
PhI(OAc)2-mediated oxidative rearrangement of allylic amides: efficient synthesis of oxazoles and β-keto amides†
Kang
Xu
,
Shuang
Yang
and
Zhenhua
Ding
 *
*
Jiangsu Key Laboratory of Drug Design and Optimization, Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 211198, China. E-mail: zhding@cpu.edu.cn
First published on 15th November 2019
Abstract
A series of 2,5-disubstituted oxazoles and β-keto amides were synthesized from allylic amides via PhI(OAc)2-mediated intramolecular cyclization and oxidation with the migration of an aryl group. These reactions were performed under mild conditions with good functional group tolerance and gave the corresponding products in good to excellent yields.
Introduction
Hypervalent iodine(III) compounds have been widely used in organic synthesis due to their attractive characteristics, such as ready availability, nontoxicity, and environmental benignity.1 These reagents are mainly employed as oxidizing reagents that replace the use of toxic heavy metal oxidants. In addition to simple oxidation, hypervalent iodine reagents have also been applied in various oxidative rearrangement reactions,2 owing to their electrophilic nature and excellent leaving-group ability. C–N and C–C migration reactions are particularly common. For C–N migration, Hoffmann rearrangement is a powerful method to convert amides into the corresponding amines.3 For C–C migration, hypervalent iodine reagents serve as electrophiles for activating double bonds in alkenes, which leads to further oxidative rearrangement through ring expansion,4 ring contraction,5 or aryl migration.6 For example, Wirth and coworkers realized a novel lactonization of unsaturated carboxylic acids with the rearrangement of an aryl group (Scheme 1a).7 The group of Zhao and Du reported the synthesis of 3-arylquinolinones from N-phenylcinnamamide via oxidative aryl migration and cyclization (Scheme 1b).8 They also disclosed an efficient access to α-ketoamide and esters from acrylic derivatives featuring a cascade oxidative rearrangement and C–C bond-cleavage process (Scheme 1c).9 The group of Fan and He developed an oxidative rearrangement of 2-allylanilines which was mediated by in situ generated iodine(III) compounds (Scheme 1d).10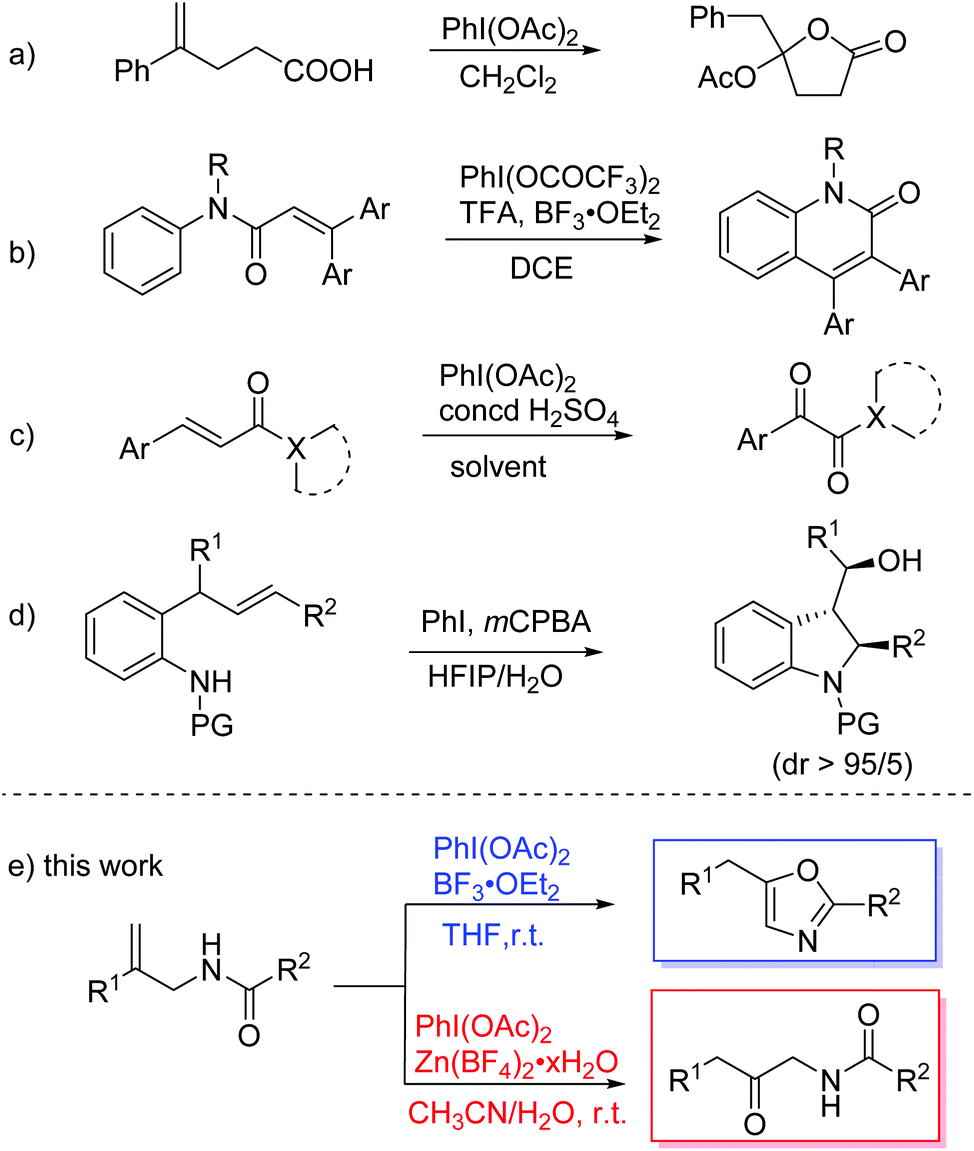 | ||
