, CK1, TK, and TKL). The results show that drugs targeting this pocket may regulate the kinase activities of these four groups. Similarly, three pockets (GSNCp2, GSNCp3, and GSNCp4) are shared by three groups (CMGC, TKL, AGC; CMGC, CK1, AGC; TK, CK1, CAMK), six pockets (GSNCp5 to GSNCp10) are shared by two groups (CMGC, AGC; TK, STE; TK, AGC; CMGC, TK; STE, CK1; AGC, CK1), four pockets (GSNCp11 to GSNCp14) are shared by only one group (CK1; TKL; CAMK; CAMK), respectively. The similarity analysis would elucidate the drug effects and side effects for different GSNC pockets. For example, the GSNCp8 pocket is shared by CMGC and TK groups, and as such, drugs targeting GSNCp8 pocket for treating brain disease (Fig. S4C†) will induce fewer side effects in comparison with ATP-competitive drugs. Similarly, because the GSNCp12 pocket is shared within the TKL group only, drugs targeting GSNCp12 pocket for treating endometriosis (Fig. S4D†), will likely have minimal side effects.
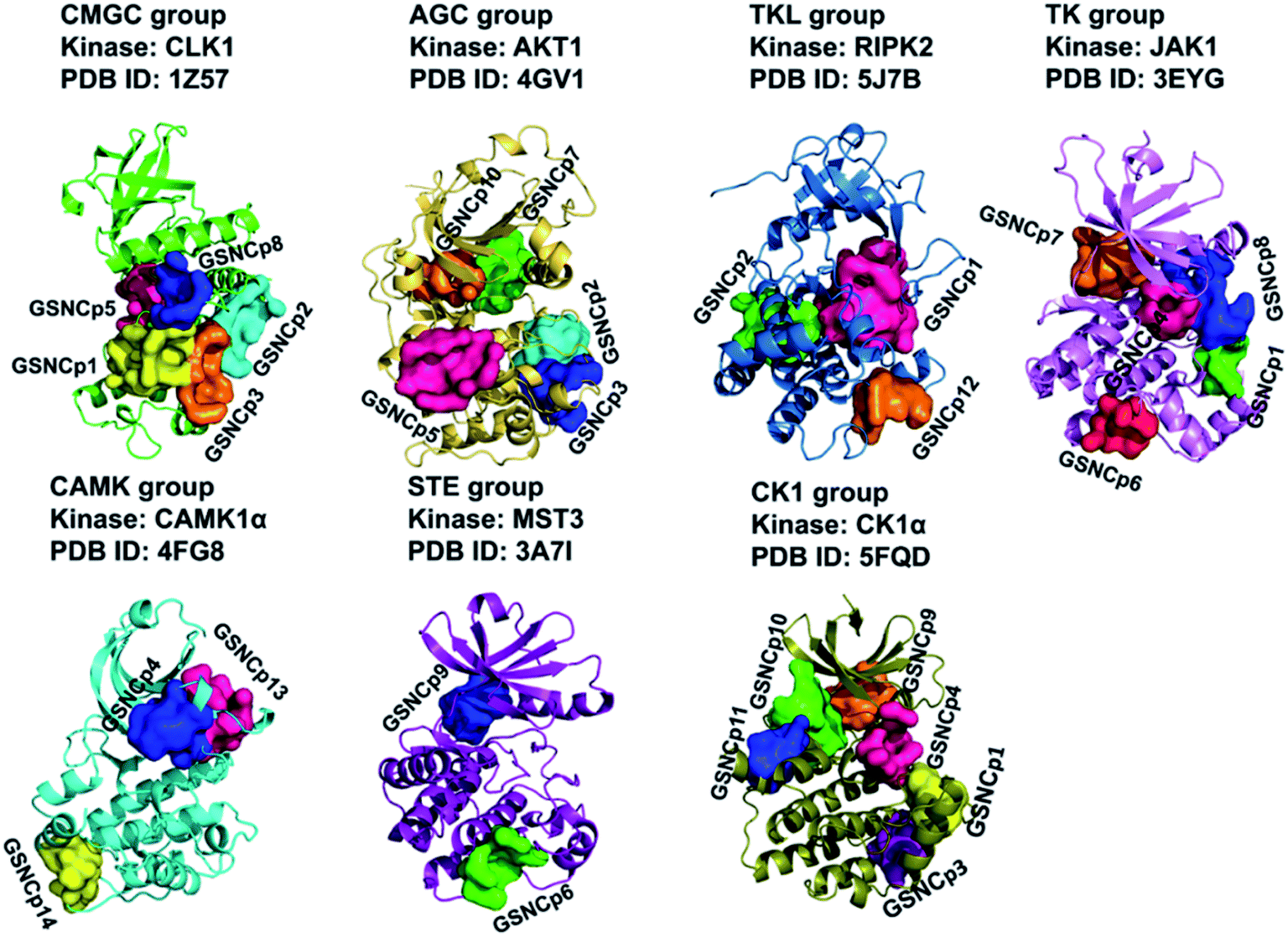 | ||
| Fig. 7 The 14 group-specific non-catalytic (GSNC) pockets (GSNCp1 to GSNCp14 pockets) were identified in the entire human kinome level. The kinase structure and identified GSNC pockets are shown as cartoon and surface, respectively. | ||
To visualize the interaction network, we constructed a force-directed graph from crystal structure using D3.JS (A JavaScript library for producing dynamic, interactive data visualizations in web browsers).46 The users can drag the nodes to achieve a dynamical effect. For example, we analyzed the p7 pocket from CLK1 kinase in the CMGC group using the protein network. The result shows that the two residues on positions 4, 12 are located at the center of the network with high closeness and degree values (Fig. 4C). These two residues are also highly conserved in the CMGC group (Fig. 4B). The result shows that the two residues can be the critical binding residues for inhibitor design. The locations of the two residues are visualized in Fig. 4D. Similarly, we analyzed other GSNC pockets in the CMGC group. The critical residues for these GSNC pockets are listed in Table S6 and Fig. S5–S8.†
