
Ruthenium/acid co-catalyzed reductive α-phosphinoylation of 1,8-naphthyridines with diarylphosphine oxides†‡
Rongqing
Guan
,
He
Zhao
,
Liang
Cao
,
Huanfeng
Jiang
 and
Min
Zhang
*
and
Min
Zhang
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Wushan Rd-381, Guangzhou 510641, People's Republic of China. E-mail: minzhang@scut.edu.cn
First published on 5th November 2020
Abstract
By an in situ coupling-interrupted transfer hydrogenation strategy, we herein report, for the first time, a ruthenium/acid co-catalyzed reductive α-phosphinoylation of 1,8-naphthyridines with diarylphosphine oxides, which offers a direct construction of novel α-phosphinoyl 1,2,3,4-tetrahydronaphthyridines with the merits of broad substrate scope, good functional group tolerance, excellent regio- and chemoselectivity, and high step and atom-efficiency. The employed strategy is anticipated to be further utilized in developing direct transformations of inert N-heteroarenes into functional frameworks.
Introduction
α-Phosphinoyl (benzo)cyclic amines represent a class of highly important N-heterocycles in organic chemistry, as they are frequently employed for the development of bioactive agents, pharmaceuticals, and agrochemicals.1 For instance, selected examples are shown in Fig. 1; compounds A and B are found to be potent inhibitors of dipeptidyl peptidase2 and antagonists of α1-adrenergic receptors,3 respectively. Compound C is utilized as a herbicide,1 and compound D is a surrogate of pipecolic acid.4 In addition, such compounds are applicable for causing further molecular complexity via phosphinoyl group-directed C–H functionalization5 and the preparation of phosphine ligands applied in transition metal catalysis.6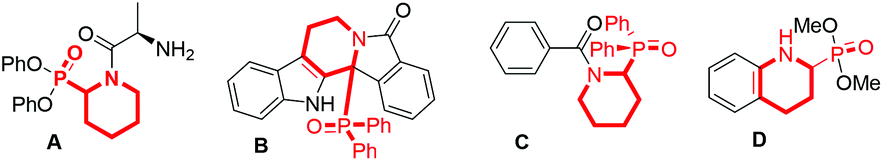 | ||
| Fig. 1 Selected functional molecules. | ||
Due to the interesting functions, the introduction of phosphinoyl groups into the α-position of cyclic amines is of important significance in the scientific community. However, there are only very limited approaches reported to date for the synthesis of α-phosphinoyl cyclic amines. In 2009, an early example was successfully demonstrated by the Li group via initial oxidation of benzocyclic amines followed by nucleophilic addition of H-phosphonates to the imino intermediates, namely a cross-dehydrogenative-coupling (CDC) strategy (Scheme 1, protocol a).7 Later, the direct addition of phosphorus nucleophiles to positions 2 and 4 of N-heteroarenes (protocol b) was also well developed to achieve the related result.8 Despite their significant utility, these synthetic protocols require preinstallation of specific coupling agents such as benzocyclic amines, and the obtained products possess an additional N-substituent or a γ-phosphinoyl group. In this context,9 the development of alternative approaches, enabling direct and selective access to N-nonsubstituted α-phosphinoyl cyclic amines from readily available but poorly reactive N-heteroarenes, would be highly desirable.
 | ||
| Scheme 1 Synthesis of an α-phosphinoyl N-heterocycle. | ||
1,2,3,4-Tetrahydronaphthyridine (THNAD) constitutes the core structure of numerous functional products that exhibit interesting biological and pharmaceutical activities.10 Moreover, THNADs also could serve as valuable building blocks for various synthetic purposes.11 In general, the catalytic hydrogenation of naphthyridines is a frequently utilized method to access THNADs.12 Recently, we have also demonstrated a hydrogen transfer-mediated synthesis of α-aryl THNADs. However, these approaches can only result in limited types of THNADs.10f It is noteworthy that although important advances in oxidative phosphinoylation of C–H bonds13 and alkenes14 have been elegantly achieved, the reductive α-phosphinoylation of naphthyridines remains an unresolved goal, yet. It is noteworthy that such a strategy would not only allow the construction of structurally novel α-phosphinoyl THNADs, but also enrich direct chemical transformation of non-activated N-heteroaromatic systems.
