
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular control of unidirectional rotary motion in a sterically overcrowded photoswitchable receptor†
Jinyu
Sheng
 a,
Stefano
Crespi
a,
Ben L.
Feringa
*a and
Sander J.
Wezenberg
*ab
a,
Stefano
Crespi
a,
Ben L.
Feringa
*a and
Sander J.
Wezenberg
*ab
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; s.j.wezenberg@lic.leidenuniv.nl
bLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands
First published on 21st October 2020
Abstract
The control of molecular motion by external stimuli has attracted major interest in recent years. In order to achieve unidirectional rotational motion, intrinsically chiral light-driven molecular motors have been shown to be particularly versatile. We recently introduced a system in which unidirectional rotation is achieved in an achiral photoswitchable receptor upon binding of a chiral guest molecule. In order to gain detailed insight into the rotary steps, we present here a modified design in which the steric crowding is increased such that interconversion between the helical isomers of the (Z)-form of the receptor becomes very slow. DFT calculations support the increase in interconversion barrier. Furthermore, two diastereomeric complexes of the chiral guest bound to the (Z)-receptor are distinguished in the 1H NMR spectrum and the ratio between them slowly changes over time as a result of thermal helix inversion. On the other hand, no enrichment in one of the diastereomeric complexes is observed in the photochemical step. Our studies therefore unequivocally confirm that net rotation, induced by the chiral guest, takes place. This study affords an improved understanding of these dynamic supramolecular systems and expands the possibilities to control chirality transfer and motion at the molecular level, spurring developments of supramolecular recognition and information transfer at the nanoscale.
Introduction
For several decades now, chemists have strived to control motion at the molecular scale using appropriate chemical stimuli.1–8 Among the different types of motion, achieving unidirectional rotations in a molecule has been a particular challenging task.9–19 Our group10,11 has developed light-driven rotary molecular motors based on sterically overcrowded alkenes, while Lehn18 and Dube19 introduced imines, and hemothioindigo motors, respectively. Light is considered particularly useful to power molecular motors, since it is non-invasive and it can be applied with high spatiotemporal control. Hence, light-driven molecular motors have been successfully applied in various functional molecular materials.20,21 Nevertheless, novel designs and functions of molecular motors are still in high demand, especially with respect to (reversible) control over rotary motion using multiple stimuli,22 allowing the future development of more sophisticated and complex molecular machinery.In light of this, we recently reported a design in which unidirectional rotation in a stiff-stilbene based photoswitchable receptor,23–26 decorated with thiourea groups, is directed supramolecularly via binding of a chiral phosphate guest (Scheme 1).27 Here, the (E)-isomer of the photoswitchable anion receptor is converted to either the right- or left-handed helical form of its (Z)-isomer upon UV irradiation. In the presence of a chiral phosphate guest, the formation of one of these helical isomers is favoured and hence, the backward photochemical step takes place predominantly from this favoured isomer. The result is a net unidirectional rotation over the central double bond. While for previous light-driven molecular motors the direction of rotation was embedded in the (pseudo-)asymmetry of the molecular structure, the direction of rotation in this case was determined by the chirality of the guest molecule and could therefore be changed at will. However, because of the fast interconversion between the possible diastereomeric complexes of the (Z)-receptor bound with the phosphate guest, it could not be determined if preferred enantiomer formation would already take place in the photochemical step, beside the thermal step, possibly impeding unidirectionality. Inspired by our work on overcrowded alkene-based molecular motors, in which increased steric hindrance slows down the thermal helix inversion process,28–30 we now introduced a xylene (R = Me, Scheme 1) instead of a benzene motif in our stiff-stilbene derived bis-thiourea receptor. This modification allowed to monitor the thermal helix inversion at room temperature and to unequivocally establish that a net unidirectional rotation takes place. Furthermore, it shows that rotary speed modulation in this new type of supramolecularly-directed molecular motors is viable.