| Scheme 1 Hypervalent iodine(III) regent mediated oxidative rearrangement. | ||
Oxazole derivatives are prevailing skeletons found in natural products and bioactive compounds.11 Consequently, a variety of approaches to oxazole compounds have been developed, such as cyclization,12 condensation,13 oxidation of oxazolines14 and transition-metal-catalyzed addition of diazole compounds to nitriles.15 Our group focused on the formation of heterocycles and recently reported hypervalent iodine(III) reagent mediated reactions of N-acetyl enamines to obtain oxazole and imidazole derivatives.16 Herein, we disclose the divergent synthesis of oxazoles and β-keto amides from allylic amides via PhI(OAc)2-mediated cyclization and oxidation with the rearrangement of an aryl group (Scheme 1e).
Results and discussion
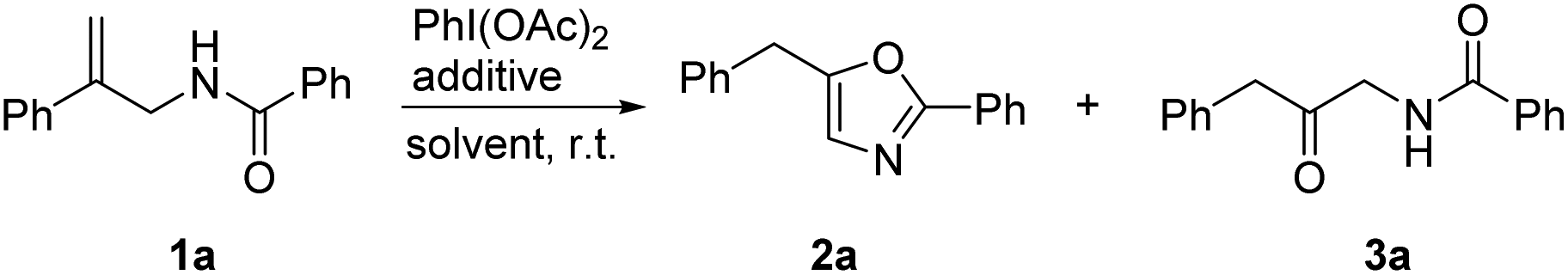
Initially, the reaction was carried out using 1a as a substrate in the presence of PhI(OAc)2 and BF3·OEt2 in CH3CN at room temperature; the oxazole 2a and β-keto amide 3a were formed in 99% total yield with a ratio of 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :19 (Table 1, entry 1). When CH2Cl2 was employed as a solvent, the yield slightly decreased and the ratio of 2a and 3a was 86:14 (entry 2). The selectivity was not improved when the reaction was performed in toluene (entry 3). When THF was used as a solvent, the ratio of 2a and 3a was improved to 96:4 (entry 4). Other additives such as [Cu(MeCN)4]BF4, AgBF4 and Zn(BF4)2 also activated the transformation with different selectivities (entries 5–7). β-Keto amide 3a was obtained as the major product when the reaction was performed using Zn(BF4)2 as an additive in CH3CN (entry 8). We also investigated the reaction in the mixed solvent of CH3CN and H2O, and a high selectivity was achieved when the ratio of CH3CN and H2O was 2/1 (entry 10).
