
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Down-regulation of the radiation-induced pEGFRThr654 mediated activation of DNA-PK by Cetuximab in cervical cancer cells
Yunxiang Qi†
*,
Jinyi Lang† *,
Xiaodong Zhu,
Jianming Huang,
Lu Li and
Guangming Yi
*,
Xiaodong Zhu,
Jianming Huang,
Lu Li and
Guangming Yi
Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610041, China. E-mail: jinyilang12@sina.com; zhang538419yang734@163.com; Tel: +86-028-85420243
First published on 6th January 2020
Abstract
The phosphorylation of EGFRThr654 is required for nuclear EGFR importing, and our previous study has shown that pEGFRThr654 is an independent prognostic factor for the low survival rate of patients with cervical squamous carcinoma. Now, we aim to examine the role of pEGFRThr654 in the activation of DNA-PK and radio resistance. Either CaSki or HeLa cells were exposed to a dose of 4 Gy with a 6 MV X-ray in the presence or absence of Cetuximab or Gefitinib, then EGFR, pEGFRThr654, DNA-PKcs and pDNA-PKThr2609 levels were determined using a western blot. DNA damage was quantified with γH2AX foci analysis and the response of CaSki and HeLa cells to irradiation was determined using a colony formation assay. In CaSki and HeLa cells, irradiation induced nuclear EGFR accumulation, and pEGFRThr654 and pDNA-PKThr2609 levels were both significantly increased. Cetuximab pre-treatment significantly reduced the expression of pEGFRThr654 and pDNA-PKThr2609 and enhanced the γH2AX foci per cell and sensitivity enhancement ratio in CaSki cells. Gefitinib pre-treatment had a similar but weaker effect. In HeLa cells, similar effects of Cetuximab and Gefitinib on pEGFRThr654 and pDNA-PKThr2609 were observed, and no significant difference was found. We found that Cetuximab had a better effect than Gefitinib on attenuating the radio resistance in cervical squamous carcinoma cells via inhibiting pEGFRThr654-mediated nuclear EGFR transport and related DNA-PKT2609-mediated DNA repair. However, in adenocarcinoma cells, both EGFR-targeted drugs had no remarkable effects on the radio sensitivity. Taken together, radiotherapy combined with Cetuximab may be a promising strategy to improve the therapeutic gain for cervical squamous carcinoma patients.
Introduction
Cervical cancer is one of the major threats to female health, has an estimated incidence of more than 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cases per year, an annual mortality around 270000, and the 5 year prevalence reaches 1.5 million.1 Routine population-based screening programs provide an effective early diagnosis for cervical cancer where surgery is used for early stage (IA and II) treatment and radiotherapy has been applied predominantly for patients with later stages of cervical cancer.2 Particularly, concurrent chemoradiation therapy (CCRT), where platinum-based chemotherapy is given concomitantly with radiotherapy, has been considered a standard of treatment for locally advanced cervical cancer (LACC) since 1999.3 The prognosis of early stage cervical cancer with CCRT is excellent, with an approximately 80–90% 5 year survival rate for International Federation of Gynecologic Oncology (FIGO) stage IIB.4 Unfortunately, though the advantages of platinum-based CCRT have been proven for over one and a half decades, the 5 year survival rate for LACC has not significantly improved and remains 40–50% over the past two decades.5 Not all the cervical cancers are sensitive to radiotherapy and radio resistance has been a major problem for LACC treatment. Deeply understanding the mechanisms of radio resistance will help to improve the prognosis of LACC.
000 cases per year, an annual mortality around 270000, and the 5 year prevalence reaches 1.5 million.1 Routine population-based screening programs provide an effective early diagnosis for cervical cancer where surgery is used for early stage (IA and II) treatment and radiotherapy has been applied predominantly for patients with later stages of cervical cancer.2 Particularly, concurrent chemoradiation therapy (CCRT), where platinum-based chemotherapy is given concomitantly with radiotherapy, has been considered a standard of treatment for locally advanced cervical cancer (LACC) since 1999.3 The prognosis of early stage cervical cancer with CCRT is excellent, with an approximately 80–90% 5 year survival rate for International Federation of Gynecologic Oncology (FIGO) stage IIB.4 Unfortunately, though the advantages of platinum-based CCRT have been proven for over one and a half decades, the 5 year survival rate for LACC has not significantly improved and remains 40–50% over the past two decades.5 Not all the cervical cancers are sensitive to radiotherapy and radio resistance has been a major problem for LACC treatment. Deeply understanding the mechanisms of radio resistance will help to improve the prognosis of LACC.