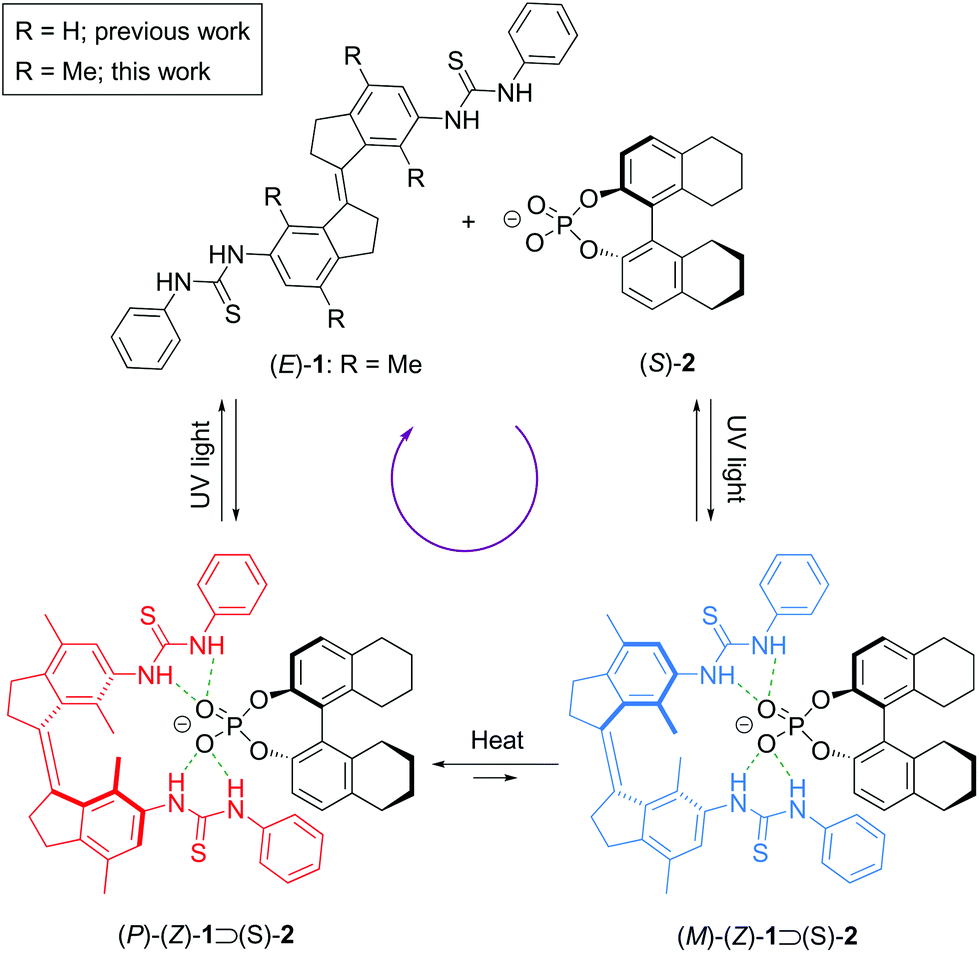 | ||
| Scheme 1 Supramolecularly-directed unidirectional rotation in a stiff-stilbene bis-thiourea receptor. | ||
Results and discussion
DFT calculations and synthesis
To assess the viability of our new design, we first conducted a series of DFT calculations at the SMD(DMSO)-ωB97X-D/def2-SVP(def2-TZVP) level of theory to predict the thermal isomerization barrier between the two helical forms of (Z)-1 (see the ESI for details†). To our delight, the predicted ΔG‡ was ca. 25 kcal mol−1 at 298.15 K for the new xylene based system (see Fig. 1), which corresponds to a half-life of over one day, much larger than that previously calculated for the compound with the benzene motif (ΔG‡ca. 4 kcal mol−1, t1/2 < 1 ns).27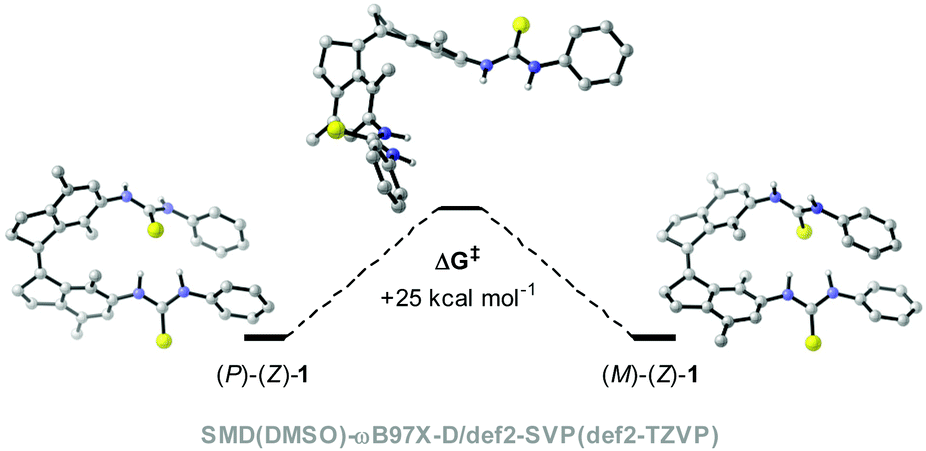 | ||
| Fig. 1 Energetic barrier for the helix inversion in (Z)-1. | ||
The (E)- and (Z)-isomer of the bis-thiourea receptor were synthesized starting from the commercially available 1,4-dibromo-2,5-dimethylbenzene 3 (Scheme 2). Indanone 5 was obtained in two steps following our previously reported protocol for a structurally related analog.31 McMurry coupling was then employed to yield (E)-6 and (Z)-6 in a 10:1 ratio, and the isomers could be separated by precipitation due to the different solubility.23 The low Z/E ratio in the McMurry coupling is ascribed to larger steric crowding in the (Z)-configuration.24,32 Subsequent Buchwald–Hartwig amination, followed by hydrolysis, afforded the bis-amine derivatives (E)-7 and (Z)-7, which were reacted with phenyl isothiocyanate to provide the corresponding bis-thioureas in good yield.