:19 (Table 1, entry 1). When CH2Cl2 was employed as a solvent, the yield slightly decreased and the ratio of 2a and 3a was 86:14 (entry 2). The selectivity was not improved when the reaction was performed in toluene (entry 3). When THF was used as a solvent, the ratio of 2a and 3a was improved to 96:4 (entry 4). Other additives such as [Cu(MeCN)4]BF4, AgBF4 and Zn(BF4)2 also activated the transformation with different selectivities (entries 5–7). β-Keto amide 3a was obtained as the major product when the reaction was performed using Zn(BF4)2 as an additive in CH3CN (entry 8). We also investigated the reaction in the mixed solvent of CH3CN and H2O, and a high selectivity was achieved when the ratio of CH3CN and H2O was 2/1 (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | Additive | Solvent | Yieldb (%) |
2a:3a |
| a The reactions were carried out using 1a (0.3 mmol, 1 equiv.), PhI(OAc)2 (1.2 equiv.) and the additive (10 mol%) in the solvent (0.1 M) for 3 h. b The yields were determined by 1H NMR using an internal standard. | ||||
| 1 | BF3·OEt2 | CH3CN | 99 | 81:19 |
| 2 | BF3·OEt2 | CH2Cl2 | 94 | 86:14 |
| 3 | BF3·OEt2 | Toluene | 99 | 84:16 |
| 4 | BF3·OEt2 | THF | 99 | 96:4 |
| 5 | [Cu(MeCN)4]BF4 | THF | 99 | 82:18 |
| 6 | AgBF4 | THF | 99 | 74:26 |
| 7 | Zn(BF4)2 | THF | 99 | 60:40 |
| 8 | Zn(BF4)2 | CH3CN | 94 | 18:82 |
| 9 | Zn(BF4)2 | CH3CN/H2O 5/1 | 99 | 11:89 |
| 10 | Zn(BF4)2 | CH3CN/H2O 2/1 | 99 | 5:95 |
| 11 | Zn(BF4)2 | CH3CN/H2O 1/1 | 99 | 15:85 |
With the optimized reaction conditions in hand, we then investigated the reactions of various allylic amides 1 in the presence of PhI(OAc)2 (1.2 equiv.) and BF3·OEt2 (10 mol%) in THF to obtain oxazole derivatives (Table 2). We first examined the substrates in which R1 is an aromatic group and R2 is a phenyl group. The reaction proceeded well when halogen substituents such as fluoro, chloro, and bromo were present at the para-position of the benzene ring and gave the products in 89–99% yields (2b–2d). Substrates with electron-donating groups participated in this reaction and gave the oxazole products in good yields (2e and 2f). Substrates with electron-withdrawing groups participated in this reaction and gave the oxazole products in slightly lower yields (2g and 2h).17 The reaction proceeded well when the methyl group was present at the meta-position of the benzene ring (2i). When the R1 group of allylic amides was 2-naphthyl, good reactivity was observed under the reactions conditions and the product was obtained in 99% yield (2j). We next explored the reaction of various allylic amide derivatives in which R1 is a phenyl group and R2 is an aromatic group. Different substituents including methyl, methoxyl, cyano, and trifluoromethyl groups and halogen at the 4-position of the benzene ring were tolerated well and provided the desired products in good yields (2k–2q). With substituents at the ortho-position or meta-position the reaction also proceeded well and provided the products in good to excellent yields (2r–2t). Heteroaromatic substituents such as 2-thienyl, 2-furyl and 3-pyridyl groups were also tolerated under these reaction conditions, and the products were obtained in good yields (2u–2w). The substrate with a benzyl substituent underwent the reaction smoothly and afforded the corresponding products in 81% yield (2x).