Previous studies have pointed out that the epidermal growth factor receptor (EGFR) plays a crucial role in the treatment resistance of various cancers, including cervical cancer.6 Normal cervical basal cells express EGFR in the cytoplasm and nucleus.7 Human papillomavirus (HPV) infection, a predominant cause of cervical cancer,8 is connected to the EGFR expression.7 Whether or not EGFR can be used as a predictor of cervical cancer therapy response is still controversial; many clinical studies have demonstrated that cervical cancer patients with highly expressed tumor EGFR have poor prognosis, with lower recurrence-free and overall survival rates.9 EGFR belongs to the HER family of receptor tyrosine kinases, containing an extracellular ligand-binding domain, a membrane-spanning domain, a nuclear localization signal (NLS), a tyrosine kinase domain, and a tyrosine-rich C-terminal tail.10 Normally, EGFR is activated by ligands, such as the epidermal growth factor, but it can also be activated in a ligand-independent way, including by exposure to genotoxic stimuli such as radiation.11 Radiation activates the EGFR partly by inducing the nuclear translocation of EGFR from the cell membrane.12 Dittmann et al. work suggests that radiation induced cellular lipid peroxidation triggers EGFR importing into the nucleus via Src activation.13 The phosphorylation of the residue Thr654 is located at the NLS domain.14
In the nucleus, the EGFR works as a transcriptional factor, a nuclear tyrosine kinase and a regulator of DNA repair.15 Nuclear EGFR regulates the expression of cyclin D1, c-myc, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2); which are related to cell proliferation, inflammation, and carcinogenesis.16 Nuclear EGFR translocation is believed to take part in the resistance of radiotherapy in cancer cells, and the inhibition of EGFR translocation leads to an improved radiation response.17 Radiation treatment induces cellular DNA damage and the most detrimental types are double-strand breaks (DSBs).18 To minimize the DSB induced damage, cancer cells repair broken chromosomes mainly via nonhomologous end-joining (NHEJ), and in this way, cancer cells are resistant to irradiation.19 Nuclear EGFR modifies the process of DNA repair against radiation-induced chromatinic DSB, thereby contributing to the radio resistance. NHEJ is the major cellular way to repair nuclear DSB damage, and DNA dependent kinase (DNA-PK) is involved in the NHEJ process.20 The activation of DNA-PK is due to the phosphorylation of residue Thr2609, which is essential for nuclear EGFR mediated DNA repair.21 Nuclear EGFR plays an important role in DNA-PK in charged DNA repairing, and the inhibition of EGFR translocation leads to reduced DNA-PK activity, thereby enhancing the radio sensitivity in cancer cells.22 EGFR nuclear localization is required for the modulation of radiation-induced DNA damage repair and binding of EGFR to DNA-PKcs, thus targeting the inhibition of nuclear EGFR transport may contribute to reducing the radio resistance in cervical cancer treatment.
Nuclear EGFR transport depends on the trafficking of the karyopherin nuclear import complex and coat protein complex I (COPI).23 In this process, the NLS domain in EGFR is essential for its correct translocation, and the residue Thr654 in NLS has been proven to play a central role in nuclear transport. The phosphorylation of EGFRThr654 has been found to lead to its transport into the nucleus in the human bronchial carcinoma cell line A549 and Chinese hamster ovary CHO-K1 cells, and the nucleus translocation was abolished by the deletion of Thr654 in CHO cells.14 Our previous study has demonstrated that phosphorylation of Thr654 is independently associated with PFS (HR = 4.092, 95% CI = 1.487–11.263; P = 0.006) and OS (HR = 2.966, 95% CI = 1.327–466.629; P = 0.008) for patients with FIGO stage III cervical cancer, and in multivariate analysis, pEGFR-Thr654 is an independent factor for poor outcome in patients with LACC.24 The critical role of phosphorylated Thr654 mediated EGFR nuclear translocation in radio resistance suggests that pEGFRThr654 may be a promising molecular target to increase the radio sensitivity in cervical cancer therapy.
In recent decades, several EGFR target drugs have been developed and applied in the clinical treatment of several cancers, such as colon cancer and lung cancer. There are two major kinds of EGFR target drugs: tyrosine kinase inhibitors, such as Gefitinib, and anti-EGFR monoclonal antibodies, such as Cetuximab. In cervical cancer, though EGFR target drugs have not yet been applied in clinical treatment, the fact that EGFR is highly expressed in poor prognosis patients suggests that Cetuximab and Gefitinib may be potential drugs to combine with radiotherapy for CCRT. In the present study, we aim to examine the effects of irradiation on nuclear EGFR transport in cervical squamous carcinoma cell line CaSki and cervical adenocarcinoma cell line HeLa. Additionally, we hope to explore the effects of Cetuximab and Gefitinib on radiation induced pEGFRThr654-mediated radio resistance in vitro. Our findings provide new information for CCRT against cervical cancer.
Materials and methods
Cell culture, drugs pre-treatment and irradiation
The human cervical cell lines CaSki and HeLa were purchased from the Cell Bank of China (Wuhan, China) and maintained in RPMI1640 supplemented with 10% FBS and antibiotics (100 units per mL penicillin G, 100 units per mL gentamicin; Gibco) at 37 °C in 5% CO2. Cells were pre-treated with Cetuximab (65 nm mL−1) or Gefitinib (2 nmol mL−1) for 16 hours and then exposed to a single dose of 4 Gy X-rays using the linear accelerator at a dose rate of 450 cGy min−1.Protein extraction and western blot
Cytoplasmic and nuclear proteins were extracted 10, 20 or 40 minutes after irradiation using the cytosolic and nuclear protein extract kit according to the manufacturer's manual. In brief, cells were harvested in lysis buffer with 1 mM phenylmethyl-sulfonyl fluoride (PMSF). The cells were allowed to swell on ice for 15 min. The homogenates were centrifuged for 5 min at 12000 × g and the supernatant was used as the cytosolic extract. The nuclear pellet was re-suspended in cold extraction buffer and the samples were centrifuged at 15000 × g for 30 min. The obtained supernatant was used as the nuclear extract.