4. Methods
As shown in Fig. 1A, the traditional method for identification of potential drug binding-pockets is to screen and evaluate protein cavities. However, the specificity of the pockets remains unknown and therefore leads to potential side effects. In our work, we first identify and compare the geometry similarity of all kinome pockets to provide group-specific information. Then, we further performed network analysis to elucidate the complex allosteric mechanism for non-catalytic pockets.4.1. Non-redundant human kinase structures
The human kinome dataset contains a total of 518 kinases (Dataset S4.xls). The non-redundant human kinase structures (structure of the highest resolution was selected if there are several experimentally determined structures for a given kinase.) were extracted from the human kinome dataset Kinome Render2 as follows.(1) Structures in the PDB database47 were extracted using the kinome UniProt ID (Dataset S4.xls).
(2) Structures of less than 250 residues were removed because 90% of kinases have more than 250 residues (Hanks and Hunter, 1995).
(3) Structures of low resolution (>4 Å)48 were removed.
(4) The structure of the highest resolution was selected if there are several experimentally determined structures for a given kinase.
(5) All structures were optimized using the template-based structure modeling tool SWISS-MODEL49 to fill in the missing atoms.
This finally resulted in 168 structures contained in the kinase structure dataset (struKin dataset, Fig. 1B).
4.2. Pocket detection and classification
Fig. S9† shows the workflow of pocket detection and classification.(1) For each of the seven groups (Table 1), a typical kinase structure was randomly selected as the reference structure to which all other structures in that group were aligned using PyMOL (http://www.pymol.org).
(2) All pockets of a given kinase structure were detected and calculated using DoGSiteScorer.25,26 DoGSiteScorer is an active site identification program that identifies all pockets on the surface of a given protein structure.
(3) The reference ATP pocket was extracted from the CDK2/ATP structure (PDB ID: 1FIN50).