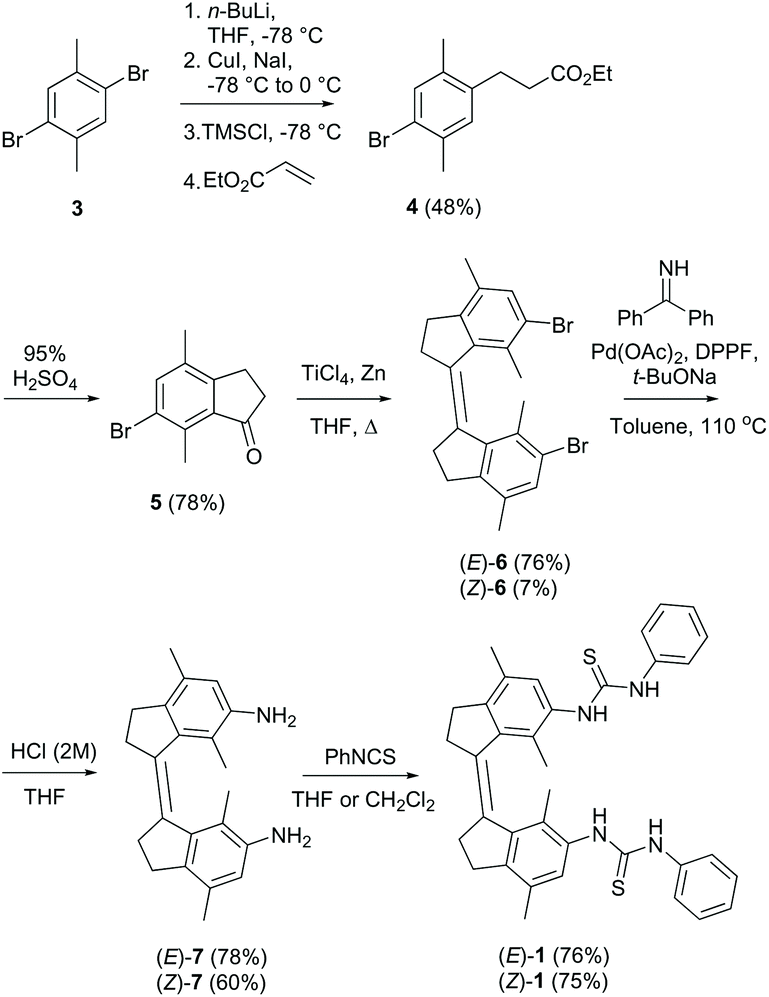 | ||
| Scheme 2 Synthesis of receptors (E)-1 and (Z)-1 (only depicted for the (Z)-isomer). | ||
Guest binding properties
Next, binding studies with [Bu4N]+[(S)-2)]− were performed in DMSO-d6/0.5% H2O using 1H NMR spectroscopy. Stepwise addition of [Bu4N]+[(S)-2)]− to a solution of (Z)-1 led to a significant downfield shift of the NH signals (Fig. 2a). Interestingly, these signals, together with the aromatic Ha and methyl signals, splitted into two sets with virtually equal integration (Fig. 2a, for full spectrum see Fig. S1 in the ESI†). This splitting is ascribed to the formation of two diastereomeric complexes, i.e. (P)-(Z)-1 ⊃ (S)-2 and (M)-(Z)-1 ⊃ (S)-2, of which the interconversion is slow on the NMR time scale. The titration data was analyzed considering the two host isomers [i.e. (P)-(Z)-1 and (M)-(Z)-1] and one guest [(S)-2] and two equilibrium constants (see Scheme S1 in the ESI†). Fitting of the data afforded Ka(1) = 4.6 × 102 M−1 and Ka(2) = 3.1 × 102 M−1 (Fig. S3 in the ESI†).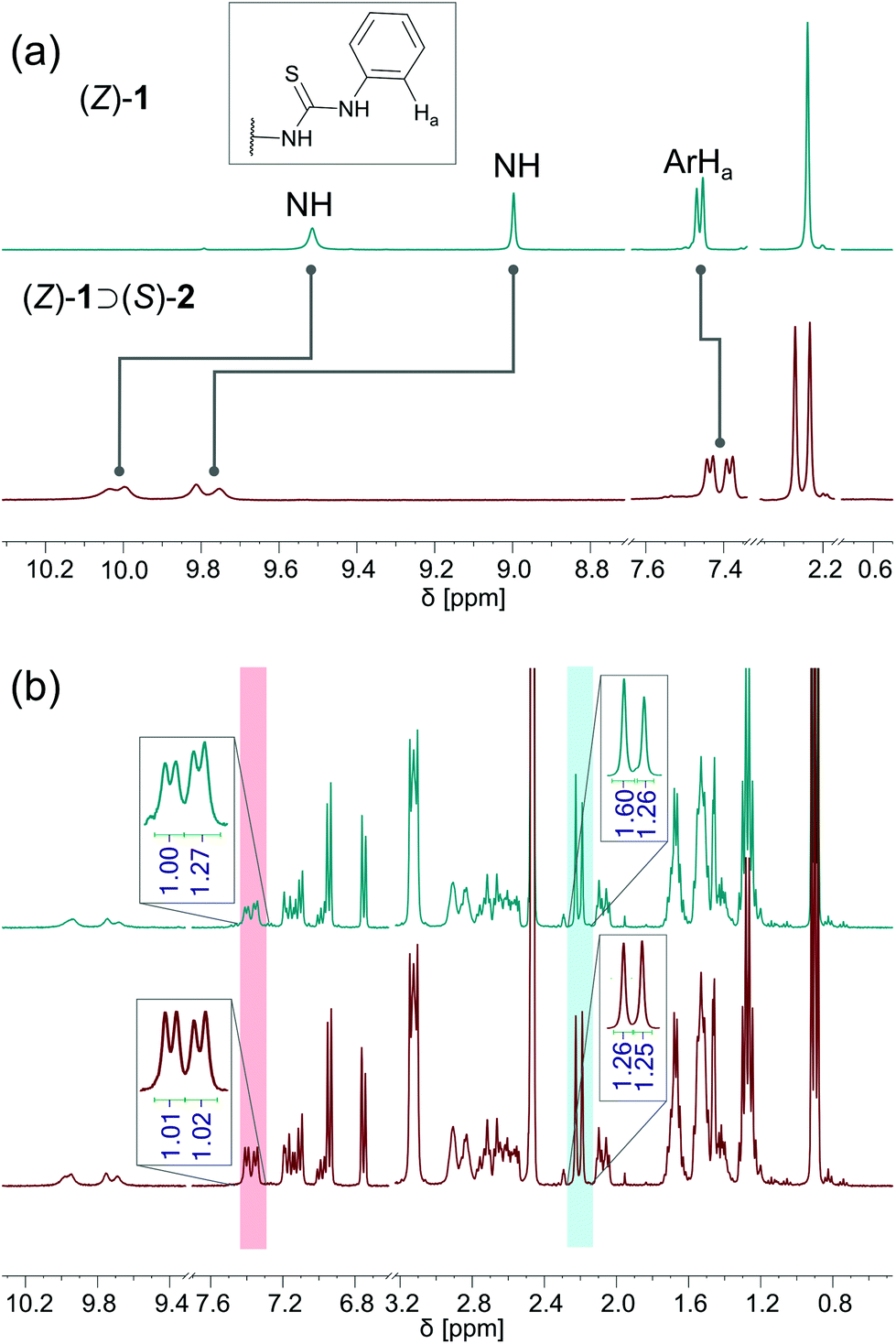 | ||
| Fig. 2 (a) 1H NMR of (Z)-1 (5 mM in DMSO-d6/0.5%H2O) before and after addition of 2 equivalents [Bu4N]+[(S)-2)]−; (b) 1H NMR changes of (Z)-1 in the presence of [Bu4N]+[(S)-2)]− freshly prepared (bottom) and after 3 days (top). | ||
Interestingly, when a mixture of (Z)-1 and [Bu4N]+[(S)-2)]− in DMSO-d6 was placed in the dark, the ratio between diastereomeric complexes had changed after 3 days from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1.27 (Fig. 2b and Fig. S4 and S5 in the ESI†), showing preferential formation of