|
|
|||
|---|---|---|---|
| R1 | R2 | Yield (%) | |
| a The reaction was carried out using allylic amide 1 (0.3 mmol, 1 equiv.), PhI(OAc)2 (0.36 mmol, 1.2 equiv.), and BF3·OEt2 (0.03 mmol, 10 mol%) in THF (0.1 M). | |||
| 2a | Ph | Ph | 82 |
| 2b | 4-FC6H4 | Ph | 89 |
| 2c | 4-ClC6H4 | Ph | 99 |
| 2d | 4-BrC6H4 | Ph | 99 |
| 2e | 4-MeC6H4 | Ph | 91 |
| 2f | 4-OMeC6H4 | Ph | 86 |
| 2g | 4-CNC6H4 | Ph | 66 |
| 2h | 4-CF3C6H4 | Ph | 85 |
| 2i | 3-MeC6H4 | Ph | 99 |
| 2j | 2-Naphthyl | Ph | 99 |
| 2k | Ph | 4-FC6H4 | 92 |
| 2l | Ph | 4-ClC6H4 | 99 |
| 2m | Ph | 4-BrC6H4 | 99 |
| 2n | Ph | 4-MeC6H4 | 80 |
| 2o | Ph | 4-OMeC6H4 | 65 |
| 2p | Ph | 4-CNC6H4 | 83 |
| 2q | Ph | 4-CF3C6H4 | 93 |
| 2r | Ph | 2-BrC6H4 | 91 |
| 2s | Ph | 2-NO2C6H4 | 99 |
| 2t | Ph | 3,5-F2C6H3 | 90 |
| 2u | Ph | 2-Thienyl | 85 |
| 2v | Ph | 2-Furanyl | 73 |
| 2w | Ph | 3-Pyridyl | 77 |
| 2x | Ph | Benzyl | 81 |
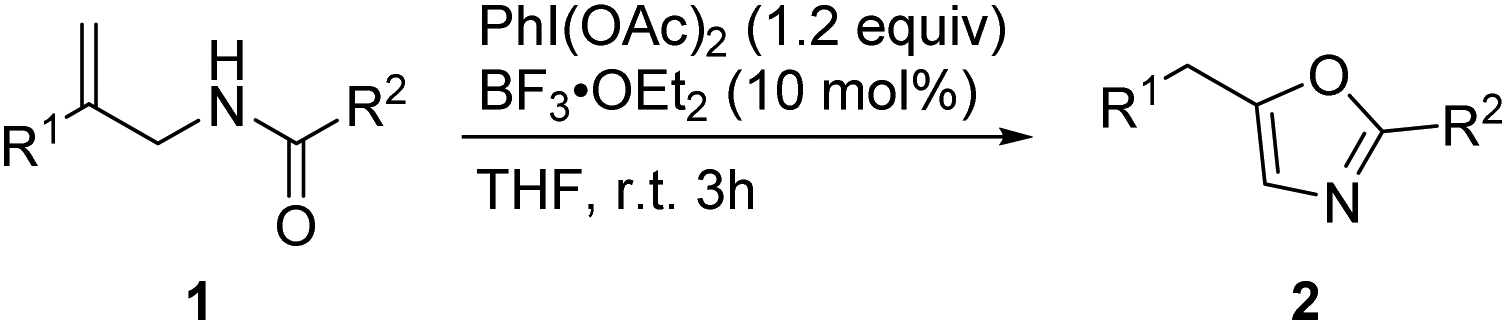
We next studied the synthesis of β-keto amides from allylic amides with the assistance of PhI(OAc)2 (1.2 equiv.) and ZnBF4 (10 mol%) in MeCN/H2O (2/1). The reaction of 1a proceeded well to give the product 3a in 89% isolated yield. We then investigated the reaction of allylic amides with different substituents of R1. The substrates bearing halogen substituents were subjected to the standard reaction conditions, affording the β-keto amide products in moderate yields (3b–3d).18 The reaction proceeded smoothly and provided the corresponding products in good to excellent yields when a methyl group was present at the para- or meta-position of the benzene ring (3e and 3i). When R1 was a 2-naphthyl group, the substrate also exhibited good reactivity under the reaction conditions and gave the product in 92% yield (3j). We