(4) To classify the non-catalytic pockets, we defined two similarity measures as location distance (LD) and shape distance (SD) since similar pockets should share a similar location and shape for the aligned kinases. The location distance (LD) calculates the separation of the geometric centers of two pockets with a low value indicating a similar position.
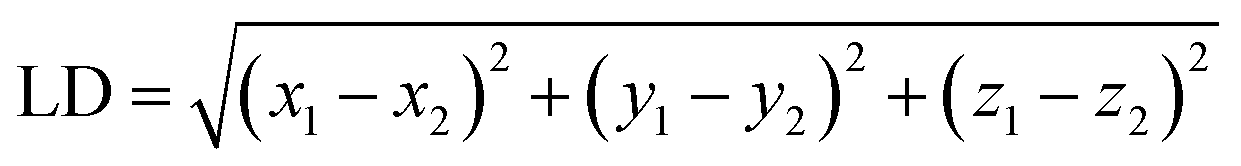 | (1) |
The shape distance computes the similarity of volumes, surfaces, and depths of two pockets with a low value indicating similar tertiary shapes.
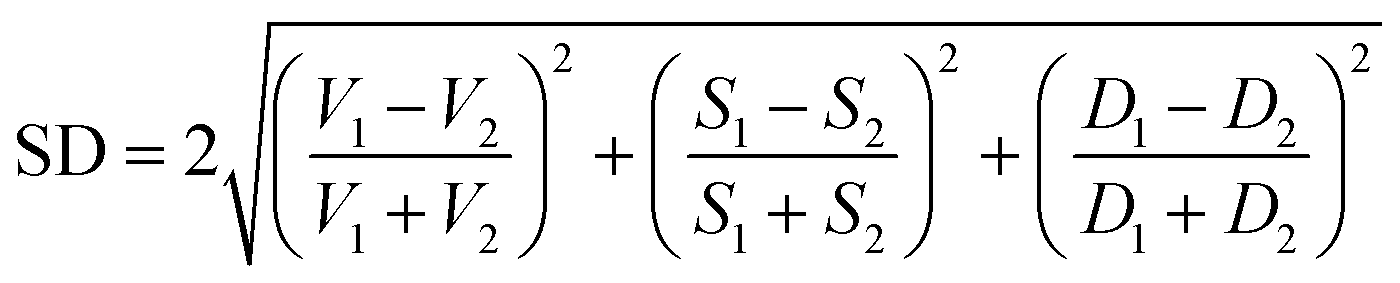 | (2) |
(5) Bandyopadhyay et al.31 inferred structure-based function using protein family-specific fingerprints that were defined as those subgraphs found in at least 80% of the family. Thus, we define the group shared non-catalytic pocket (GSNC pocket for short) if more than 80% kinases of one group have a shared pocket.
4.3. Sequence conservation analysis
The sequence conservation analysis was performed to infer the critical residues for pocket structure and function. The multiple sequence alignment of 168 kinase sequences was obtained using MAFFT.52 The evolutionary conservation scores were calculated using the ConSurf.27,28 The pocket conservation is defined by average scores of all residues in the pocket. The LigPlot53,54 was used to identify the hydrogen bonds and hydrophobic interactions between ATP and residues in ATP pocket. The conservation was visualized by WebLogo.38,394.4. Network analysis
We performed a closeness analysis to identify drug-binding pocket based on our previous work.13,42 First, a given kinase was transformed into a connected network. A node in the network denotes a single residue of the kinase. Two nonconsecutive residues in a sequence are connected by an edge if they contain a pair of heavy atoms, one from each residue, less than 8 Å apart. Second, the closeness value of each node in PDB structures of human kinase's network was calculated to identify the drug-binding sites. In the construct of the network, the closeness of a node is defined as the inverse of the sum of its shortest distances to all other (n − 1) nodes as the following:
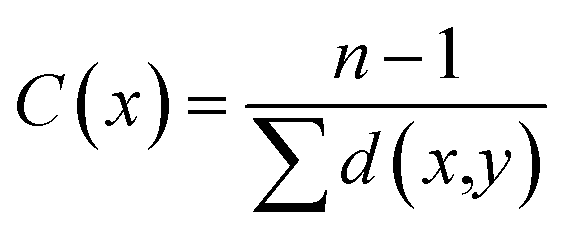 | (3) |
In addition, the correlations between non-catalytic pockets and the DFG (Asp–Phe–Gly) residues were calculated. For a given pocket, the correlation between the pocket and a residue is defined as the average shortest path of all residues in the pocket to the residue. Thus, the correlations between a given pocket and the DFG residues is defined as the average shortest path of all residues in the pocket to the three residues (DFG) as the following:
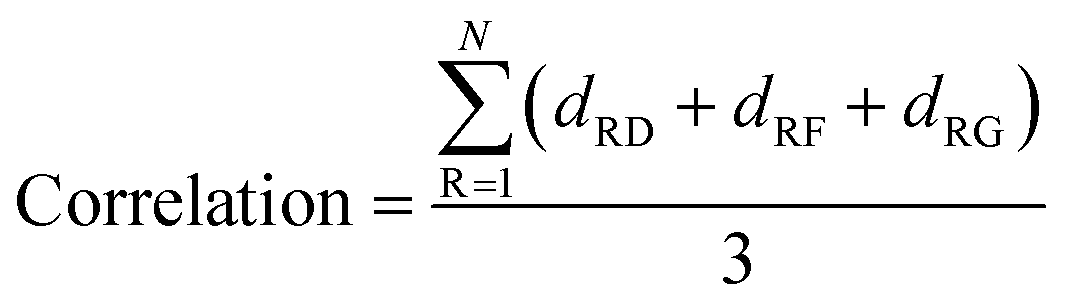 | (4) |
5. Conclusions
In summary, we systematically analyzed the pockets of the structurally well-covered kinome to define the group-specific non-catalytic pockets for designing the specific drugs. We also proposed a practical hybrid approach of sequence, structure and network analysis to pinpoint the druggable non-catalytic pockets and the corresponding key residues of each pocket that may interact strongly with inhibitors targeting the pocket. In addition, the network analysis was performed to elucidate the complex allosteric mechanism. This system analysis method for pockets of a class of proteins and the features of the 14 non-catalytic pockets will benefit the research related to human kinase drug design.Author contributions
H. W. performed most computational analysis under the supervision of Y. Z., Y. J., and C. Z.; Z. G. developed the network visualization tool; Z. G. and J. Q. helped with pocket analysis; Y. Z. supervised the overall study, analyzed the data and wrote the paper. All authors edited the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China 11704140, Natural Science Foundation of Hubei 2017CFB116, and self-determined research funds of CCNU from the colleges' basic research and operation of MOE CCNU19QD008 to YZ.References
- V. Reiterer, P. A. Eyers and H. Farhan, Trends Cell Biol., 2014, 24, 489–505 CrossRef CAS PubMed
.