Proteins were separated by sodium dodecyl sulfatepoly-acrylamide gel electrophoresis (SDS-PAGE) through 12.5% gel and transferred onto PVDF membranes. The gels were run and transferred under the same experimental conditions. The membranes were blocked with 3% bovine serum albumin in TBS containing 1% Tween-20 (TBST) for 2 h at the room temperature, followed by incubation with the appropriate primary antibodies for EGFR (BD Biosciences), pEGFRThr654 (Millipore), DNA-PKcs/PRKDC (Epitmics) or pDNA-PKcsThr2609 (Abcam) overnight in 4 °C, and either horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibodies for 1 h in room temperature. GAPDH and Lamin B1 (Epitmics) were served as the loading controls for non-nuclear protein and nuclear protein, respectively.
Antibody-bound proteins were detected using an ECL Stable Peroxide solution. All protein bands were visualized by a FluroChem MI imaging system (Alpha Innotech Santa Clara, CA) under the room temperature.
Clonogenic survival assay
Cells were seeded into six-well plates at 200–2000 cells per well depending on the dose of radiation. The cells were irradiated with 0, 1, 2, 4, 6 or 8 Gy X-ray irradiation by linear accelerators at a dose rate of 450 cGy min−1. After the irradiation, the cells were grown for 12–14 days to allow for colony formation and were subsequently fixed and stained using crystal violet. Colonies consisting of 50 or more cells were counted as a clone. The radiation sensitivity enhancement ratio (SER) was measured according to the multi-target single hit model, and the linear-quadratic model was accessed.Quantification of γ-H2AX foci formation
After irradiation, cells were incubated for another 24 hours and then fixed with 4% paraformaldehyde for 10 min, treated with 1% Triton X-100 for 10 min, and then incubated with blocking serum for 1 h at room temperature. After washing with PBS, samples were incubated with a rabbit monoclonal against γH2AX antibody (ABcam, UK) overnight at 4 °C, followed by FITC-conjugated goat-anti-rabbit secondary antibody for 2 h. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO). The cells were observed using an Olympus fluorescence microscope (Olympus, Tokyo, Japan). γH2AX foci were counted in each cell. At least 100 cells were counted from 10 randomly chosen fields of view.Statistical data analyses
Data were expressed as the means ± SD of at least three independent experiments. The data were then analyzed by one-way analysis of variance (ANOVA). The sensitizer enhancement ratios (SER) were measured according to the multi-target single hit model. Differences were considered statistically significant when p < 0.05. SPSS Statistical software (version 19.0) was used for statistical analysis.Results
Irradiation stimulated nuclear EGFR accumulation in both CaSki and HeLa cells
To illustrate the expression change levels of EGFR in cervical cancer, we first examined the cytosolic and nuclear EGFR protein pattern in both CaSki and HeLa cells before and after irradiation treatment (Fig. 1). Immunoblotting illustrated that in CaSki cells, the protein levels of cytosolic and nuclear EGFR were both elevated after 4 Gy of irradiation in a time-dependent way. Forty minutes after the irradiation, the nuclear EGFR levels were significantly higher than that of cells with no irradiation treatment, simultaneously, the cytosolic EGFR was increased as well. Similar results were observed in HeLa cells, where irradiation did not increase the nuclear EGFR protein amounts, but no significant effects were observed on the cytosolic EGFR level. | ||
| Fig. 1 The expression levels of cytosolic EGFR (cEGFR) and nuclear EGFR (nEGFR) in CaSki and HeLa cells after 10, 20 or 40 minutes irradiation treatment. | ||
Irradiation induced EGFRThr654 phosphorylation and downstream DNA-PKThr2609 expression and phosphorylation
We then examined the phosphorylation level of EGFRThr654 in CaSki cells and HeLa cells using immunoblotting. The results showed that 20 minutes after irradiation, the pEGFRThr654 level in CaSki cell nuclei was significantly increased, and another 20 minutes later, the phosphorylation level of EGFRThr654 was further elevated to around 3 times higher than that of the control cells with no irradiation treatment (Fig. 2), suggesting that the irradiation induces EGFRThr654 in a time-dependent way. Similar results were found in HeLa cells. | ||
| Fig. 2 The expression levels of pEGFR(T654), nDNA-PK and pDNA-PK(T2609) in CaSki and HeLa cells. **p < 0.01, ***p < 0.001, vs. 0 min. | ||
Additionally, we examined the nuclear DNA-PK and pDNA-PKThr2609 level in irradiation treated CaSki and HeLa cells. In both cell lines, after forty minutes of 4 Gy irradiation, both nDNA-PK and pDNA-PKThr2609 levels significantly elevated compared with that of the control cells. These findings suggest that irradiation induced not only EGFR expression and nuclear translocation, but also the activation of DNA-PK in cervical cancer cells.