one of the diastereomeric complexes. DFT calculations at the SMD(DMSO)-ωB97X-D/def2-TZVPP//SMD(DMSO)-ωB97X-D/def2-SVP level of theory revealed that the calculated ΔG for the binding process of the (P)-(Z)-1 ⊃ (S)-2 complex is −13.4 kcal mol−1, which is larger than that calculated for (M)-(Z)-1 ⊃ (S)-2 (−9.7 kcal mol−1). Hence, the first complex is 3.7 kcal mol−1 lower in energy than the latter (Fig. 3). In the (P)-(Z)-1 ⊃ (S)-2 complex, the formation of a stabilizing π-stacking interaction between the phenyl substituent of the guest and the receptor is allowed, most likely accounting for the higher stability. This is reflected in the NH⋯O bond lengths being somewhat shorter (1.79 Å and 1.80 Å for (P)-(Z)-1 ⊃ (S)-2vs. 1.78 Å and 1.88 Å for (M)-(Z)-1 ⊃ (S)-2.
:1 to 1:1.27 (Fig. 2b and Fig. S4 and S5 in the ESI†), showing preferential formation of one of the diastereomeric complexes. DFT calculations at the SMD(DMSO)-ωB97X-D/def2-TZVPP//SMD(DMSO)-ωB97X-D/def2-SVP level of theory revealed that the calculated ΔG for the binding process of the (P)-(Z)-1 ⊃ (S)-2 complex is −13.4 kcal mol−1, which is larger than that calculated for (M)-(Z)-1 ⊃ (S)-2 (−9.7 kcal mol−1). Hence, the first complex is 3.7 kcal mol−1 lower in energy than the latter (Fig. 3). In the (P)-(Z)-1 ⊃ (S)-2 complex, the formation of a stabilizing π-stacking interaction between the phenyl substituent of the guest and the receptor is allowed, most likely accounting for the higher stability. This is reflected in the NH⋯O bond lengths being somewhat shorter (1.79 Å and 1.80 Å for (P)-(Z)-1 ⊃ (S)-2vs. 1.78 Å and 1.88 Å for (M)-(Z)-1 ⊃ (S)-2.
