
Ruthenium(II)-catalyzed reductive N–O bond cleavage of N-OR (R = H, alkyl, or acyl) substituted amides and sulfonamides†
Tingjie
You
,
Maosheng
Zhang
,
Jianhui
Chen
 *,
Hongmei
Liu
and
Yuanzhi
Xia
*
*,
Hongmei
Liu
and
Yuanzhi
Xia
*
College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, China. E-mail: cjh@wzu.edu.cn; xyz@wzu.edu.cn
First published on 13th November 2020
Abstract
With a commercially available ruthenium(II) catalyst and a mixture of HCOOH/NEt3 as the hydride source under an air atmosphere, a convenient method for the reductive cleavage of N–O bonds was described. This catalytic system was applicable for a variety of N-oxygen-substituted amides, as well as N-alkoxy sulfonamides, efficiently delivering the corresponding amide or primary sulfonamide products with good functional group tolerance in moderate to good yields.
Introduction
The development of an efficient strategy to access primary amides is an important topic in synthetic organic chemistry, which has been widely used in the fields of materials, pesticides and pharmaceuticals.1 Several classical methods such as Schmidt-type reaction,2 hydration of nitrile compounds3 and amidation reaction of carboxylic acid derivatives4 have been developed over the past few years. On the other hand, transition metal-catalyzed C–H activation of amides represented a powerful methodology to increase the molecular complexity of amide derivatives.5 However, these transformations were generally realized by employing N-oxygen-substituted amide functionalities as the directing groups (Fig. 1) owing to the low reactivity of the free amide group in such reactions.6 While the N–O moiety in the directing group may act as an internal oxidant in certain systems,7 it is still retained in the products in many cases.8 Thus, the development of an effective method for removing the activating oxygen-containing substituents would be very useful to produce the value-added primary amide. | ||
| Fig. 1 Selected examples of directing groups containing an N-oxygen-substituted amide moiety for C–H activation. | ||
Traditional methods for the reductive cleavage of N–O bonds usually featured the use of stoichiometric amounts of metal reagents including titanium(III),9 samarium diiodide,10 lithium powder,11 sodium amalgam12 and Cu(II) ions.13 In 2008, Murphy and co-workers developed a metal-free reductive cleavage of N–O bonds in Weinreb amides with 1.5 equivalents of an organic neutral super-electron donor, delivering the corresponding secondary amides as the products.14 Very recently, the Du group disclosed an elemental sulfur-mediated conversion of N-alkoxyamides to primary amides.15 Although various methods have been reported, the problems associated with the formation of a lot of solid waste and limited substrate scope are yet to be solved. Hence, the development of a novel catalytic process16 for efficient reductive cleavage of N–O bonds is highly desirable.
Transition metal-catalyzed transfer hydrogenation is regarded as a convenient and powerful method to access target hydrogenated products, and has been widely adopted in recent years.17 As an attractive alternative protocol to direct hydrogenation with hydrogen gas, transfer hydrogenation has recently become the center of research in hydrogenation science. Using readily available and inexpensive hydrogen donors including alcohols, acids and amines, catalytic transfer hydrogenation and asymmetric transfer hydrogenation reactions of unsaturated compounds, such as ketones,18 imines,19 as well as alkenes20 and alkynes21 have been well established (Scheme 1a).22 This methodology is also applicable for the reductive cleavage of single polar bonds, and has been employed as an efficient approach for transfer hydro-dehalogenation of organic halides (Scheme 1b).23 However, little attention has been paid to the reduction of other single polar bonds.24 During the course of our previous investigation on transfer hydro-dehalogenation of organic halides, we found the Ru(II) catalyst to be efficient at transforming C–X bonds (X = Cl, Br, or I) into C–H bonds using the iso-propanol solvent as the hydride source.23h This encouraged us to extend the transfer hydrogenation catalysis to the other single polar bonds. Herein, we report a Ru-catalyzed reductive cleavage of N–O bonds in N-oxygen-substituted amides and sulfonamides using HCOOH/NEt3 as the hydride source without any additives under air (Scheme 1c).
 | ||
| Scheme 1 The applications of transition metal-catalyzed transfer hydrogenation. | ||
Results and discussion
We initiated our study by identifying reliable conditions for this Ru-catalyzed reductive cleavage of N–O bonds starting from N-methoxybenzamide (1a). After screening the reaction parameters, we found that 1a could be converted to the desired product 2a in almost quantitative yield (92% isolated yield) using 2.5 mol% of [RuCl2(p-cymene)]2 as the catalyst, and H2O/HCOOH/NEt3 as a co-solvent and hydride source (Table 1, entry 1). The metal catalyst was proven to be essential (entry 2), and when Ru(PPh3)3Cl2 was used as the catalyst, the yield of 2a decreased to 22% (entry 3). Employing [RhCp*Cl2]2 instead of [RuCl2(p-cymene)]2 also led to an excellent yield of 2a (entry 4). In the absence of HCOOH/NEt3 or H2O, the reaction gave 2a in 6% and 57% yields, respectively (entries 5 and 6). On using MeOH, EtOH or iPrOH instead of the co-solvent mixture, the yield of 2a decreased obviously (entries 7–9). The yield of 2a decreased dramatically when the reaction was run at a relatively lower temperature, indicating that heating at 100 °C is necessary for high yield (entry 10).|
|
||
|---|---|---|
| Entry | Deviation of standard conditionsa | Yieldb/% |
a Reaction conditions: 1a (0.2 mmol), catalyst (2.5 mol%), HCOOH/Et3N (molar ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.5 mL), and H2O (0.5 mL) at 100 °C, 12 h.
b Yields were determined by 1H NMR analysis using mesitylene as an internal standard.