Cetuximab treatment significantly decreased irradiation-induced nEGFR accumulation and pEGFRThr654 phosphorylation in CaSki and HeLa cells, but Gefitinib only worked for HeLa cells
In order to reveal the role of pEGFRThr654 in EGFR targeted therapy enhanced irradiation induced DNA repair, we then examined the nuclear EGFR expression level and phosphorylation level of pEGFRThr654 in Cetuximab or Gefitinib treated CaSki cells and HeLa cells using immunoblotting. In CaSki cells, irradiation induced nuclear EGFR accumulation, whereas Cetuximab pre-treatment reduced the nuclear EGFR accumulation after irradiation, compared with that of cells treated with irradiation alone (Fig. 3). Additionally, the phosphorylated EGFRThr654 level in Cetuximab pre-treated cells was significantly lower than in irradiation alone cells, which was coincident with the nuclear EGFR level. No significant effects on the nuclear EGFR or phosphorylation of EGFRThr654 were observed in Gefitinib pre-treated CaSki cells. Simultaneously, the cytoplasmic EGFR level in Cetuximab pre-treated CaSki cells had no difference compared with that of Gefitinib pre-treated cells or irradiation alone cells. These findings suggest that Cetuximab may increase the DNA repair rate of irradiation treated CaSki via inhibiting the pEGFRThr654-mediated nuclear EGFR translocation. In HeLa cells, similar effects of Cetuximab on the nuclear EGFRThr654 were observed. After 4 Gy of irradiation treatment, the phosphorylated EGFRThr654 level in Cetuximab pre-treated cells and Gefitinib pre-treated were both significantly lower than that of irradiation treated only cells, which were coincident with the nuclear EGFR amounts. These findings suggested that in both CaSki and HeLa cells, Cetuximab could inhibit the EGFR nuclear translocation via reducing the phosphorylated EGFRThr654 level, but Gefitinib only inhibited the EGFR nuclear transport in HeLa cells.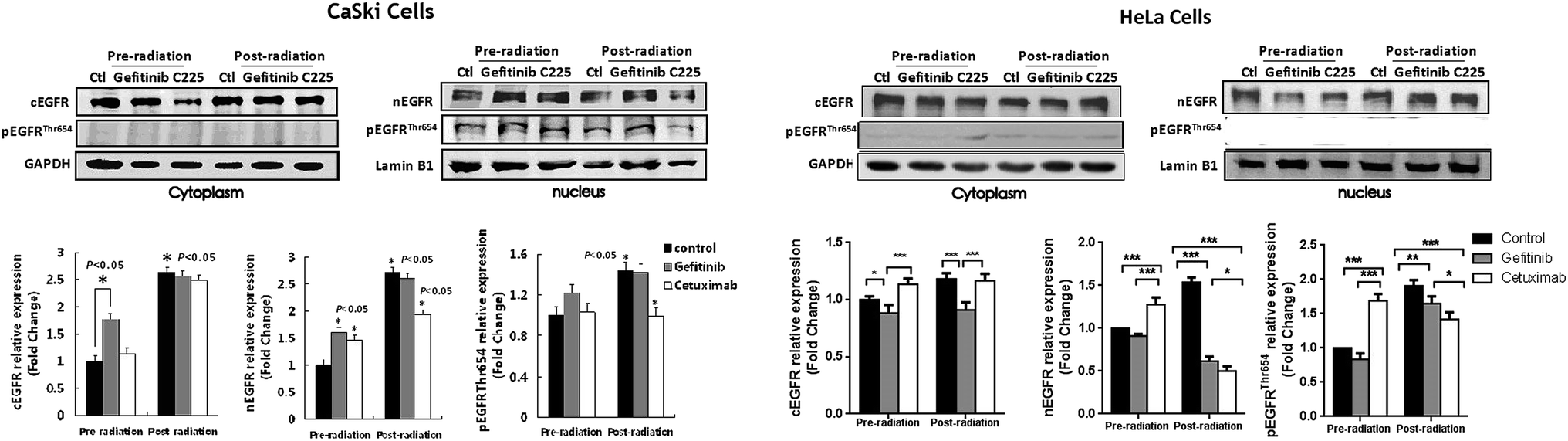 | ||
| Fig. 3 The expression levels of cEGFR, nEGFR and pEGFR(T654) in un-irradiated or irradiated CaSki and HeLa cells with Gefitinib or Cetuximab treatment. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Cetuximab and Gefitinib inhibited irradiation induced DNA-PK expression and phosphorylation in both CaSki and HeLa cells
To investigate if the Cetuximab induced decrease in nuclear EGFR accumulation contributes to the enhanced radio sensitivity in a DNA-PK dependent way, we examined the expression and phosphorylation of DNA-PK in CaSki and HeLa cells using immunoblotting. In CaSki cells, western blotting illustrated that DNA-PK was only expressed in the nucleus, irradiation slightly stimulated the expression of DNA-PK, and the phosphorylated DNA-PKT2609 amount was significantly increased (Fig. 4). Cells treated with Cetuximab for 16 hours before irradiation significantly reduced the irradiation induced DNA-PK expression and phosphorylation at residue T2609, and pre-treatment of Gefitinib had similar but weaker effects with significance. Similar results were observed in HeLa cells, in which both Cetuximab and Gefitinib reduced the phosphorylated DNA-PKT2609 in post-irradiation cells. | ||
| Fig. 4 The expression levels of DNA-PK and pDNA-PK(T2609) in un-irradiated or irradiated CaSki and HeLa cells with Gefitinib or Cetuximab treatment. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Cetuximab raised the radio sensitivity in CaSki cells, whereas Gefitinib showed no effects on CaSki cell lines, and both drugs showed no effects on HeLa cells
Nuclear EGFR plays a critical role in radio resistance, so in order to elucidate the effects of anti-EGFR drugs on the radio sensitivity of cervical cancer, we performed clonogenic survival assays to investigate the impacts of Cetuximab and Gefitinib on radio sensitivity in cervical squamous carcinoma cell line CaSki and adenocarcinoma cell line HeLa. Cells were treated with Cetuximab or Gefitinib 16 hours prior to irradiation at 0, 1, 2, 4, 6 and 8 Gy. A multi-target single hit model was adopted to obtain the major parameters in dose–survival curves. In the CaSki cells, the mean lethal dose (D0) of the irradiation alone group (I), irradiation plus Cetuximab group (C) and irradiation plus Gefitinib group (G) was 2.612, 1.572 and 2.396 Gy, respectively. The quasi-threshold doses (Dq) were 1.475, 0.630 and 1.047 Gy, respectively, and the surviving fraction of 2 Gy irradiation (SF2) was 59.86%, 31.03% and 50.58%, respectively (Fig. 5). The SER (D0) value of Cetuximab was 1.66, whereas that of Gefitinib was 1.09. The Cetuximab treatment induced reduction of D0 and Dq in CaSki cells were both significant, whereas Gefitinib treatment presented no significant effects on D0 and Dq. The linear-quadratic model was also performed, in which the ratio of the α/β in group I, C and G was 6.331 Gy, 49.3735 Gy and 11.3456 Gy, respectively. These findings suggest that Cetuximab has a much better radio sensitizing effect than Gefitinib in CaSki cells. In HeLa cells, the D0 of I, C, and G groups was 2.915, 2.872 and 2.889 Gy, respectively, while the Dq values were 1.791, 0.900 and 1.173 Gy, respectively. The SF2 were 72.5%, 62.7% and 65.4%, respectively. The SER (D0) of Cetuximab and Gefitinib in HeLa cells was 1.015 and 1.009, respectively. Consistently, the linear-quadratic model analysis indicated that in HeLa cells, the α/β in group I, C and G was 3.884 Gy, 9.201 Gy and 7.200 Gy, respectively. Cetuximab treatment significantly decreased the Dq compared with that of the irradiation alone group in HeLa cells, implying that Cetuximab could enhance the radio sensitivity in HeLa cells. | ||