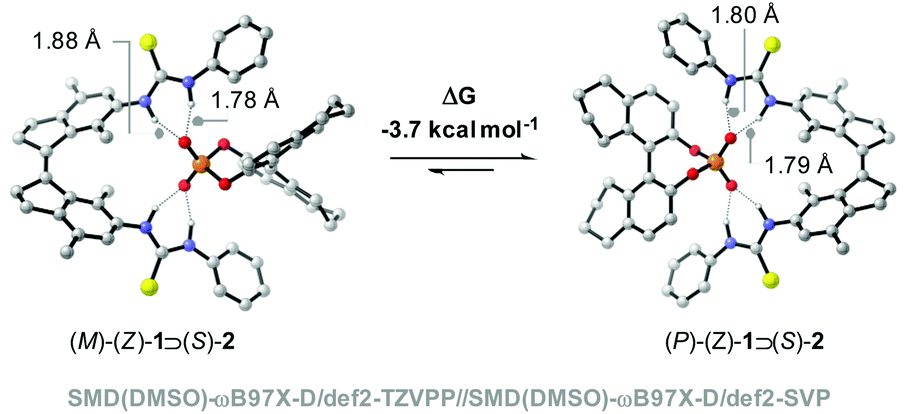 | ||
| Fig. 3 DFT-calculated structures of (M)-(Z)-1 ⊃ (S)-2 and (P)-(Z)-1 ⊃ (S)-2. | ||
Similar 1H NMR spectral changes were observed when [Bu4N]+[(R)-2)]− was added (Fig. S6 in the ESI†) and, based on the computational results, we predict that (S)-2 imposes a preference for (P)-(Z)-1 ⊃ (S)-2 and that (R)-2 favours the corresponding enantiomeric complex (M)-(Z)-1 ⊃ (R)-2. Consequently, the largest binding constant determined (vide supra) can be ascribed to binding of (S)-2 to (P)-(Z)-1 and the lower one to binding to (M)-(Z)-1. A titration of [Bu4N]+[(S)-2)]− to (E)-1 was also performed, but resulted in minor spectral changes, illustrative of very weak binding (see Fig. S2 in the ESI†). Additionally, the diastereomeric complexes were prepared in CD2Cl2 and a similar change in ratio was observed (Fig. S7 and S8 in the ESI†). However, equilibrium was reached faster (ca. 12 h) and the diastereomeric ratio (1:1.46) was higher than in DMSO. Furthermore, a change in CD absorption was observed with a freshly prepared solution of (Z)-1 in CH2Cl2 with either (S)-2 or (R)-2 after several hours (Fig. S10 in the ESI†).
Photoswitching behavior
The photoinduced interconversion between the (E)- and (Z)-form of the receptor was monitored by UV-vis and NMR spectroscopy in DMSO. Irradiation of (E)-1 with 312 nm light at room temperature resulted in a decrease of intensity of the absorption maximum at λ = 282 nm, and a new absorption band appeared at longer wavelength (Fig. 4a). These spectral changes are indicative of the formation of (Z)-1.23 Starting from (Z)-1, the opposite transformation could be triggered by irradiation with 365 nm light, affording the opposite spectral changes (Fig. S11 in the ESI†). In both cases, irradiation was continued until no further changes were observed, meaning that the photostationary states (PSS) had been reached. The presence of a clear isosbestic point revealed the unimolecular nature of the photoisomerization process.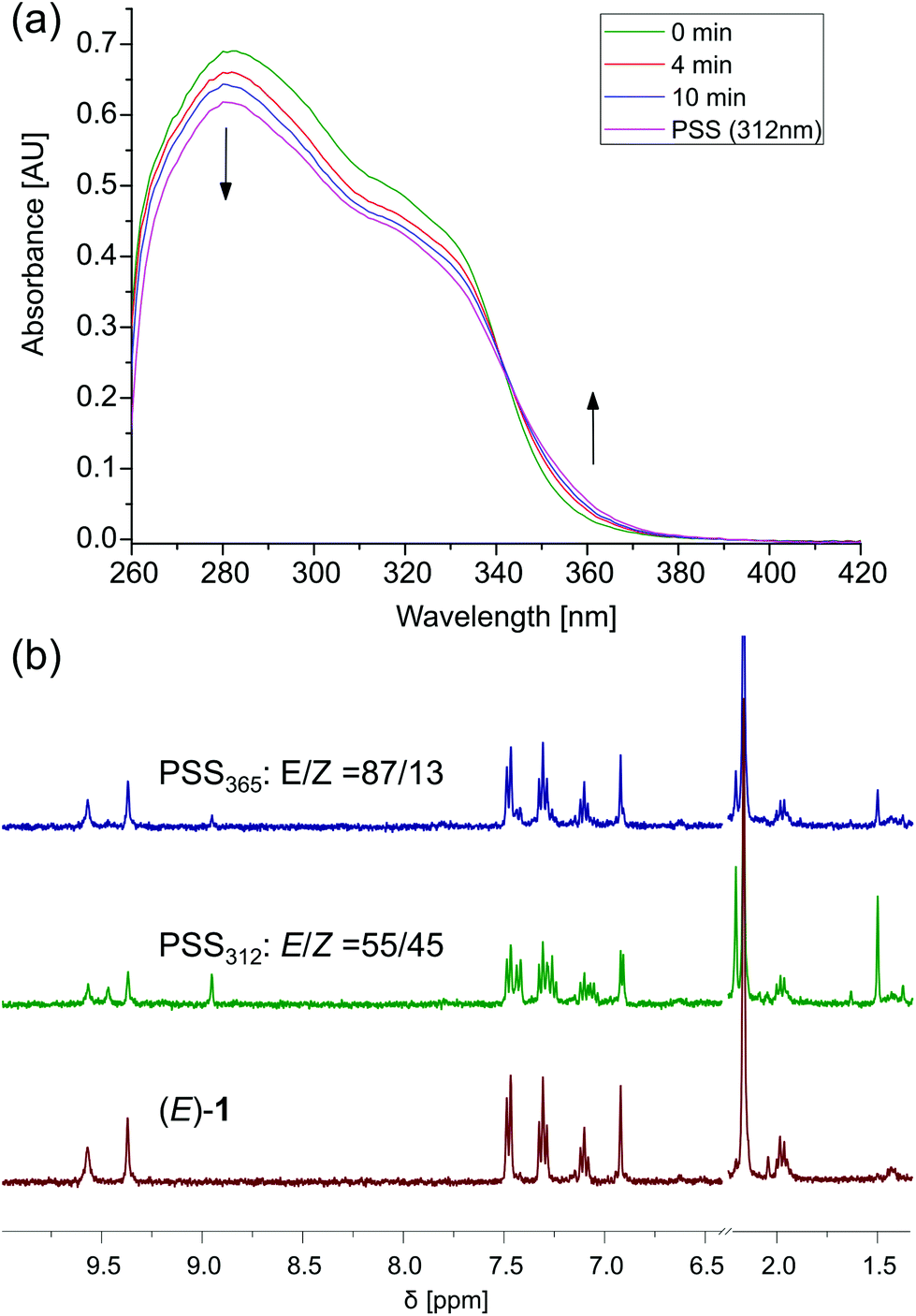 | ||