c Isolated yield is shown in brackets. :1, 0.5 mL), and H2O (0.5 mL) at 100 °C, 12 h.
b Yields were determined by 1H NMR analysis using mesitylene as an internal standard.
c Isolated yield is shown in brackets.
|
||
| 1 | None | 99 (92)c |
| 2 | Without [RuCl2(p-cymene)]2 | 0 |
| 3 | Ru(PPh3)3Cl2 instead of [RuCl2(p-cymene)]2 | 22 |
| 4 | [RhCp*Cl2]2 instead of [RuCl2(p-cymene)]2 | 99 |
| 5 | H2O instead of a co-solvent mixture | 6 |
| 6 | HCOOH/Et3N (1/1) instead of a co-solvent mixture | 57 |
| 7 | MeOH instead of a co-solvent mixture | 16 |
| 8 | EtOH instead of a co-solvent mixture | 8 |
| 9 | iPrOH instead of a co-solvent mixture | 45 |
| 10 | At 80 °C | 65 |

The scope of this catalytic system turned out to be broad. With the optimized conditions, the substrate scope of N-alkoxy amides is illustrated in Table 2. Various electron-donating or -withdrawing groups at the phenyl ring of N-methoxybenzamides, such as methyl, alkoxy, chloro and phenyl groups, were well tolerated, affording the corresponding primary amides in good to excellent yields (1a–1i). A slight decrease in the yield was observed when the substrate contained a free phenolic hydroxyl group (1j). The reactions of N-methoxybenzamides bearing two substituents on the phenyl ring resulted in moderate yields (1k and 1l). N-Methoxy poly- and heterocycle substituted amides, such as 1-naphthyl (1m), 2-naphthyl (1n), 4-pyridinyl (1o), 2-furoyl (1p) and 2-thienyl (1q) amides, were quite suitable for this catalytic transformation, yielding the desired amides in 33–99% yields. Methoxy aliphatic amides (1r–1t) were also compatible with this method, showing a similar reactivity under the standard conditions. N-Alkoxy-benzamides containing ethoxy (1u) and benzyloxy (1v) substituents proceeded smoothly to give benzamides in 91% and 88% yields, respectively. Benzohydroxamic acids (1w–1y) were then investigated, and they afforded the desired products in 78–83% yields. The reactivities were relatively lower in the case of acyloxy benzamides (1aa–1ae). The nitro group could also be well tolerated in this reaction, delivering the desired product in moderate yield (1ac). The N–O bond cleavage catalytic system could be applied to a tertiary amide (1ad), giving the corresponding secondary amide in 64% yield. In addition, N-(benzoyloxy)succinimide (1ae) was also compatible with this catalytic system, producing benzoic acid as the product in excellent yield. We found that the substrates containing alkyl halide, cyano, alkyne, and alkene moieties led to complex mixtures, indicating that these groups are not compatible in the current transformation.

Next, we extended this N–O bond cleavage methodology to N-alkoxy sulfonamides (Scheme 2). N-Methoxy sulfonamides were quite suitable for this catalytic system, producing the corresponding primary sulfonamides in excellent yields under the standard conditions (4a–4d).
 | ||
| Scheme 2 Ru(II)-catalyzed N–O bond cleavage of N-alkoxy sulfonamides. | ||
To further showcase the synthetic utility, a gram-scale reaction was performed smoothly to give the desired product without compromising the yield [eqn. (1)].
 | (1) |
On the basis of previous reports,23h,25 a plausible mechanism for this Ru(II)-catalyzed N–O bond cleavage reaction is proposed in Scheme 3. First, a ligated ruthenium hydride species (A) is formed by treating [RuCl2(cymene)]2 with HCOOH/NEt3. Then oxidative addition of the N–O bond to the Ru(II) center generates a Ru(IV) complex (B), which would undergo reductive elimination to give the desired product and a Ru(II) alkoxy species (C). Subsequently, a ligand exchange occurred between C and HCOO− to deliver a Ru(II) formate complex (D). Finally, D released a CO2 molecule to regenerate A.
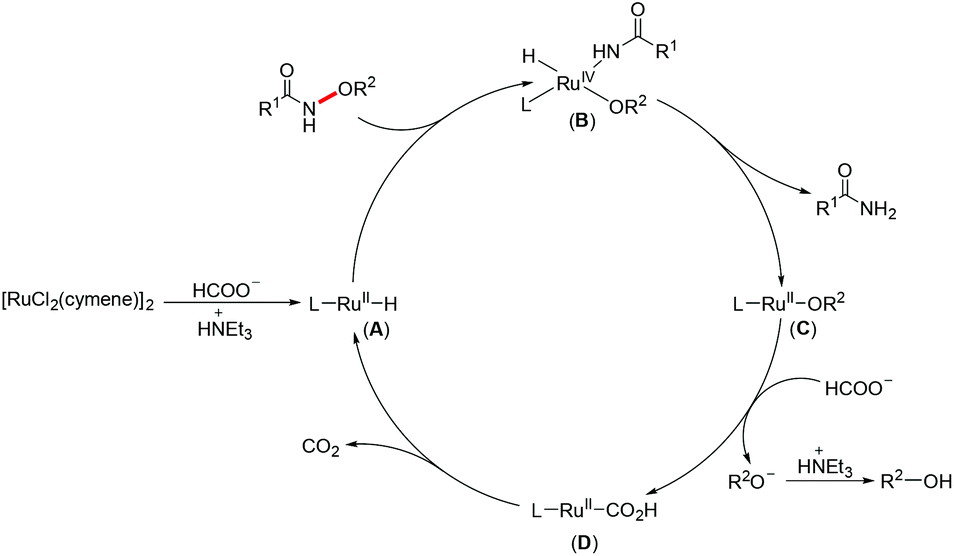 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, we have developed a Ru-catalyzed N–O bond cleavage of N-oxygen-substituted amides and sulfonamides employing the transfer hydrogenation methodology using H2O/HCOOH/NEt3 as the co-solvent and hydride source. This catalytic system was applicable for a variety of substituted amides and sulfonamides, delivering the corresponding amides and primary sulfonamides in moderate to excellent yields. The reaction could be scaled up to gram scale smoothly under air. Featuring a commercially available catalyst, simple and easy operation, and readily available starting materials with good functional group compatibility, this is a practical method to access valuable primary amides. Further investigations on the reductive cleavage of the other single polar bonds with the transfer hydrogenation methodology are undergoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by NSFC (No. 21801191, 21572163, and 21873074) and Wenzhou Science & Technology Bureau (G20180016).Notes and references
- (a) V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS; (b) J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Expanding the medicinal chemistry synthetic toolbox, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef.
