
Recent developments in 1,6-addition reactions of para-quinone methides (p-QMs)
 *a
and
Bo
Jiang
*a
*a
and
Bo
Jiang
*a
Abstract
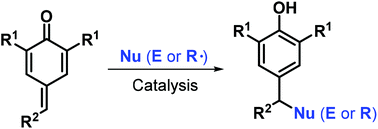
In recent years, para-quinone methides (p-QMs) have emerged as attractive and versatile synthons in organic synthesis owing to their high reactivity. Consequently, p-QM chemistry has attracted increasing attention and remarkable advances have been achieved. Among the numerous transformations involving p-QMs, catalytic reactions play a pivotal role, and a variety of catalytic systems mediated by Lewis acids, Brønsted acids, bases, transition metals, N-heterocyclic carbenes, and other catalysts have been established for performing 1,6-conjugate addition reactions. Various molecular scaffolds have been constructed using p-QMs to obtain the core structures of numerous natural and synthetic substances of chemical and biomedical relevance. In this review, we provide a comprehensive overview of recent progress in this rapidly growing field by summarizing the 1,6-conjugate addition and annulation reactions of p-QMs with consideration of their mechanisms and applications.
 Please wait while we load your content...
Please wait while we load your content...