| Fig. 4 (a) UV/Vis spectral changes starting from (E)-1 upon 312 nm irradiation (1 × 10−5 M in degassed DMSO-d6/0.5%H2O); (b) 1H NMR changes upon 312 nm irradiation followed by 365 nm irradiation (1 × 10−3 M in degassed DMSO-d6/0.5%H2O). | ||
Irradiation with 312 nm light of a solution of (E)-1 in DMSO-d6/0.5%H2O led to the appearance of a new set of signals in the 1H NMR spectrum, which could be ascribed to (Z)-1 (Fig. 4b). Subsequent irradiation with 365 nm light led to partial recovery of the initial 1H NMR spectrum of (E)-1. The PSS312 and PSS365 ratios were determined by average integration of the urea and aromatic proton signals, affording (E/Z) 55:45 and 87:13, respectively. Similar results were observed when (Z)-1 was first irradiated with 365 nm light and successively with 312 nm light (see Fig. S12 in the ESI†).
To probe the photoisomerization behaviour in the presence of the chiral phosphate anion, a 1:1 mixture of (E)-1 and [Bu4N]+[(R)-2)]− in DMSO was prepared. Subsequently, irradiation with 312 nm light led to the emergence of two new sets of signals ascribed to the formation of the two possible diastereomeric complexes [i.e. (P)-(Z)-1 ⊃ (S)-2 and (M)-(Z)-1 ⊃ (S)-2] (Fig. 5a). Again, the PSS ratio was determined by the integration of aromatic proton signals and the ratio of 60:40 (E:Z) is similar to the ratio in absence of the anion. Very importantly, a 1:1 ratio of the diastereomeric complexes was observed immediately after irradiation. This result, combined with our finding that the thermal interconversion between (P)-(Z)-1 and (M)-(Z)-1 is slow on the NMR time scale in this design, reveals that no preferred formation of either the (P)- or (M)-helical (Z)-isomer takes place in the photochemical step. To further explore this aspect, we irradiated a 1:1 mixture of (Z)-1 and [Bu4N]+[(R)-2)]− with 365 nm light, showing consumption of both of the two diastereomeric complexes (PSS365 = 82:18, as determined by the integration of aromatic proton signals, see Fig. 5b). To this end, the enrichment in one of the helical (Z)-isomers was confirmed to occur only in the thermal step.