- For selected examples, see: (a) K. F. Schmidt, Über die Einwirkung von NH auf organische Verbindungen, Angew. Chem., 1923, 36, 511 Search PubMed; (b) J.-J. Shie and J.-M. Fang, Direct conversion of aldehydes to amides, tetrazoles, and triazines in aqueous media by one-pot tandem reactions, J. Org. Chem., 2003, 68, 1158–1160 CrossRef CAS; (c) G. E. Veitch, K. L. Bridgwood and S. V. Ley, Magnesium nitride as a convenient source of ammonia: preparation of primary amides, Org. Lett., 2008, 10, 3623–3625 CrossRef CAS; (d) J. Liu, C. Zhang, Z. Zhang, X. Wen, X. Dou, J. Wei, X. Qiu, S. Song and N. Jiao, Nitromethane as a nitrogen donor in Schmidt-type formation of amides and nitriles, Science, 2020, 367, 281–285 CrossRef CAS.
- For selected examples, see: (a) K. Yamaguchi, M. Matsushita and N. Mizuno, Efficient hydration of nitriles to amides in water, catalyzed by ruthenium hydroxide supported on alumina, Angew. Chem., Int. Ed., 2004, 43, 1576–1580 CrossRef CAS; (b) A. Goto, K. Endo and S. Saito, RhI-catalyzed hydration of organonitriles under ambient conditions, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS; (c) P. Marcé, J. Lynch, A. J. Blacker and J. M. J. Williams, A mild hydration of nitriles catalysed by copper(II) acetate, Chem. Commun., 2016, 52, 1436–1438 RSC; (d) E. Liardo, R. González-Fernández, N. Ríos-Lombardía, F. Morís, J. García-Álvarez, V. Cadierno, P. Crochet, F. Rebolledo and J. González-Sabín, Strengthening the combination between enzymes and metals in aqueous medium: concurrent ruthenium-catalyzed nitrile hydration-asymmetric ketone bioreduction, ChemCatChem, 2018, 10, 4676–4682 CrossRef CAS; (e) K. Wang, P. Jiang, M. Yang, P. Ma, J. Qin, X. Huang, L. Ma and R. Li, Metal-free nitrogen -doped carbon nanosheets: a catalyst for the direct synthesis of imines under mild conditions, Green Chem., 2019, 21, 2448–2461 RSC.
- For selected examples, see: (a) Y. Liu, C. Lei, X.-Y. Xu, X.-S. Shao and Z. Li, Design, synthesis and insecticidal activity of biphenyl-diamide derivatives, Chin. Chem. Lett., 2016, 27, 321–324 CrossRef CAS; (b) X. Qi, H.-J. Ai, C.-X. Cai, J.-B. Peng, J. Ying and X.-F. Wu, A convenient palladium-catalyzed aminocarbonylation of aryl iodides to primary amides under gas-free conditions, Eur. J. Org. Chem., 2017, 7222–7225 CrossRef CAS; (c) A. M. Veatch and E. J. Alexanian, Cobalt-catalyzed aminocarbonylation of (hetero) aryl halides promoted by visible light, Chem. Sci., 2020, 11, 7210–7213 RSC; (d) J. Chen, Y. Xia and S. Lee, Transamidation for the synthesis of primary amides at room temperature, Org. Lett., 2020, 22, 3504–3508 CrossRef CAS.
- Selected recent reviews for C-H activation of amides, see: (a) C. Zhu, R. Wang and J. R. Falck, Amide-directed tandem C–C/C–N bond formation through C–H Activation, Chem. – Asian J., 2012, 7, 1502–1514 CrossRef CAS; (b) R. Das, G. S. Kumar and M. Kapur, Amides as weak coordinating groups in proximal C–H bond activation, Eur. J. Org. Chem., 2017, 5439–5459 CrossRef CAS; (c) J. Kalepu and L. T. Pilarski, Weinreb amides as directing groups for transition metal-catalyzed C-H functionalizations, Molecules, 2019, 24, 830–851 CrossRef; (d) Q. Zheng, C.-F. Liu, J. Chen and G.-W. Rao, C–H functionalization of aromatic amides, Adv. Synth. Catal., 2020, 362, 1406–1446 CrossRef CAS.