Enlightened by the step-wise hydrogenation in N-heteroaromatic reduction15 and our recent work on direct functionalization of N-heterocycles,16,10f we envisioned that the utilization of initially hydrogenated naphthyridines as the coupling partners for the next step would offer new avenues to achieve molecular diversity of THNADs. Ideally, such a concept benefits from the streamlined synthesis of the desired molecules that are difficult to access with the conventional approaches. As shown in Scheme 1, the ipso-formed active metal hydride species [MH2], arising from the interaction of a compatible transition metal catalyst (M) and a hydrogen donor (HD), initially leads to the formation of intermediate A and its tautomer Bvia the first transfer hydrogenation (1st TH) of naphthyridine 1. Then, the capture of A by phosphine oxide nucleophile 2 with the assistance of an acidic catalyst would give rise to the new α-phosphorus THNAD 3. Being different from quinolines and the related derivatives having 2,4-bisphosphinoylation selectivity,8 the higher reduction reactivity of naphthyridine 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10f in the presence of the metal catalyst and HD would preferentially result in the first transfer hydrogenation prior to direct nucleophilic addition of phosphine oxide 2 to naphthyridine 1, and only generate mono-phosphinoyl compound 3. However, to achieve such a synthetic purpose, there remains a key issue to be addressed, that is, the capture of imine B by nucleophiles 2 should be much faster than the second hydrogenation of imine B, thus suppressing the formation of undesired non-coupling THNADs 3′.
10f in the presence of the metal catalyst and HD would preferentially result in the first transfer hydrogenation prior to direct nucleophilic addition of phosphine oxide 2 to naphthyridine 1, and only generate mono-phosphinoyl compound 3. However, to achieve such a synthetic purpose, there remains a key issue to be addressed, that is, the capture of imine B by nucleophiles 2 should be much faster than the second hydrogenation of imine B, thus suppressing the formation of undesired non-coupling THNADs 3′.
Results and discussion
The above idea prompted us to choose the reductive cross-coupling of 2-phenyl-1,8-naphthyridine 1a and diphenylphosphine oxide 2a as a model system to screen an efficient catalytic system. Initially, five catalysts (Cat. 1–Cat. 5) employed frequently for hydrogen transfer reactions17 were tested by performing the reaction in p-xylene at 120 °C for 16 h by using p-toluenesulfonic acid (p-TSA) as the acidic catalyst and formic acid as the hydrogen donor (HD). Gratifyingly, Ru3(CO)12 (Cat. 1) exhibited the best catalytic performance and afforded product 3aa in 43% isolated yield (entries 1–5, Table 1 and Table S2 in the ESI‡ for details). Then a series of acids in combination with Ru3(CO)12 (entries 6–8) were tested, and D-Camphorsulfonic acid (CSA) improved the product yield to 75%, which implies that, in comparison with HCOOH, relatively strong acidity is beneficial for the reaction. However, the absence of HCO2H, Ru, and CSA, and the use of HCO2Na or i-propanol as the HD led to either no product formation or low product yield (entries 9–13), showing that the combination of Ru3(CO)12/HCOOH/CSA constitutes the most efficient system for the reaction. The screening of polar and less-polar solvents showed that they were inferior to p-xylene (entry 14). Furthermore, the increase or decrease of the amount of hydrogen donors, substrate 2a, and CSA, or the reaction temperatures diminished the product yields (entries 15–18). Hence, the optimal conditions are as shown in entry 6 of Table 1.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | [HD] | Additive |