| Fig. 5 Survival fraction of CaSki cells (up) and HeLa cells (down) under Gefitinib or Cetuximab treatment. (Left) Multitarget-single hitting model: dose–survival curve in CaSki cells. (Right) Linear-quadratic model: dose–survival curve in CaSki cells. | ||
Cetuximab and Gefitinib increased the irradiation induced DNA repair rate in CaSki cells and HeLa cells
Stimulated DSB repairing in cancer cells leads to radio resistance. We examined the effects of Cetuximab and Gefitinib on DSB repair after irradiation induced DNA damage. The formation of γ-H2AX foci in the nuclei has been widely used as a marker of DSB present in radiation treated cells.25 In the present study, we illustrated the γ-H2AX foci formation in CaSki cells and HeLa cells using immunofluorescence staining. CaSki and HeLa cells were treated with either Cetuximab or Gefitinib for 16 hours and then treated with 4 Gy irradiation. Treated cells were then cultured for 2 hours to recover from DNA damage and then stained with γ-H2AX antibody to assess the residual nuclear γ-H2AX foci formation. In CaSki cells, both Cetuximab and Gefitinib treatment elevated the percentage of cells containing residual γ-H2AX nuclear foci to around 20%, compared with 10% of the γ-H2AX foci positive cells in irradiation treated only CaSki cells (Fig. 6). The mean number of foci in each positive cell was significantly increased from 2 in irradiation only cells to either 5 in irradiation plus Gefitinib treated cells or 6 in irradiation plus Cetuximab treated cells. These findings demonstrated that anti-EGFR drug treatment increased the irradiation induced DNA damage in CaSki cells. Moreover, Cetuximab had a greater effect than Gefitinib. Consistently, in HeLa cells, Cetuximab and Gefitinib treatment increased the percentage of γ-H2AX foci positive cells from 2 to 4.13 and 3.27, respectively, and the mean foci numbers per cell were also raised from 8.20 to 13.03 and 10.33, respectively. | ||
| Fig. 6 The formation of γ-H2AX foci in the nuclei in different groups of cells. *p < 0.05 compared with the control; #p < 0.05 compared with the Gefitinib group. | ||
Discussion
Cervical cancer threatens female health, and radiotherapy combined with platinum-based chemotherapy, known as CCRT, has been developed to be the golden-standard for non-surgery treatment of cervical cancer.3 However, the radio resistance of cervical cancer cells reduces the therapeutic effects of radiotherapy, thereby becoming a major problem in clinical treatment. Many studies have indicated that nuclear EGFR takes part in the radio resistance in various cancers and radiotherapy combined with anti-EGFR treatment has been believed to be an auspicious strategy for cancer therapy.26 In the present study, we aimed to explore the potential mechanisms of nuclear EGFR translocation and its effects on radio resistance in cervical cancer. Additionally, we planned to examine the effects of anti-EGFR-targeted therapies in enhancing the radio sensitivity of cervical cancer in vitro.A number of studies have indicated that EGFR plays a critical role in the etiology of epithelial tumors, such as lung, head and neck, bladder and oesophageal cancers.27 Several studies have elucidated that in cervical cancer, the expression level of EGFR shows a correlation with the poor prognosis, and our studies further revealed that the nuclear EGFRThr654 level is negatively correlated to cervical carcinoma prognosis.28 The classic role of EGFR is known as a cellular membrane-bound receptor tyrosine kinase, activating the MAPK, PI3K/AKT, PKC and STAT pathways, and the dysregulation of EGFR leads to carcinogenesis.29 Besides the membrane-bound tyrosine kinase activity, subcellular translocation of EGFR takes part in tumor growth, progression and therapy-resistance.15 Particularly, EGFR shuttles into the nucleus are connected to radio resistance.30 Recent studies have pointed out that nuclear EGFR levels, but not the cytosolic EGFR level, could be used as a negative indicator for the clinical treatment of cancers.15 Radiation is one of the major reasons for EGFR nuclear translocation in bronchial cancer cells.12 However, whether radiotherapy induces the nuclear transport of EGFR in cervical cancer remains unclear. In the present study, immunoblotting illustrated that 4 Gy of irradiation elevated the nuclear EGFR amounts in a time-dependent way in CaSki cells, as well as in HeLa cells. Thus, irradiation moderately stimulates the nuclear accumulation of EGFR (Fig. 1). The nuclear transport of EGFR depends on the interaction of importin proteins and the EGFR NLS domain.31 Forty minutes after the irradiation treatment, the phosphorylation level of nuclear EGFRThr654 significantly increased in CaSki cells. Our findings revealed that in both cervical squamous carcinoma and adenocarcinoma cells, radiation induces nuclear EGFR transport at least partly depending on the phosphorylation of residue T654.