- For selected references, see: (a) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, Pd(II)-catalyzed cross-coupling of sp3 C–H bonds with sp2 and sp3 boronic acids using air as the oxidant, J. Am. Chem. Soc., 2008, 130, 7190–7191 CrossRef CAS; (b) M. Wasa and J.-Q. Yu, Synthesis of β-, γ-, and δ-lactams via Pd(II)-catalyzed C–H activation reactions, J. Am. Chem. Soc., 2008, 130, 14058–14059 CrossRef CAS; (c) N. Guimond, C. Gouliaras and K. Fagnou, Rhodium(III)-catalyzed isoquinolone synthesis: the N–O bond as a handle for C–N bond formation and catalyst turnover, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS; (d) G.-W. Wang, T.-T. Yuan and D.-D. Li, One-pot formation of C–C and C–N bonds through palladium-catalyzed dual C–H activation: synthesis of phenanthridinones, Angew. Chem., Int. Ed., 2011, 50, 1380–1383 CrossRef CAS; (e) J. Karthikeyan and C.-H. Cheng, Synthesis of phenanthridinones from N-methoxybenzamides and arenes by multiple palladium-catalyzed C–H activation steps at room temperature, Angew. Chem., Int. Ed., 2011, 50, 9880–9883 CrossRef CAS; (f) L. Ackermann and S. Fenner, Ruthenium-catalyzed C–H/N–O bond functionalization: green isoquinolone syntheses in water, Org. Lett., 2011, 13, 6548–6551 CrossRef CAS; (g) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, Mild Rh(III)-catalyzed C–H activation and annulation with alkyne MIDA boronates: short, efficient synthesis of heterocyclic boronic acid derivatives, J. Am. Chem. Soc., 2012, 134, 19592–19595 CrossRef CAS; (h) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Biotinylated Rh(III) complexes in engineered streptavidin for accelerated asymmetric C–H activation, Science, 2012, 338, 500–503 CrossRef CAS; (i) B. Ye and N. Cramer, Chiral cyclopentadienyl ligands as stereocontrolling element in asymmetric C–H functionalization, Science, 2012, 338, 504–506 CrossRef CAS; (j) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Ruthenium-catalyzed oxidative C–H bond olefination of N-methoxybenzamides using an oxidizing directing group, Org. Lett., 2012, 14, 736–739 CrossRef CAS; (k) R. Zeng, S. Wu, C. Fu and S. Ma, Room-temperature synthesis of trisubstituted allenylsilanes via regioselective C–H functionalization, J. Am. Chem. Soc., 2013, 135, 18284–18287 CrossRef CAS; (l) R.-Y. Zhu, M. E. Farmer, Y.-Q. Chen and J.-Q. Yu, A simple and versatile amide directing group for C–H functionalizations, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS.
- For selected examples, see: (a) F. W. Patureau and F. Glorius, Oxidizing directing groups enable efficient and innovative C–H activation reactions, Angew. Chem., Int. Ed., 2011, 50, 1977–1979 CrossRef CAS; (b) L. Xu, Q. Zhu, G. Huang, B. Cheng and Y. Xia, Computational elucidation of the internal oxidant-controlled reaction pathways in Rh(III)-catalyzed aromatic C–H functionalization, J. Org. Chem., 2012, 77, 3017–3024 CrossRef CAS; (c) H. Huang, X. Ji, W. Wu and H. Jiang, Transition metal-catalyzed C–H functionalization of N-oxyenamine internal oxidants, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (d) J. Mo, L. Wang, Y. Liu and X. Cui, Transition-metal-catalyzed direct C–H functionalization under external-oxidant-free conditions, Synthesis, 2015, 439–459 CAS; (e) T. Zhou, W. Guo and Y. Xia, RhV-nitrenoid as a key intermediate in RhIII-catalyzed heterocyclization by C–H activation: a computational perspective on the cycloaddition of benzamide and diazo compounds, Chem. – Eur. J., 2015, 21, 9209–9218 CrossRef CAS; (f) W. Guo, T. Zhou and Y. Xia, Mechanistic understanding of the aryl-dependent ring formations in Rh(III)-catalyzed C–H activation/cycloaddition of benzamides and methylenecyclopropanes by DFT calculations, Organometallics, 2015, 34, 3012–3020 CrossRef CAS; (g) W. Guo and Y. Xia, Mechanistic understanding of the divergent reactivity of cyclopropenes in Rh(III)-catalyzed C–H activation/cycloaddition reactions of N-phenoxyacetamide and N-pivaloxybenzamide, J. Org. Chem., 2015, 80, 8113–8121 CrossRef CAS; (h) J. Chen, W. Guo and Y. Xia, Computational revisit to the β-carbon elimination step in Rh(III)-catalyzed C–H activation/cycloaddition reactions of N-phenoxyacetamide and cyclopropenes, J. Org. Chem., 2016, 81, 2635–2638 CrossRef CAS; (i) Z. Wang, P. Xie and Y. Xia, Recent progress in Ru(II)-catalyzed C–H activations with oxidizing directing groups, Chin. Chem. Lett., 2018, 29, 47–53 CrossRef CAS; (j) J.-L. Pan, P. Xie, C. Chen, Y. Hao, C. Liu, H.-Y. Bai, J. Ding, L.-R. Wang, Y. Xia and S.-Y. Zhang, Rhodium(III)-catalyzed redox-neutral cascade [3 + 2] annulation of N-phenoxyacetamides with propiolates via C–H functionalization/isomerization/lactonization, Org. Lett., 2018, 20, 7131–7136 CrossRef CAS.
- For selected examples, see: (a) G.-W. Wang and T.-T. Yuan, Palladium-catalyzed alkoxylation of N-methoxybenzamides via direct sp2 C–H bond activation, J. Org. Chem., 2010, 75, 476–479 CrossRef CAS; (b) R. Zeng, C. Fu and S. Ma, Highly selective mild stepwise allylation of N-methoxybenzamides with allenes, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef CAS; (c) B. Ye and N. Cramer, A tunable class of chiral Cp ligands for enantioselective rhodium(III)-catalyzed C–H allylations of benzamides, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS; (d) S.-S. Zhang, J.-Q. Wu, Y.-X. Lao, X.-G. Liu, Y. Liu, W.-X. Lv, D.-H. Tan, Y.-F. Zeng and H. Wang, Mild rhodium(III)-catalyzed C–H allylation with 4-vinyl-1,3-dioxolan-2-ones: direct and stereoselective synthesis of (E)-allylic alcohols, Org. Lett., 2014, 16, 6412–6415 CrossRef CAS; (e) B. Ye and N. Cramer, Chiral cyclopentadienyl ligands enable a rhodium(III)-catalyzed enantioselective access to hydroxychromanes and phthalides, Synlett, 2015, 26, 1490–1495 CrossRef CAS; (f) Z. Chen, L. Hu, F. Zeng, R. Zhu, S. Zheng, Q. Yu and J. Huang, Selective mono-alkylation of N-methoxybenzamides, Chem. Commun., 2017, 53, 4258–4261 RSC.