3aab (%) |
| a Conditions: unless otherwise stated, all the reactions were conducted with 1a (0.25 mmol), 2a (0.30 mmol), catalyst (2 mol%), hydrogen donor (4.0 equiv.), additive (0.1 equiv.), and p-xylene (1.0 mL) at 120 °C for 16 h. b Isolated yield. c Yields obtained with DMSO, t-amyl alcohol, 1,4-dioxane, DMF, and toluene as the solvents, respectively. d Yields obtained with 3 and 5 equiv. of HCOOH, respectively. e Yields obtained with 0.25 and 0.375 mmol of 2a, respectively. f Yields obtained with 0.05 and 0.15 eq. CSA, respectively. g Yields obtained at 100 °C, 110 °C, and 130 °C, respectively. | ||||
| 1 | Cat. 1 | HCOOH | p-TSA | 43 |
| 2 | Cat. 2 | HCOOH | p-TSA | 11 |
| 3 | Cat. 3 | HCOOH | p-TSA | <5 |
| 4 | Cat. 4 | HCOOH | p-TSA | <10 |
| 5 | Cat. 5 | HCOOH | p-TSA | 0 |
| 6 | Cat. 1 | HCOOH | CSA | 75 |
| 7 | Cat. 1 | HCOOH | Benzoic acid | 21 |
| 8 | Cat. 1 | HCOOH | CF3COOH | 15 |
| 9 | Cat. 1 | — | CSA | 0 |
| 10 | — | HCOOH | CSA | 0 |
| 11 | Cat. 1 | HCOOH | — | 10 |
| 12 | Cat. 1 | HCOONa | CSA | 0 |
| 13 | Cat. 1 | i-Propanol | CSA | 0 |
| 14 | Cat. 1 | HCOOH | CSA | (0, 30, 21, 0, 39)c |
| 15 | Cat. 1 | HCOOH | CSA | (58, 63)d |
| 16 | Cat. 1 | HCOOH | CSA | (70, 71)e |
| 17 | Cat. 1 | HCOOH | CSA | (66, 67)f |
| 18 | Cat. 1 | HCOOH | CSA | (62, 69, 73)g |

With the availability of the optimal conditions, we first tested the reductive coupling of a wide array of 1,8-naphthyridines 1 with phosphine oxide 2a. As shown in Scheme 2, all the reactions proceeded smoothly and furnished the desired products in moderate to excellent yields upon isolation (3aa–3za). Significantly, various functionalities (–Me, –OMe, –F, –Cl, –Br, –I, –(hetero)aryl, –CF3, and –OH) on the 1,8-naphthyridines were well tolerated, and their electronic properties significantly influenced the product yields. In general, 1,8-naphthyridines bearing strong electron-withdrawing groups (3ia, 3ka, 3la, and 3na) afforded the products in relatively higher yields than those of electron-rich group substituted substrates (3ba–3ha), presumably because the electron-withdrawing groups could enhance the reactivity of 1,8-naphthyridines. It is noteworthy that although nonsubstituted 1,8-naphthyridine has two reactive α-sites, only one site took part in the reaction (3va). In addition, heteroaryl substituted 1,8-naphthyridines also underwent smooth reductive α-phosphinoylation, and the obtained products have the potential to be applied as hemilabile ligands (3wa and 3xa).18
 | ||
| Scheme 2 Substrate scope of 1,8-naphthyridines. | ||
Furthermore, we turned our attention to the variation of both coupling partners. Thus, different combinations of diarylphosphine oxides 2 and naphthyridines 1 were examined. As shown in Scheme 3, all the reactions underwent efficient reductive cross-coupling, affording the α-phosphinoyl 1,2,3,4-tetrahydro-1,8-naphthalidines in good to excellent yields upon isolation (3ab–3ad, 3bd, 3rb, 3db, and 3md). Similar to the results described in Scheme 2, different functional groups on both coupling agents were well tolerated, and the retention of these functionalities (Schemes 2 and 3) would offer the potential for the elaboration of complex molecules via further chemical transformations.