- M. Chartier, T. Chénard, J. Barker and R. Najmanovich, PeerJ, 2013, 1, e126 CrossRef PubMed
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS PubMed
- X. Ma, H. Meng and L. Lai, J. Chem. Inf. Model., 2016, 56, 1725–1733 CrossRef CAS PubMed
- C. N. Hancock, A. T. Macias, A. D. MacKerell Jr and P. Shapiro, Med. Chem., 2006, 2, 213–222 CrossRef CAS PubMed
- M. Sonoshita, A. P. Scopton, P. M. Ung, M. A. Murray, L. Silber, A. Y. Maldonado, A. Real, A. Schlessinger, R. L. Cagan and A. C. Dar, Nat. Chem. Biol., 2018, 14, 291–298 CrossRef CAS PubMed
- P. A. Jänne, N. Gray and J. Settleman, Nat. Rev. Drug Discovery, 2009, 8, 709–723 CrossRef PubMed
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed
- A. Volkamer, S. Eid, S. Turk, S. Jaeger, F. Rippmann and S. Fulle, J. Chem. Inf. Model., 2015, 55, 538–549 CrossRef PubMed
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed
- T. Stout, P. Foster and D. Matthews, Curr. Pharm. Des., 2004, 10, 1069–1082 CrossRef CAS PubMed
- F. Carles, S. Bourg, C. Meyer and P. Bonnet, Molecules, 2018, 23, 908 CrossRef PubMed
- Z. Zhao and P. E. Bourne, Drug Discovery Today, 2018, 23, 727–735 CrossRef CAS PubMed
- C. H. Yang, W. C. Lin, C. K. Chuang, Y. C. Chang, S. T. Pang, Y. C. Lin, T. T. Kuo, J. J. Hsieh and J. Chang, Br. J. Dermatol., 2008, 158, 592–596 CrossRef CAS PubMed
- L. S. Wood, Clin. J. Oncol. Nurs., 2009, 13, 13–18 CrossRef PubMed
- X. Zhao, T. Lwin, A. Silva, B. Shah, J. Tao, B. Fang, L. Zhang, K. Fu, C. Bi and J. Li, Nat. Commun., 2017, 8, 14920 CrossRef CAS PubMed
- M. Bührmann, B. M. Wiedemann, M. P. Müller, J. Hardick, M. Ecke and D. Rauh, PLoS One, 2017, 12, e0184627 CrossRef PubMed
- E. F. Choo, J. Ly, J. Chan, S. K. Shahidi-Latham, K. Messick, E. Plise, C. M. Quiason and L. Yang, Mol. Pharm., 2014, 11, 4199–4207 CrossRef CAS PubMed
- H. Chen, Y. Zhao, H. Li, D. Zhang, Y. Huang, Q. Shen, R. V. Duyne, F. Kashanchi, C. Zeng and S. Liu, PLoS One, 2014, 9, e109154 CrossRef PubMed
- Y. Hu, S. Li, F. Liu, L. Geng, X. Shu and J. Zhang, Bioorg. Med. Chem. Lett., 2015, 25, 4069–4073 CrossRef CAS PubMed
- A. A. Wylie, J. Schoepfer, W. Jahnke, S. W. Cowan-Jacob, A. Loo, P. Furet, A. L. Marzinzik, X. Pelle, J. Donovan, W. Zhu, S. Buonamici, A. Q. Hassan, F. Lombardo, V. Iyer, M. Palmer, G. Berellini, S. Dodd, S. Thohan, H. Bitter, S. Branford, D. M. Ross, T. P. Hughes, L. Petruzzelli, K. G. Vanasse, M. Warmuth, F. Hofmann, N. J. Keen and W. R. Sellers, Nature, 2017, 543, 733–737 CrossRef CAS PubMed
- S. Müller, A. Chaikuad, N. S. Gray and S. Knapp, Nat. Chem. Biol., 2015, 11, 818–821 CrossRef PubMed
- K. D. Barnash, L. I. James and S. V. Frye, Nat. Chem. Biol., 2017, 13, 1053 CrossRef CAS PubMed
- H. Chen, R. Van Duyne, N. Zhang, F. Kashanchi and C. Zeng, Proteins, 2009, 74, 122–132 CrossRef CAS PubMed
- A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann and M. Rarey, J. Chem. Inf. Model., 2012, 52, 360–372 CrossRef CAS PubMed