- (a) P. G. Mattingly and M. J. Miller, Titanium trichloride reduction of substituted N-hydroxy-2-azetidinones and other hydroxamic acids, J. Org. Chem., 1980, 45, 410–415 CrossRef CAS; (b) L. E. Fisher, J. M. Caroon, Jahangir, S. R. Stabler, S. Lundberg and J. M. Muchowski, O-Methylarenehydroxamates as ortho-lithiation directing groups. Titanium(III)-mediated conversion of O-methyl hydroxamates to primary amides, J. Org. Chem., 1993, 58, 3643–3647 CrossRef CAS; (c) C. Cesario, L. P. Tardibono Jr. and M. J. Miller, Titanocene(III) chloride-mediated reductions of oxazines, hydroxamic acids, and N-hydroxy carbamates, J. Org. Chem., 2009, 74, 448–451 CrossRef CAS.
- (a) G. E. Keck, S. F. McHardy and T. T. Wager, Reductive cleavage of N-O bonds in hydroxylamine and hydroxamic acid derivatives using SmI2/THF, Tetrahedron Lett., 1995, 36, 7419–7422 CrossRef CAS; (b) J. L. Chiara, C. Destabel, P. Gallego and J. Marco-Contelles, Cleavage of N-O bonds promoted by samarium diiodide: reduction of free or N-acylated O-alkylhydroxylamines, J. Org. Chem., 1996, 61, 359–360 CrossRef CAS; (c) Y. Ishido, N. Kanbayashi, T. Okamura and K. Onitsuka, Synthesis of nonnatural helical polypeptide via asymmetric polymerization and reductive cleavage of N–O bond, Macromolecules, 2017, 50, 5301–5307 CrossRef CAS.
- M. Yus, G. Radivoy and F. Alonso, Lithium/DTBB-induced reduction of N-alkoxyamides and acyl azides, Synthesis, 2001, 914–918 CrossRef CAS.
- J. E. Jackson, B. N. O'Brien, S. K. Kedzior, G. R. Fryz, F. S. Jalloh, A. Banisafar, M. A. Caldwell, M. B. Braun, B. M. Dunyak and J. L. Dye, Reductive N–O cleavage of Weinreb amides by sodium in alumina and silica gels: synthetic and mechanistic studies, Tetrahedron Lett., 2015, 56, 6227–6230 CrossRef CAS.
- S. P. Wathen and A. W. Czarnik, Cu(II)-catalysis in the reduction of N,O-Dibenzoylhydroxylamine by a tertiary amine, Bioorg. Med. Chem. Lett., 1993, 3, 1245–1246 CrossRef CAS.
- S. P. Y. Cutulic, J. A. Murphy, H. Farwaha, S.-Z. Zhou and E. Chrystal, Metal-free reductive cleavage of N–O bonds in Weinreb amides by an organic neutral super-electron donor, Synlett, 2008, 2132–2136 CAS.
- S. Wang, X. Zhao, D. Zhang-Negrerie and Y. Du, Reductive cleavage of the N–O bond: elemental sulfur-mediated conversion of N-alkoxyamides to amides, Org. Chem. Front., 2019, 6, 347–351 RSC.
- Recently, several transformations mediated via N–O bond cleavage with transition metal catalysis have been reported; for selected examples, see: (a) N. Asao, K. Sato and Y. Yamamoto, AuBr3-catalyzed cyclization of o-(alkynyl)nitrobenzenes. Efficient synthesis of isatogens and anthranils, Tetrahedron Lett., 2003, 44, 5675–5677 CrossRef CAS; (b) C. V. Ramana, P. Patel, K. Vanka, B. Miao and A. Degterev, A Combined Experimental and Density Functional Theory Study on the Pd-Mediated Cycloisomerization of o-Alkynylnitrobenzenes-Synthesis of Isatogens and Their Evaluation as Modulators of ROS-Mediated Cell Death, Eur. J. Org. Chem., 2010, 5955–5966 CrossRef CAS; (c) L. Ackermann, A. V. Lygin and N. Hofmann, Ruthenium-Catalyzed Oxidative Annulation by Cleavage of C–H/N–H Bonds, Angew. Chem., Int. Ed., 2011, 50, 6379–6382 CrossRef CAS; (d) C. Kornhaaß, J. Li and L. Ackermann, Cationic Ruthenium Catalysts for Alkyne Annulations with Oximes by C–H/N–O Functionalizations, J. Org. Chem., 2012, 77, 9190–9198 CrossRef; (e) F. Yang and L. Ackermann, Dehydrative C–H/N–OH Functionalizations in H2O by Ruthenium(II) Catalysis: Subtle Effect of Carboxylate Ligands and Mechanistic Insight, J. Org. Chem., 2014, 79, 12070–12082 CrossRef CAS; (f) Y. Li, S. Liu, Z. Qi, X. Qi, X. Li and Y. Lan, The Mechanism of N–O Bond Cleavage in Rhodium-Catalyzed C–H Bond Functionalization of Quinoline N-oxides with Alkynes: A Computational Study, Chem. – Eur. J., 2015, 21, 10131–10137 CrossRef CAS; (g) C. V. Suneel Kumar and C. V. Ramana, Ru-Catalyzed Redox-Neutral Cleavage of the N–O Bond in Isoxazolidines: Isatogens to Pseudoindoxyls via a One-Pot [3 + 2]Cycloaddition/N–O Cleavage, Org. Lett., 2015, 17, 2870–2873 CrossRef CAS; (h) S. Scholz and B. Plietker, Fe-Catalyzed reductive NO-bond cleavage – a route to the diastereoselective 1,4-aminohydroxylation of 1,3-dienes, Org. Chem. Front., 2016, 3, 1295–1298 RSC.