 | ||
| Scheme 3 Variation of both coupling partners. | ||
To gain mechanistic insights into the reaction, several control experiments were performed under the optimal conditions (Scheme 4). First, interruption of the model reaction at 30 min produced product 3aa in 10% yield along with dihydro-1,8-naphthyridine (1a-1 or 1a-2) in 1% combined yield and THNAD 1a-3 in 3% yield (eqn (1)). Furthermore, the reaction of 1a-3 and 2a failed to yield product 3aa (eqn (2)), whereas prolonging the model reaction time to 16 h led to full consumption of the dihydro-1,8-naphthyridine and resulted in 3aa in 75% yield (Table 1, entry 6), the results show that dihydro-1,8-naphthyridine, instead of THNAD 1a-3, is the reaction intermediate. Furthermore, the model reaction in the absence of reductant HCOOH failed to yield the coupling adduct 3aa′ (eqn (3)), suggesting that the first transfer hydrogenation of 1,8-naphthyridine 1a occurs prior to the addition of 2a to the α-site of 1a. Then, the deuterium-labelling experiment of 1a and 2a using deuterated formic acid as the reductant (eqn (4)) generated product 3aa-dn with different H/D ratios on the naphthyridyl skeleton, which reveals that both pyridyl rings undergo transfer hydrogenation, and the coupling selectively occurs on the sterically less-hindered side. All these findings are in good agreement with the reaction pathway depicted in Scheme 1.
 | ||
| Scheme 4 Control experiments. | ||
Furthermore, we were interested in demonstrating the synthetic utility of the developed chemistry. As shown in Scheme 5, the gram-scale synthesis of α-phosphinoyl tetra-hydro-1,8-naphthyridine 3aa was successfully achieved by scaling up substrates 1a and 2a to 4.85 mmol and 5.82 mmol, respectively, and a 51% yield was obtained (Scheme 5, eqn (1)). In addition, the reaction of compound 3aa with diiso-butyl aluminum hydride and potassium phosphate in THF solution generated a new α-phosphine-substituted tetrahydro-1,8-naphthyridine 4aa in moderate yield (eqn (2)). This example demonstrates the potential of the obtained products in the development of N, P-based ligands. Furthermore, treating compound 3aa in acetonitrile under aerobic copper catalysis resulted in a dehydroaromatization product 5aa in 60% yield (eqn (3)), which offers a way for the preparation of α-phosphinoyl 1,8-naphthyridines.
 | ||
| Scheme 5 Synthetic utility. | ||
Conclusions
In summary, by an in situ coupling-interrupted transfer hydrogenation strategy, we have established a new ruthenium/acid co-catalyzed reductive α-phosphinoylation of 1,8-naphthyridines with diarylphosphine oxides, which offers a practical platform for direct construction of novel α-phosphinoyl tetrahydro-1,8-naphthyridines with the merits of broad substrate and functional group compatibility, excellent regio- and chemoselectivity, and high step and atom-efficiency. In consideration of the significant utility of α-phosphinoyl cyclic amines, the developed chemistry has the potential to be applied in various fields. In addition, the employed strategy will spur others to further develop new chemical transformations of inert N-aromatic systems into functional frameworks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key Research and Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (21971071), and the foundation of the Department of Education of Guangdong Province (2017KZDXM085) for financial support.Notes and references
-
R. Bansal, Phosphorous Heterocycles I, Springer Berlin Heidelberg, Berlin, Germany, 2009 Search PubMed
.