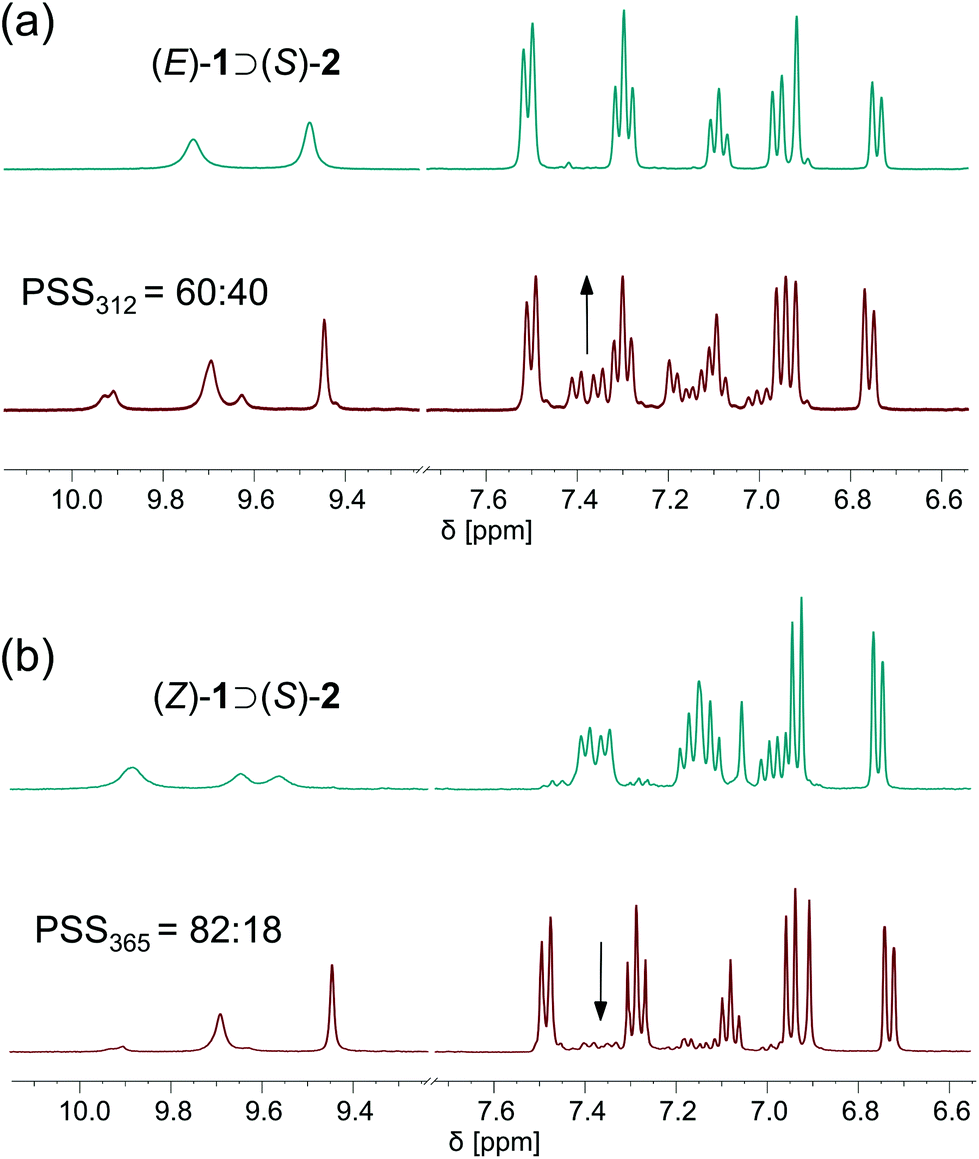 | ||
| Fig. 5 (a) 1H NMR spectra of (E)-1 (10 mM in DMSO-d6/0.5%H2O) with [Bu4N]+[(S)-2)]− before (top) and after (bottom) 312 nm light irradiation; (b) 1H NMR of (Z)-1 with [Bu4N]+[(S)-2)]− before (top) and after (bottom) 365 nm light irradiation. | ||
Conclusions
In summary, by synthesizing a photoaddressable stiff-stilbene receptor bearing a xylene core we have unequivocally established that supramolecularly induced unidirectional rotation takes place in the presence of a chiral phosphate guest. Binding of a chiral phosphate anion, e.g. (S)-2, to (Z)-1 led to the formation of two diastereomeric complexes in equal amount, which could be clearly distinguished in the 1H NMR spectrum. Over time, however, the (M)-(Z)-1 ⊃ (S)-2 complex slowly converted by helix inversion at ambient temperature to (P)-(Z)-1 ⊃ (S)-2 and vice versa when (R)-2 was used. That is, with this design, the helix inversion process can be observed, while in our previous work, this process was too fast at room temperature. Furthermore, UV light irradiation of (E)-1 in the presence of the chiral phosphate guest resulted in equal formation of (P)-(Z)-1 and (M)-(Z)-1, illustrating that enrichment in one of the helical isomers takes place only in the thermally activated step, and not in the photochemical step. This work therefore provides direct evidence that the non-covalent chiral phosphate binding induces unidirectional rotary motion since the chirality induction was observed in the ground state rather than in the excited state. These findings and new insights will further stimulate the use of supramolecular chirality transfer to control unidirectional rotary motion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the China Scholarship Council (CSC PhD Fellowship No. 201808330459 to J.S.), the Horizon 2020 Framework Programme (ERC Advanced Investigator Grant No. 694345 to B.L.F., ERC Starting Grant No. 802830 to S.J.W., and Marie Skłodowska-Curie Grant No. 838280 to S.C.) is gratefully acknowledged.Notes and references
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Artificial Molecular Machines, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef CAS.
- K. Kinbara and T. Aida, Toward Intelligent Molecular Machines: Directed Motions of Biological and Artificial Molecules and Assemblies, Chem. Rev., 2005, 105, 1377–1400 CrossRef CAS.
- B. L. Feringa, The Art of Building Small: From Molecular Switches to Molecular Motors, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS.
- B. Champin, P. Mobian and J. P. Sauvage, Transition metal complexes as molecular machine prototypes, Chem. Soc. Rev., 2007, 36, 358–366 RSC.