- For selected reviews, see: (a) R. Noyori and S. Hashiguchi, Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (b) A. Robertson, T. Matsumoto and S. Ogo, The development of aqueous transfer hydrogenation catalysts, Dalton Trans., 2011, 40, 10304–10310 RSC; (c) P. E. Sues, K. Z. Demmans and R. H. Morris, Rational development of iron catalysts for asymmetric transfer hydrogenation, Dalton Trans., 2014, 43, 7650–7667 RSC; (d) J. Ito and H. Nishiyama, Recent topics of transfer hydrogenation, Tetrahedron Lett., 2014, 55, 3133–3146 CrossRef; (e) D. Wang and D. Astruc, The golden age of transfer hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS; (f) X. Wu, C. Wang and J. Xiao, Transfer hydrogenation in water, Chem. Rec., 2016, 16, 2772–2786 CrossRef; (g) A. Matsunami and Y. Kayaki, Upgrading and expanding the scope of homogeneous transfer hydrogenation, Tetrahedron Lett., 2018, 59, 504–513 CrossRef CAS; (h) Z. Zhang, N. A. Butt, M. Zhou, D. Liu and W. Zhang, Asymmetric transfer and pressure hydrogenation with earth-abundant transition metal catalysts, Chin. J. Chem., 2018, 36, 443–454 CrossRef CAS.
- For selected reviews on transfer hydrogenation of ketones, see: (a) X. Wu and J. Xiao, Aqueous-phase asymmetric transfer hydrogenation of ketones – a greener approach to chiral alcohols, Chem. Commun., 2007, 2449–2466 RSC; (b) R. Malacea, R. Poli and E. Manoury, Asymmetric hydrosilylation, transfer hydrogenation and hydrogenation of ketones catalyzed by iridium complexes, Coord. Chem. Rev., 2010, 254, 729–752 CrossRef CAS; (c) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Iron-, cobalt-, and nickel-catalyzed asymmetric transfer hydrogenation and asymmetric hydrogenation of ketones, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS; (d) F. Foubelo, C. Najera and M. Yus, Catalytic asymmetric transfer hydrogenation of ketones: recent advances, Tetrahedron: Asymmetry, 2015, 26, 769–790 CrossRef CAS. For selected examples, see: (e) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (f) X.-D. Du, L.-X. Dai, X.-L. Hou, L.-J. Xia and M.-H. Tang, Highly efficient catalysts derived from planar chiral ferrocenes for asymmetric transfer hydrogenation of ketones, Chin. J. Chem., 1998, 16, 90–93 CAS; (g) Y.-X. Chen, Y.-M. Li, K.-H. Lam and A. S. C. Chan, New chiral ligand N-toluenesulfonyl-2,2′-dimethoxy-6,6′-di-aminobiphenyl for catalytic asymmetric transfer hydrogenation of ketones, Chin. J. Chem., 2002, 20, 606–609 CrossRef CAS; (h) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Amine(imine)diphosphine iron catalysts for asymmetric transfer hydrogenation of ketones and imines, Science, 2013, 342, 1080–1083 CrossRef CAS; (i) M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Molecularly defined manganese pincer complexes for selective transfer hydrogenation of ketones, ChemSusChem, 2017, 10, 83–86 CrossRef CAS; (j) Y.-M. Zhang, M.-L. Yuan, W.-P. Liu, J.-H. Xie and Q.-L. Zhou, Iridium-catalyzed asymmetric transfer hydrogenation of alkynyl ketones using sodium formate and ethanol as hydrogen sources, Org. Lett., 2018, 20, 4486–4489 CrossRef CAS; (k) L.-S. Zheng, P. Phansavath and V. Ratovelomanana-Vidal, Ruthenium-catalyzed dynamic kinetic asymmetric transfer hydrogenation: stereoselective access to syn 2-(1,2,3,4-tetrahydro-1-isoquinolyl)ethanol derivatives, Org. Chem. Front., 2018, 5, 1366–1370 RSC; (l) L. De Luca, A. Passera and A. Mezzetti, Asymmetric transfer hydrogenation with a bifunctional iron(II) hydride: experiment meets computation, J. Am. Chem. Soc., 2019, 141, 2545–2556 CrossRef.