- A. Volkamer, A. Griewel, T. Grombacher and M. Rarey, J. Chem. Inf. Model., 2010, 50, 2041–2052 CrossRef CAS PubMed
- H. Ashkenazy, E. Erez, E. Martz, T. Pupko and N. Bental, Nucleic Acids Res., 2010, 38, 529–533 CrossRef PubMed
- A. Armon, G. Dan and N. Ben-Tal, J. Mol. Biol., 2001, 307, 447–463 CrossRef CAS PubMed
- J. F. Ohren, H. Chen, A. Pavlovsky, C. Whitehead, E. Zhang, P. Kuffa, C. Yan, P. Mcconnell, C. Spessard and C. Banotai, Nat. Struct. Mol. Biol., 2004, 11, 1192–1197 CrossRef CAS PubMed
- S. Eid, S. Turk, A. Volkamer, F. Rippmann and S. Fulle, BMC Bioinf., 2017, 18, 16 CrossRef PubMed
- D. Bandyopadhyay, J. Huan, J. Liu, J. Prins, J. Snoeyink, W. Wang and A. Tropsha, Protein Sci., 2006, 15, 1537–1543 CrossRef CAS PubMed
- J. D. Durrant, C. A. F. D. Oliveira and J. A. Mccammon, J. Mol. Graphics Modell., 2011, 29, 773–776 CrossRef CAS PubMed
- F. Cramer, Pharm. Acta Helv., 1995, 69, 193–203 CrossRef CAS
- J. K. Awino, L. Hu and Y. Zhao, Org. Lett., 2016, 18, 1650–1653 CrossRef CAS PubMed
- K. M. Comess, C. Sun, C. Abadzapatero, E. R. Goedken, R. J. Gum, D. W. Borhani, M. Argiriadi, D. R. Groebe, Y. Jia and J. E. Clampit, ACS Chem. Biol., 2011, 6, 234–244 CrossRef CAS PubMed
- J. Yang, N. Campobasso, M. P. Biju, K. Fisher, X. Q. Pan, J. Cottom, S. Galbraith, T. Ho, H. Zhang, X. Hong, P. Ward, G. Hofmann, B. Siegfried, F. Zappacosta, Y. Washio, P. Cao, J. Qu, S. Bertrand, D. Y. Wang, M. S. Head, H. Li, S. Moores, Z. Lai, K. Johanson, G. Burton, C. Erickson-Miller, G. Simpson, P. Tummino, R. A. Copeland and A. Oliff, Chem. Biol., 2011, 18, 177–186 CrossRef CAS PubMed
- J. A. Capra, R. A. Laskowski, J. M. Thornton, M. Singh and T. A. Funkhouser, PLoS Comput. Biol., 2009, 5, e1000585 CrossRef PubMed
- T. D. Schneider and R. M. Stephens, Nucleic Acids Res., 1990, 18, 6097–6100 CrossRef CAS PubMed
- G. E. Crooks, G. Hon, J. M. Chandonia and S. E. Brenner, Genome Res., 2004, 14, 1188–1190 CrossRef CAS PubMed
- J. Yang, J. Wu, J. M. Steichen, A. P. Kornev, M. S. Deal, S. Li, B. Sankaran, V. L. Woods Jr and S. S. Taylor, J. Mol. Biol., 2012, 415, 666–679 CrossRef CAS PubMed
- S. Berg, M. Bergh, S. Hellberg, K. Högdin, Y. Loalfredsson, P. Söderman, S. B. Von, T. Weigelt, M. Ormö, Y. Xue, J. Tucker, J. Neelissen, E. Jerning, Y. Nilsson and R. Bhat, J. Med. Chem., 2012, 55, 9107–9119 CrossRef CAS PubMed
- Y. Zhao, Y. Jian, Z. Liu, H. Liu, Q. Liu, C. Chen, Z. Li, L. Wang, H. H. Huang and C. Zeng, Sci. Rep., 2017, 7, 2876 CrossRef PubMed
- G. Amitai, A. Shemesh, E. Sitbon, M. Shklar, D. Netanely, I. Venger and S. Pietrokovski, J. Mol. Biol., 2004, 344, 1135–1146 CrossRef CAS PubMed
- M. Iwatani, H. Iwata, A. Okabe, R. J. Skene, N. Tomita, Y. Hayashi, Y. Aramaki, D. J. Hosfield, A. Hori, A. Baba and H. Miki, Eur. J. Med. Chem., 2013, 61, 49–60 CrossRef CAS PubMed
- A. P. Kornev, N. M. Haste, S. S. Taylor and L. F. Eyck, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17783–17788 CrossRef CAS PubMed