Nuclear EGFR participated in DNA damage repair via binding to DNA-PK and activating the NHEJ process. The activation of DNA-PK is due to the phosphorylation of residue Thr2609, which is essential for nuclear EGFR mediated DNA repair.21 Not surprisingly, the nuclear total DNA-PK and pDNA-PKthr2609 levels elevated significantly in irradiation treated cervical cancer cells, which was coincident with the nuclear EGFR accumulation. These findings elucidated that the radio resistance in cervical cancer radiotherapy may be partly attributed to pEGFRThr654 mediated EGFR nuclear transport and downstream DNA-PK activation.
As the EGFR plays important roles in epithelial tumor growth, several strategies have been developed to inhibit the activity of EGFR. One strategy is to use monoclonal antibodies such as Cetuximab (C225) to block the ligand-binding domain and reduce the receptor activity.32 Another one utilizes the tyrosine kinase inhibitors such as Gefitinib to inhibit the kinase activity via binding the ATP-binding domain. Although Liao et al. indicated that Cetuximab treatment itself induces EGFR nuclear translocation,33 Dittamm and colleagues have demonstrated that Cetuximab could reduce the radiation induced nuclear EGFR accumulation in bronchial carcinoma and breast adenoma cells, thereby enhancing the cancer cells radio sensitivity.34 In the current study, we proved that in cervical cancer cell lines CaSki and HeLa, Cetuximab provided similar effects and rescued the radiation induced nuclear EGFR translocation, while Gefitinib showed no effects on the nuclear EGFR level in post-irradiation CaSki cells. Only HeLa cells presented the sensitivity to Gefitinib (Fig. 3). Coincident with the nuclear EGFR level, Cetuximab treatment decreased the phosphorylated EGFRThr654 level in irradiation treated CaSki and HeLa cells, suggesting that Cetuximab regulates the EGFR nuclear translocation at least partly via modifying the EGFRThr654 phosphorylation.
Nuclear EGFR contributes to radio resistance and the effects of Cetuximab may lead to enhanced radio sensitivity. Indeed, in CaSki cells, clonogenic survival assays revealed that Cetuximab treatment remarkably increased the radio sensitivity and the SER of Cetuximab reached 1.66, whereas the Gefitinib treatment only slightly enhanced the radio sensitivity with a SER of 1.09. These findings suggest that in cervical squamous carcinoma cells, Cetuximab may attenuate the radio resistance via reducing the nuclear EGFR translocation. However, in HeLa cells, both Cetuximab and Gefitinib had no influence on radio sensitivity, the SER of Cetuximab and Gefitinib were only 1.015 and 1.009, respectively, though the SF2 and Dq of irradiation combined with Cetuximab treated cells were moderately improved compared with that of irradiation treated alone cells. These results imply that in cervical adenocarcinoma, the effects of Cetuximab are much weaker than in squamous carcinoma.
Nuclear EGFR affects DNA repair, thereby contributing to radio resistance. Interestingly, in our experiments, though only Cetuximab treatment inhibited nuclear EGFR transport induced by irradiation, both Cetuximab and Gefitinib treatment decreased the efficiency of DNA repair. In CaSki and HeLa cells, γ-H2AX staining illustrated that Cetuximab treatment significantly elevated both the γ-H2AX positive cell ratios and foci numbers per cell, and Gefitinib provided a similar but weaker effect. γ-H2AX is an excellent marker for the nuclear DSB repair complex,25 and our findings implied that 24 hours after Cetuximab or Gefitinib treatment, the cervical cancer cells exhibited more unrepaired DNA damage than irradiation alone cells. However, since Gefitinib treatment showed no effect on the nuclear EGFR level, how Gefitinib treatment inhibited the DNA repair process in irradiation treated cells is still unclear. Gefitinib can inhibit the activation of the AKT pathway, which may partly explain the reduced DNA repair efficiency.35
As mentioned above, nuclear EGFR regulates the DNA repair in a DNA-PK dependent way, and Dittmann et al. revealed that Cetuximab can inhibit the nuclear transport of EGFR and DNA-PK activation in A549 and MDA MB 231 cells.34 In the present study, we elucidated that in both cervical cancer cells CaSki and HeLa, Cetuximab treatment significantly decreased the total and phosphorylated DNA-PKthr2609 level in irradiation treated cells, and Gefitinib showed a weaker effect. These findings implied that Cetuximab had a greater effect than Gefitinib to reduce the expression of pEGFRThr654 and DNA-PKT2609in cervical cancer cells, which may contribute to impaired DNA repair processes, thereby enhancing the radio sensitivity.