- For selected examples on transfer hydrogenation of imines, see: (a) N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, Asymmetric transfer hydrogenation of imines, J. Am. Chem. Soc., 1996, 118, 4916–4917 CrossRef CAS; (b) A. A. Mikhailine, M. I. Maishan and R. H. Morris, Asymmetric transfer hydrogenation of ketimines using well-defined iron(II)-based precatalysts containing a PNNP ligand, Org. Lett., 2012, 14, 4638–4641 CrossRef CAS; (c) Z. Wu, M. Perez, M. Scalone, T. Ayad and V. Ratovelomanana-Vidal, Ruthenium-catalyzed asymmetric transfer hydrogenation of 1-aryl-substituted dihydroisoquinolines: access to valuable chiral 1-aryl-tetrahydroisoquinoline scaffolds, Angew. Chem., Int. Ed., 2013, 52, 4925–4928 CrossRef CAS; (d) G. Zhang and S. K. Hanson, Cobalt-catalyzed transfer hydrogenation of C=O and C=N bonds, Chem. Commun., 2013, 49, 10151–10153 RSC; (e) H. Xu, P. Yang, P. Chuanprasit, H. Hirao and J. Zhou, Nickel-catalyzed asymmetric transfer hydrogenation of hydrazones and other ketimines, Angew. Chem., Int. Ed., 2015, 54, 5112–5116 CrossRef CAS; (f) V. S. Shende, S. H. Deshpande, S. K. Shingote, A. Joseph and A. A. Kelkar, Asymmetric transfer hydrogenation of imines in water by varying the ratio of formic acid to triethylamine, Org. Lett., 2015, 17, 2878–2881 CrossRef CAS; (g) M. Wu, T. Cheng, M. Ji and G. Liu, Ru-catalyzed asymmetric transfer hydrogenation of α-trifluoromethylimines, J. Org. Chem., 2015, 80, 3708–3713 CrossRef CAS; (h) H.-J. Pan, Y. Zhang, C. Shan, Z. Yu, Y. Lan and Y. Zhao, Asymmetric transfer hydrogenation of imines using alcohol: efficiency and selectivity are influenced by the hydrogen donor, Angew. Chem., Int. Ed., 2016, 55, 9615–9619 CrossRef CAS; (i) Y. Li, M. Lei, W. Yuan, E. Meggers and L. Gong, An N-heterocyclic carbene iridium catalyst with metal-centered chirality for enantioselective transfer hydrogenation of imines, Chem. Commun., 2017, 53, 8089–8092 RSC; (j) E. de Julián, E. Menéndez-Pedregal, M. Claros, M. Vaquero, J. Díez, E. Lastra, P. Gamasa and A. Pizzano, Practical synthesis of enantiopure benzylamines by catalytic hydrogenation or transfer hydrogenation reactions in isopropanol using a Ru-pybox catalyst, Org. Chem. Front., 2018, 5, 841–849 RSC.
- For selected examples on transfer hydrogenation of alkenes, see: (a) O. Soltani, M. A. Ariger and E. M. Carreira, Transfer hydrogenation in water: enantioselective, catalytic reduction of (E)-β,β-disubstituted nitroalkenes, Org. Lett., 2009, 11, 4196–4198 CrossRef CAS; (b) S. Horn and M. Albrecht, Transfer hydrogenation of unfunctionalised alkenes using N-heterocyclic carbeneruthenium catalyst precursors, Chem. Commun., 2011, 47, 8802–8804 RSC; (c) G. K. Zielinski, C. Samojlowicz, T. Wdowik and K. Grela, In tandem or alone: a remarkably selective transfer hydrogenation of alkenes catalyzed by ruthenium olefin metathesis catalysts, Org. Biomol. Chem., 2015, 13, 2684–2688 RSC; (d) V. H. Mai and G. I. Nikonov, Transfer hydrogenation of nitriles, olefins, and N-heterocycles catalyzed by an N-heterocyclic carbene-supported half-Sandwich complex of ruthenium, Organometallics, 2016, 35, 943–949 CrossRef CAS; (e) Y. Wang, Z. Huang, X. Leng, H. Zhu, G. Liu and Z. Huang, Transfer hydrogenation of alkenes using ethanol catalyzed by a NCP pincer iridium complex: scope and mechanism, J. Am. Chem. Soc., 2018, 140, 4417–4429 CrossRef CAS.
- For selected examples on transfer hydrogenation of alkynes, see: (a) P. Hauwert, G. Maestri, J. W. Sprengers, M. Catellani and C. J. Elsevier, Transfer semihydrogenation of alkynes catalyzed by a zero-valent palladium N-heterocyclic carbene complex, Angew. Chem., Int. Ed., 2008, 47, 3223–3226 CrossRef CAS; (b) J. Li, R. Hua and T. Liu, Highly chemo- and stereoselective palladium-catalyzed transfer semihydrogenation of internal alkynes affording cis-alkenes, J. Org. Chem., 2010, 75, 2966–2970 CrossRef CAS; (c) P. Hauwert, R. Boerleider, S. Warsink, J. J. Weigand and C. J. Elsevier, Mechanism of Pd(NHC)-catalyzed transfer hydrogenation of alkynes, J. Am. Chem. Soc., 2010, 132, 16900–16910 CrossRef CAS; (d) C. Belger, N. M. Neisius and B. Plietker, A selective Ru-catalyzed semireduction of alkynes to Z olefins under transfer-hydrogenation conditions, Chem. – Eur. J., 2010, 16, 12214–12220 CrossRef CAS; (e) G. Wienhöfer, F. A. Westerhaus, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Selective iron-catalyzed transfer hydrogenation of terminal alkynes, Chem. Commun., 2012, 48, 4827–4829 RSC; (f) R. M. Drost, T. Bouwens, N. P. van Leest, B. de Bruin and C. J. Elsevier, Convenient transfer semihydrogenation methodology for alkynes using a PdII-NHC precatalyst, ACS Catal., 2014, 4, 1349–1357 CrossRef CAS; (g) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, Ligand-controlled cobalt-catalyzed transfer hydrogenation of alkynes: stereodivergent synthesis of Z- and E-alkenes, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS; (h) J. Yang, C. Wang, Y. Sun, X. Man, J. Li and F. Sun, Ligand-controlled iridium-catalyzed semihydrogenation of alkynes with ethanol: highly stereoselective synthesis of E- and Z-alkenes, Chem. Commun., 2019, 55, 1903–1906 RSC.