- S. Teller, Data visualization with d3.js, Packt Publishing Ltd, 2013 Search PubMed
- J. L. Sussman, D. Lin, J. Jiang, N. O. Manning, J. Prilusky, O. Ritter and E. E. Abola, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1998, 54, 1078–1084 CrossRef CAS PubMed
- M. J. Bick, V. Lamour, K. R. Rajashankar, Y. Gordiyenko, C. V. Robinson and S. A. Darst, J. Mol. Biol., 2009, 386, 163–177 CrossRef CAS PubMed
- K. Arnold, L. Bordoli and T. Schwede, Bioinformatics, 2006, 22, 195–201 CrossRef CAS PubMed
- P. D. Jeffrey, A. A. Russo, K. Polyak, E. Gibbs, J. Hurwitz, J. Massagué and N. P. Pavletich, Nature, 1995, 376, 313–320 CrossRef CAS PubMed
- F. Morcos, T. Hwa, J. N. Onuchic and M. Weigt, Methods Mol. Biol., 2014, 1137, 55–70 CrossRef CAS PubMed
- K. Katoh and D. M. Standley, Mol. Biol. Evol., 2013, 30, 772–780 CrossRef CAS PubMed
- R. A. Laskowski and M. B. Swindells, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127–134 CrossRef CAS PubMed
- R. Fraczkiewicz and W. Braun, J. Comput. Chem., 2015, 19, 319–333 CrossRef
- A. N. Bullock, S. Das, J. E. Debreczeni, P. Rellos, O. Fedorov, F. H. Niesen, K. Guo, E. Papagrigoriou, A. L. Amos, S. Cho, B. E. Turk, G. Ghosh and S. Knapp, Structure, 2009, 17, 352–362 CrossRef CAS PubMed
- S. B. Patel, P. M. Cameron, B. Frantz-Wattley, E. O'Neill, J. W. Becker and G. Scapin, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1696, 67–73 CrossRef CAS PubMed
- N. K. Williams, R. S. Bamert, O. Patel, C. Wang, P. M. Walden, A. F. Wilks, E. Fantino, J. Rossjohn and I. S. Lucet, J. Mol. Biol., 2009, 387, 219–232 CrossRef CAS PubMed
- M. Addie, P. Ballard, D. Buttar, C. Crafter, G. Currie, B. R. Davies, J. Debreczeni, H. Dry, P. Dudley, R. Greenwood, P. D. Johnson, J. G. Kettle, C. Lane, G. Lamont, A. Leach, R. W. Luke, J. Morris, D. Ogilvie, K. Page, M. Pass, S. Pearson and L. Ruston, J. Med. Chem., 2013, 56, 2059–2073 CrossRef CAS PubMed
- P. A. Haile, B. J. Votta, R. W. Marquis, M. J. Bury, J. F. Mehlmann, R. Singhaus Jr, A. K. Charnley, A. S. Lakdawala, M. A. Convery, D. B. Lipshutz, B. M. Desai, B. Swift, C. A. Capriotti, S. B. Berger, M. K. Mahajan, M. A. Reilly, E. J. Rivera, H. H. Sun, R. Nagilla, A. M. Beal, J. N. Finger, M. N. Cook, B. W. King, M. T. Ouellette, R. D. Totoritis, M. Pierdomenico, A. Negroni, L. Stronati, S. Cucchiara, B. Ziokowski, A. Vossenkamper, T. T. MacDonald, P. J. Gough, J. Bertin and L. N. Casillas, J. Med. Chem., 2016, 59, 4867–4880 CrossRef CAS PubMed
- M. Zha, C. Zhong, Y. Ou, L. Han, J. Wang and J. Ding, PLoS One, 2012, 7, e44828 CrossRef CAS PubMed
- T. P. Ko, W. Y. Jeng, C. I. Liu, M. D. Lai, C. L. Wu, W. J. Chang, H. L. Shr, T. J. Lu and A. H. J. Wang, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 145–154 CrossRef CAS PubMed
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07471f |
| This journal is © The Royal Society of Chemistry 2020 |