In conclusion, our present study indicated that in cervical cancer cells, pEGFRThr654 mediates EGFR nuclear translocation, and nuclear EGFR activates DNA repair in a DNA-PKT2609 dependent way, which leads to radiation resistance. Cetuximab attenuated the radio resistance at least partly via inhibiting the accumulation of nuclear pEGFRThr654 and DNA-PKT2609 in cervical cancer cells. Our findings suggest that radiotherapy combined with anti-EGFR-targeted therapy such as Cetuximab may be a promising strategy for cervical cancer treatment, particularly for the cervical squamous carcinoma. Further studies are required for clinical trials.
Abbreviations
| CCRT | Concurrent chemoradiation therapy |
| LACC | Locally advanced cervical cancer |
| FIGO | Federation of gynecologic oncology |
| EGFR | Epidermal growth factor receptor |
| HPV | Human papillomavirus |
| NLS | Nuclear localization signal |
| iNOS | Inducible nitric oxide synthase |
| COX-2 | Cyclooxygenase-2 |
| DSBs | Double-strand breaks |
| NHEJ | Nonhomologous end-joining |
| DNA-PK | DNA dependent kinase |
| COPI | Coat protein complex I |
| CHO | Chinese hamster ovary |
| PMSF | Phenylmethyl-sulfonyl fluoride |
| SDS-PAGE | Sulfatepoly-acrylamide gel electrophoresis |
| TBST | TBS containing 1% Tween-20 |
| SER | Sensitivity enhancement ratio |
| ANOVA | Analysis of variance |
| cEGFR | Cytosolic EGFR |
| nEGFR | Nuclear EGFR |
Conflicts of interest
There are no conflicts to declare.References
- GLOBOCAN, Cancer Fact Sheets: Cervical Cancer, http://globocaniarcfr/Pages/fact_sheets_canceraspx, accessed December 31, 2015 Search PubMed.
- D. S. Dizon, H. J. Mackay, G. M. Thomas, T. L. Werner, E. C. Kohn and D. Hess, et al., State of the science in cervical cancer: where we are today and where we need to go, Cancer, 2014, 120(15), 2282–2288, DOI:10.1002/cncr.28722.
- P. G. Rose, B. N. Bundy, E. B. Watkins, J. T. Thigpen, G. Deppe and M. A. Maiman, et al., Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer, N. Engl. J. Med., 1999, 340(15), 1144–1153, DOI:10.1056/NEJM199904153401502.
- H. M. Keys, B. N. Bundy, F. B. Stehman, L. I. Muderspach, W. E. Chafe and C. L. Suggs 3rd, et al., Cisplatin, radiation, and adjuvant hysterectomy compared with radiation and adjuvant hysterectomy for bulky stage IB cervical carcinoma, N. Engl. J. Med., 1999, 340(15), 1154–1161, DOI:10.1056/NEJM199904153401503.
- M. Morris, P. J. Eifel, J. Lu, P. W. Grigsby, C. Levenback and R. E. Stevens, et al., Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer, N. Engl. J. Med., 1999, 340(15), 1137–1143, DOI:10.1056/NEJM199904153401501.
- T. M. Brand, M. Iida, N. Luthar, M. M. Starr, E. J. Huppert and D. L. Wheeler, Nuclear EGFR as a molecular target in cancer, Radiother. Oncol., 2013, 108(3), 370–377, DOI:10.1016/j.radonc.2013.06.010.
- W. B. Chapman, A. T. Lorincz, G. D. Willett, V. C. Wright and R. J. Kurman, Epidermal growth factor receptor expression and the presence of human papillomavirus in cervical squamous intraepithelial lesions, Int. J. Gynecol. Pathol., 1992, 11(3), 221–226 CrossRef CAS PubMed.
- N. Munoz, F. X. Bosch, S. de Sanjose, R. Herrero, X. Castellsague and K. V. Shah, et al., Epidemiologic classification of human papillomavirus types associated with cervical cancer, N. Engl. J. Med., 2003, 348(6), 518–527, DOI:10.1056/NEJMoa021641.
- R. I. Nicholson, J. M. Gee and M. E. Harper, EGFR and cancer prognosis, Eur. J. Cancer, 2001, 37(suppl. 4), S9–S15 CrossRef CAS PubMed.
- T. Soonthornthum, H. Arias-Pulido, N. Joste, L. Lomo, C. Muller and T. Rutledge, et al., Epidermal growth factor receptor as a biomarker for cervical cancer, Ann. Oncol., 2011, 22(10), 2166–2178, DOI:10.1093/annonc/mdq723.
- C. Lu, L. Z. Mi, M. J. Grey, J. Zhu, E. Graef and S. Yokoyama, et al., Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor, Mol. Cell. Biol., 2010, 30(22), 5432–5443, DOI:10.1128/MCB.00742-10.
- R. K. Schmidt-Ullrich, R. B. Mikkelsen, P. Dent, D. G. Todd, K. Valerie and B. D. Kavanagh, et al., Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation, Oncogene, 1997, 15(10), 1191–1197, DOI:10.1038/sj.onc.1201275.
- K. Dittmann, C. Mayer, R. Kehlbach and H. P. Rodemann, Radiation-induced caveolin-1 associated EGFR internalization is linked with nuclear EGFR transport and activation of DNA-PK, Mol. Cancer, 2008, 7, 69, DOI:10.1186/1476-4598-7-69.
- K. Dittmann, C. Mayer, R. Kehlbach, M. C. Rothmund and H. Peter Rodemann, Radiation-induced lipid peroxidation activates src kinase and triggers nuclear EGFR transport, Radiother. Oncol., 2009, 92(3), 379–382, DOI:10.1016/j.radonc.2009.06.003.