- For selected references on transfer hydrogenation of other unsaturated compounds, see: (a) Y.-C. Chen, J.-G. Deng, T.-F. Wu, X. Cui, Y.-Z. Jiang, M. C. K. Choi and A. S. C. Chan, Facile preparation of chiral 1, 3-diols via stereoselective transfer hydrogenation of 1, 3-diones, Chin. J. Chem., 2001, 19, 807–810 CrossRef CAS; (b) C.-Y. Wang, C.-F. Fu, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Synthesis of iridium pyridinyl N-heterocyclic carbene complexes and their catalytic activities on reduction of nitroarenes, Inorg. Chem., 2007, 46, 5779–5786 CrossRef CAS; (c) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, pH-Regulated asymmetric transfer hydrogenation of quinolines in water, Angew. Chem., Int. Ed., 2009, 48, 6524–6528 CrossRef CAS; (d) O. Soltani, M. A. Ariger and E. M. Carreira, Transfer hydrogenation in water: enantioselective, catalytic reduction of (E)-β,β-disubstituted nitroalkenes, Org. Lett., 2009, 11, 4196–4198 CrossRef CAS; (e) D. Spasyuk, S. Smith and D. G. Gusev, Replacing phosphorus with sulfur for the efficient hydrogenation of esters, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS; (f) P. Yang, H. Xu and J. Zhou, Nickel-catalyzed asymmetric transfer hydrogenation of olefins for the synthesis of α– and β–amino acids, Angew. Chem., Int. Ed., 2014, 53, 12210–12213 CrossRef CAS; (g) M. Liu, F. Zhou, Z. Jia and C.-J. Li, A silver-catalyzed transfer hydrogenation of aldehyde in air and water, Org. Chem. Front., 2014, 1, 161–166 RSC; (h) N. Dai, R. Shang, M. Fu and Y. Fu, Transfer hydrogenation of ethyl Levulinate to γ-Valerolactone catalyzed by iron complexes, Chin. J. Chem., 2015, 33, 405–408 CrossRef CAS; (i) Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Mild and selective cobalt-catalyzed chemodivergent transfer hydrogenation of nitriles, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS.
- For selected examples on transfer hydro-dehalogenation of organic halides, see: (a) A. C. Bényei, S. Lehel and F. Joó, Transfer hydrodehalogenation of organic halides catalyzed by water soluble ruthenium(II) phosphine complexes, J. Mol. Catal. A: Chem., 1997, 116, 349–354 CrossRef; (b) M. E. Cucullu, S. P. Nolan, T. R. Belderrain and R. H. Grubbs, Catalytic dehalogenation of aryl chlorides mediated by ruthenium(II) phosphine complexes, Organometallics, 1999, 18, 1299–1304 CrossRef CAS; (c) K. Fujita, M. Owaki and R. Yamaguchi, Chemoselective transfer hydrodechlorination of aryl chlorides catalyzed by Cp*Rh complexes, Chem. Commun., 2002, 2964–2965 RSC; (d) S. Kuhl, R. Schneider and Y. Fort, Catalytic carbon-fluorine bond activation with monocoordinated nickel-carbene complexes: reduction of fluoroarenes, Adv. Synth. Catal., 2003, 345, 341–344 CrossRef CAS; (e) O. Navarro, N. Marion, Y. Oonishi, R. A. Kelly III and S. P. Nolan, Suzuki–Miyaura, α-ketone arylation and dehalogenation reactions catalyzed by a versatile N-heterocyclic carbene–palladacycle complex, J. Org. Chem., 2006, 71, 685–692 CrossRef CAS; (f) J. Yang and M. Brookhart, Iridium-catalyzed reduction of alkyl halides by triethylsilane, J. Am. Chem. Soc., 2007, 129, 12656–12657 CrossRef CAS; (g) J. Chen, Y. Zhang, L. Yang, X. Zhang, J. Liu, L. Li and H. Zhang, A practical palladium catalyzed dehalogenation of aryl halides and α-haloketones, Tetrahedron, 2007, 63, 4266–4270 CrossRef CAS; (h) T. You, Z. Wang, J. Chen and Y. Xia, Transfer hydro-dehalogenation of organic halides catalyzed by ruthenium(II) complex, J. Org. Chem., 2017, 82, 1340–1346 CrossRef CAS; (i) I. Parveen, D. Khan and N. Ahmed, Regioselective hydrodehalogenation of aromatic α– and β–halo carbonyl compounds by CuI in isopropanol, Eur. J. Org. Chem., 2019, 759–764 CrossRef CAS; (j) C. Chen, H. Zuo and K. S. Chan, Catalytic hydrodebromination of aryl bromides by cobalt tetra-butyl porphyrin complexes with EtOH, Tetrahedron, 2019, 75, 510–517 CrossRef CAS.
- For an example of the reductive cleavage of N-C bonds with transfer hydrogenation catalysis, see: (a) D. Pu, Y. Zhou, F. Yang, G. Shen, Y. Gao, W. Sun, R. Khan and B. Fan, Asymmetric ring-opening reactions of azabenzonorbornadienes through transfer hydrogenation using secondary amines as hydrogen sources: tuning of absolute configuration by acids, Org. Chem. Front., 2018, 5, 3077–3082 RSC. For an example of the reductive cleavage of N-O bonds with transfer hydrogenation catalysis, see: (b) H. Fukuzawa, Y. Ura and Y. Kataoka, Ruthenium-catalyzed reduction of N-alkoxy- and N-hydroxyamides, J. Organomet. Chem., 2011, 696, 3643–3648 CrossRef CAS.
- W. Leitner, J. M. Brown and H. Brunner, Mechanistic Aspects of the Rhodium-Catalyzed Enantioselective Transfer Hydrogenation of α,β-Unsaturated Carboxylic Acids Using Formic Acid/Triethylamine (5:2) as the Hydrogen Source, J. Am. Chem. Soc., 1993, 115, 152–159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qo01093f |
| This journal is © the Partner Organisations 2021 |