-
(a) A. Mucha, P. Kafarski and Y. Berlicki, Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS
-
(a) F. Qi, F. Fang and P. Li, Catalyst-Free Phospha-Nucleophilic Substitution of Hydroxylactams by Diarylphosphine Oxide, Curr. Organocatal., 2018, 5, 145–149 CrossRef CAS
- M. Ordóñez, A. Arizpe, F. Sayago, A. Jiménez and C. Cativiela, Practical and Efficient Synthesis of α-Aminophosphonic Acids Containing 1,2,3,4-Tetrahydroquinoline or 1,2,3,4-Tetrahydroisoquinoline Heterocycles, Molecules, 2016, 21, 1140–1153 CrossRef
-
(a) Y. Ma, S. Li and S. Yang, New Approaches for Biaryl-Based Phosphine Ligand Synthesis via P=O Directed C–H Functionalizations, Acc. Chem. Res., 2017, 50, 1480–1492 CrossRef CAS
-
(a) L. Bialy and H. Waldmann, Inhibitors of Protein Tyrosine Phosphatases: Next-Generation Drugs?, Angew. Chem., Int. Ed., 2005, 44, 3814–3839 CrossRef CAS
- O. Basle and C. Li, Copper-Catalyzed Aerobic Phosphonation of sp3C–H Bonds, Chem. Commun., 2009, 4124–4126 RSC
-
(a) Y. Gao, H. Deng, S. Zhang, W. Xue, Y. Wu, H. Qiao, P. Xu and Y. Zhao, Nickel-Catalyzed One-Pot Tandem 1,4-1,2-Addition of P(O)H Compounds to 1,10-Phenanthrolines, J. Org. Chem., 2015, 80, 1192–1199 CrossRef CAS
-
(a) S. Van Der Jeught and C. Stevens, Direct Phosphonylation of Aromatic Azaheterocycles, Chem. Rev., 2009, 109, 2672–2702 CrossRef CAS
-
(a) J. Wang, M. Breslin, P. Coleman, M. Duggan, C. Hunt, J. Hutchinson, C. Leu, S. Rodan, G. Rodan, L. Duong and G. Hartman, Non-peptide α,vβ3 antagonists. Part 7: 3-Substituted Tetrahydro-1, 8naphthyridine Derivatives, Bioorg. Med. Chem. Lett., 2004, 14, 1049–1052 CrossRef CAS
- Selected examples on the use of THNADs as synthetic building blocks:
(a) Z. Tan, Z. Fu, J. Yang, Y. Wu, L. Cao, H. Jiang, J. Li and M. Zhang, Hydrogen Transfer-Mediated Multicomponent Reaction for Direct Synthesis of Quinazolines by a Naphthyridine-Based Iridium Catalyst, iScience, 2020, 23, 101003–101015 CrossRef CAS
-
(a)
H. Kroth, W. Froestl, A. Pfeifer and A. Muhs, PCT Int, 045383, 2011 Search PubMed
-
(a) Q. Chen, X. Wang, G. Yu, C. Wen and Y. Huo, DDQ-Mediated Direct C(sp3)–H Phosphorylation of Xanthene Derivatives, Org. Chem. Front., 2018, 5, 2652–2656 RSC
- selected examples, see:
(a) S. Chen, P. Zhang, W. Shu, Y. Gao, G. Tang and Y. Zhao, Cascade Phosphinoylation/Cyclization/Isomerization Process for the Synthesis of 2-Phosphinoyl-9H-pyrrolo[1,2-a]indoles, Org. Lett., 2016, 18, 5712–5715 CrossRef CAS
-
(a) K. Murugesan, V. Chandrashekhar, C. Kreyenschulte, M. Beller and R. Jagadeesh, A General Catalyst Based on Cobalt Core–Shell Nanoparticles for the Hydrogenation of N–Heteroarenes Including Pyridines, Angew. Chem., 2020, 59, 17408–17412 CrossRef CAS
-
(a) R. Xie, F. Xie, C. Zhou, H. Jiang and M. Zhang, Hydrogen Transfer-Mediated Selective Dual C–H Alkylations of 2-Alkylquinolines by Doped TiO2-Supported Nanocobalt Oxides, J. Catal., 2019, 377, 449–454 CrossRef CAS
-
(a) D. Wang and D. Astruc, The Golden Age of Transfer Hydrogenation, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS
-
(a) Z. Tan, H. Jiang and M. Zhang, A Novel Iridium/Acid Co-Catalyzed Transfer Hydrogenative C(sp3)–H bond Alkylation to Access Functionalized N-heteroaromatics, Chem. Commun., 2016, 52, 9359–9363 RSC
Footnotes |
| † This work is dedicated to Prof. Pierre. H. Dixneuf for his outstanding contribution in organometallic chemistry and catalysis. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and NMR spectra for all compounds. See DOI: 10.1039/d0qo01284j |
| This journal is © the Partner Organisations 2021 |