- P. M. Harari and S. M. Huang, Radiation response modification following molecular inhibition of epidermal growth factor receptor signaling, Semin. Radiat. Oncol., 2001, 11(4), 281–289 CrossRef CAS PubMed.
- K. Rothkamm, M. Kuhne, P. A. Jeggo and M. Lobrich, Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks, Cancer Res., 2001, 61(10), 3886–3893 CAS.
- J. E. Haber, Partners and pathways repairing a double-strand break, Trends Genet., 2000, 16(6), 259–264 CrossRef CAS PubMed.
- P. S. Harris, S. Venkataraman, I. Alimova, D. K. Birks, A. M. Donson and J. Knipstein, et al., Polo-like kinase 1 (PLK1) inhibition suppresses cell growth and enhances radiation sensitivity in medulloblastoma cells, BMC Cancer, 2012, 12, 80, DOI:10.1186/1471-2407-12-80.
- D. Bandyopadhyay, M. Mandal, L. Adam, J. Mendelsohn and R. Kumar, Physical interaction between epidermal growth factor receptor and DNA-dependent protein kinase in mammalian cells, J. Biol. Chem., 1998, 273(3), 1568–1573 CrossRef CAS PubMed.
- K. Dittmann, C. Mayer, B. Fehrenbacher, M. Schaller, U. Raju and L. Milas, et al., Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase, J. Biol. Chem., 2005, 280(35), 31182–31189, DOI:10.1074/jbc.M506591200.
- Y. N. Wang, H. Wang, H. Yamaguchi, H. J. Lee, H. H. Lee and M. C. Hung, COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport, Biochem. Biophys. Res. Commun., 2010, 399(4), 498–504, DOI:10.1016/j.bbrc.2010.07.096.
- H. J. Liao and G. Carpenter, Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression, Mol. Biol. Cell, 2007, 18(3), 1064–1072, DOI:10.1091/mbc.E06-09-0802.
- K. Dittmann, C. Mayer, B. Fehrenbacher, M. Schaller, R. Kehlbach and H. P. Rodemann, Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654, FEBS Lett., 2010, 584(18), 3878–3884, DOI:10.1016/j.febslet.2010.08.005.
- J. Lang, X. Lai, J. Huang, W. Wang and P. Wang, Expression of mEGFR, pEGFRThr654, and pPKN1Thr774 Predict Response to Chemoradiation and Prognosis in Patients With Cervical Cancer, Int. J. Radiat. Oncol., Biol., Phys., 2013, 87(2 suppl.), S663–S664, DOI:10.1016/j.ijrobp.2013.06.1758.
- A. Celeste, O. Fernandez-Capetillo, M. J. Kruhlak, D. R. Pilch, D. W. Staudt and A. Lee, et al., Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks, Nat. Cell Biol., 2003, 5(7), 675–679, DOI:10.1038/ncb1004.
- S. M. Huang and P. M. Harari, Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis, Clin. Cancer Res., 2000, 6(6), 2166–2174 CAS.
- Y. L. Yu, R. H. Chou, C. H. Wu, Y. N. Wang, W. J. Chang and Y. J. Tseng, et al., Nuclear EGFR suppresses ribonuclease activity of polynucleotide phosphorylase through DNAPK-mediated phosphorylation at serine 776, J. Biol. Chem., 2012, 287(37), 31015–31026, DOI:10.1074/jbc.M112.358077.
- G. Liccardi, J. A. Hartley and D. Hochhauser, EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment, Cancer Res., 2011, 71(3), 1103–1114, DOI:10.1158/0008-5472.CAN-10-2384.
- L. J. Willmott and B. J. Monk, Cervical cancer therapy: current, future and anti-angiogenesis targeted treatment, Expert Rev. Anticancer Ther., 2009, 9(7), 895–903, DOI:10.1586/era.09.58.
- D. J. Chen and C. S. Nirodi, The epidermal growth factor receptor: a role in repair of radiation-induced DNA damage, Clin. Cancer Res., 2007, 13(22 pt 1), 6555–6560, DOI:10.1158/1078-0432.CCR-07-1610.
- W. Han and H. W. Lo, Landscape of EGFR signaling network in human cancers: biology and therapeutic response in relation to receptor subcellular locations, Cancer Lett., 2012, 318(2), 124–134, DOI:10.1016/j.canlet.2012.01.011.
- G. Carpenter and H. J. Liao, Trafficking of receptor tyrosine kinases to the nucleus, Exp. Cell Res., 2009, 315(9), 1556–1566, DOI:10.1016/j.yexcr.2008.09.027.
- H. J. Liao and G. Carpenter, Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor, Cancer Res., 2009, 69(15), 6179–6183, DOI:10.1158/0008-5472.CAN-09-0049.
- K. Dittmann, C. Mayer and H. P. Rodemann, Inhibition of radiation-induced EGFR nuclear import by C225 (Cetuximab) suppresses DNA-PK activity, Radiother. Oncol., 2005, 76(2), 157–161, DOI:10.1016/j.radonc.2005.06.022.
- T. Kawamoto, J. D. Sato, A. Le, J. Polikoff, G. H. Sato and J. Mendelsohn, Growth stimulation of A431 cells by epidermal growth factor: identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody, Proc. Natl. Acad. Sci. U. S. A., 1983, 80(5), 1337–1341 CrossRef CAS PubMed.
Footnote |
| † First author: Yunxiang Qi and Jinyi Lang are joint first author. |
| This journal is © The Royal Society of Chemistry 2020 |