- V. Balzani, A. Credi and M. Venturi, Light powered molecular machines, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, Controlling Motion at the Nanoscale: Rise of the Molecular Machines, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS.
- S. Kassem, T. Van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Artificial molecular motors, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS.
- T. R. Kelly, H. De Silva and R. A. Silva, Unidirectional rotary motion in a molecular system, Nature, 1999, 401, 150–152 CrossRef CAS.
- N. Koumura, R. W. J. Zijistra, R. A. Van Delden, N. Harada and B. L. Feringa, Light-driven monodirectional molecular rotor, Nature, 1999, 401, 152–155 CrossRef CAS.
- N. Koumura, E. M. Geertsema, A. Meetsma and B. L. Feringa, Light-Driven Molecular Rotor: Unidirectional Rotation Controlled by a Single Stereogenic Center, J. Am. Chem. Soc., 2000, 122, 12005–12006 CrossRef CAS.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Unidirectional rotation in a mechanically interlocked molecular rotor, Nature, 2003, 424, 174–179 CrossRef CAS.
- J. V. Hernández, E. R. Kay and D. A. Leigh, A reversible synthetic rotary molecular motor, Science, 2004, 306, 1532–1537 CrossRef.
- S. P. Fletcher, F. Dumur, M. M. Pollard and B. L. Feringa, A reversible, unidirectional molecular rotary motor driven by chemical energy, Science, 2005, 310, 80–82 CrossRef CAS.
- J. C. M. Kistemaker, P. Štacko, J. Visser and B. L. Feringa, Unidirectional rotary motion in achiral molecular motors, Nat. Chem., 2015, 7, 890–896 CrossRef CAS.
- B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, A chemically powered unidirectional rotary molecular motor based on a palladium redox cycle, Nat. Chem., 2016, 8, 860–866 CrossRef CAS.
- M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, An autonomous chemically fuelled small-molecule motor, Nature, 2016, 534, 235–240 CrossRef CAS.
- L. Greb and J. M. Lehn, Light-Driven Molecular Motors: Imines as Four-Step or Two-Step Unidirectional Rotors, J. Am. Chem. Soc., 2014, 136, 13114–13117 CrossRef CAS.
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor, Nat. Commun., 2015, 6, 8406 CrossRef CAS.
- T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Dynamic control of function by light-driven molecular motors, Nat. Rev. Chem., 2017, 1, 0096 CrossRef CAS.
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors, Chem. Rev., 2020, 120, 310–433 CrossRef CAS.
- A. Faulkner, T. Van Leeuwen, B. L. Feringa and S. J. Wezenberg, Allosteric Regulation of the Rotational Speed in a Light-Driven Molecular Motor, J. Am. Chem. Soc., 2016, 138, 13597–13603 CrossRef CAS.
- S. J. Wezenberg and B. L. Feringa, Photocontrol of Anion Binding Affinity to a Bis-urea Receptor Derived from Stiff-Stilbene, Org. Lett., 2017, 19, 324–327 CrossRef CAS.
- D. Villarón and S. J. Wezenberg, Stiff–Stilbene Photoswitches: From Fundamental Studies to Emergent Applications, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef.
- S. Lee and A. H. Flood, Photoresponsive receptors for binding and releasing anions, J. Phys. Org. Chem., 2013, 26, 79–86 CrossRef CAS.
- D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Photoresponsive Host–Guest Functional Systems, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS.
- S. J. Wezenberg and B. L. Feringa, Supramolecularly directed rotary motion in a photoresponsive receptor, Nat. Commun., 2018, 9, 1984 CrossRef.
- J. Vicario, A. Meetsma and B. L. Feringa, Controlling the speed of rotation in molecular motors. Dramatic acceleration of the rotary motion by structural modification, Chem. Commun., 2005, 116, 5910–5912 RSC.
- J. Vicario, M. Walko, A. Meetsma and B. L. Feringa, Fine Tuning of the Rotary Motion by Structural Modification in Light-Driven Unidirectional Molecular Motors, J. Am. Chem. Soc., 2006, 128, 5127–5135 CrossRef CAS.
- W. Li, C. Jiao, X. Li, Y. Xie, K. Nakatani, H. Tian and W. Zhu, Separation of Photoactive Conformers Based on Hindered Diarylethenes: Efficient Modulation in Photocyclization Quantum Yields, Angew. Chem., Int. Ed., 2014, 53, 4603–4607 CrossRef CAS.
- T. M. Neubauer, T. Van Leeuwen, D. Zhao, A. S. Lubbe, J. C. M. Kistemaker and B. L. Feringa, Asymmetric Synthesis of First Generation Molecular Motors, Org. Lett., 2014, 16, 4220–4223 CrossRef CAS.
- D. Lenoir and P. Lemmen, Sterisch gehinderte Olefine, VIII. Reaktion von 1−Indanonen mit niedrigwertigen Titanverbindungen. Sterischer Einfluß von Methylsubstituenten bei 1−(1−Indanyliden)indan und 9−(9−Fluorenyliden)fluoren, Chem. Ber., 1980, 113, 3112–3119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization of new compounds, 1H NMR, UV-vis and CD studies as well as theoretical calculations. See DOI: 10.1039/d0qo01154a |
| This journal is © the Partner Organisations 2020 |