then explored the reaction of various allylic amide derivatives in which R1 is a phenyl group and R2 is an aryl substituent. Halogen substituents were tolerated, as the C–X bond remained intact (3k–3m). Substrates with electron-donating or electron-withdrawing groups at the 4-position of the benzene ring showed excellent reactivities for this transformation (3n–3q). The reaction also proceeded well and provided the products in good to excellent yields when substrates with substituents at the ortho-position or meta-position of the benzene ring (3r–3t) were used. Substrates with different heteroaromatic substituents also underwent the oxidative rearrangement efficiently and afforded the corresponding products in moderate to good yields (3u–3w). The substrate 1y in which R2 is an aliphatic substituent reacted as well as 1a and gave the corresponding product in 88% yield (3x) (Table 3).
|
|
|||
|---|---|---|---|
| R1 | R2 | Yield (%) | |
| a The reaction was carried out using allylic amide 1 (0.2 mmol, 1 equiv.), PhI(OAc)2 (0.24 mmol, 1.2 equiv.), and Zn(BF4)2·xH2O (0.02 mmol, 10 mol%) in MeCN/H2O = 2/1 (3 mL). | |||
| 3a | Ph | Ph | 89 |
| 3b | 4-FC6H4 | Ph | 63 |
| 3c | 4-ClC6H4 | Ph | 64 |
| 3d | 4-BrC6H4 | Ph | 61 |
| 3e | 4-MeC6H4 | Ph | 98 |
| 3i | 3-MeC6H4 | Ph | 73 |
| 3j | 2-Naphthyl | Ph | 92 |
| 3k | Ph | 4-FC6H4 | 73 |
| 3l | Ph | 4-ClC6H4 | 69 |
| 3m | Ph | 4-BrC6H4 | 97 |
| 3n | Ph | 4-MeC6H4 | 77 |
| 3o | Ph | 4-OMeC6H4 | 78 |
| 3p | Ph | 4-CNC6H4 | 73 |
| 3q | Ph | 4-CF3C6H4 | 84 |
| 3r | Ph | 2-BrC6H4 | 64 |
| 3s | Ph | 2-NO2C6H4 | 67 |
| 3t | Ph | 3,5-F2C6H3 | 54 |
| 3u | Ph | 2-Thienyl | 79 |
| 3v | Ph | 2-Furanyl | 69 |
| 3w | Ph | 3-Pyridyl | 48 |
| 3x | Ph | Benzyl | 88 |
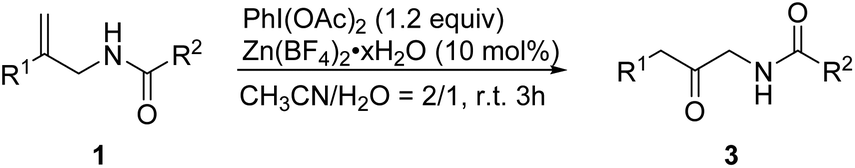
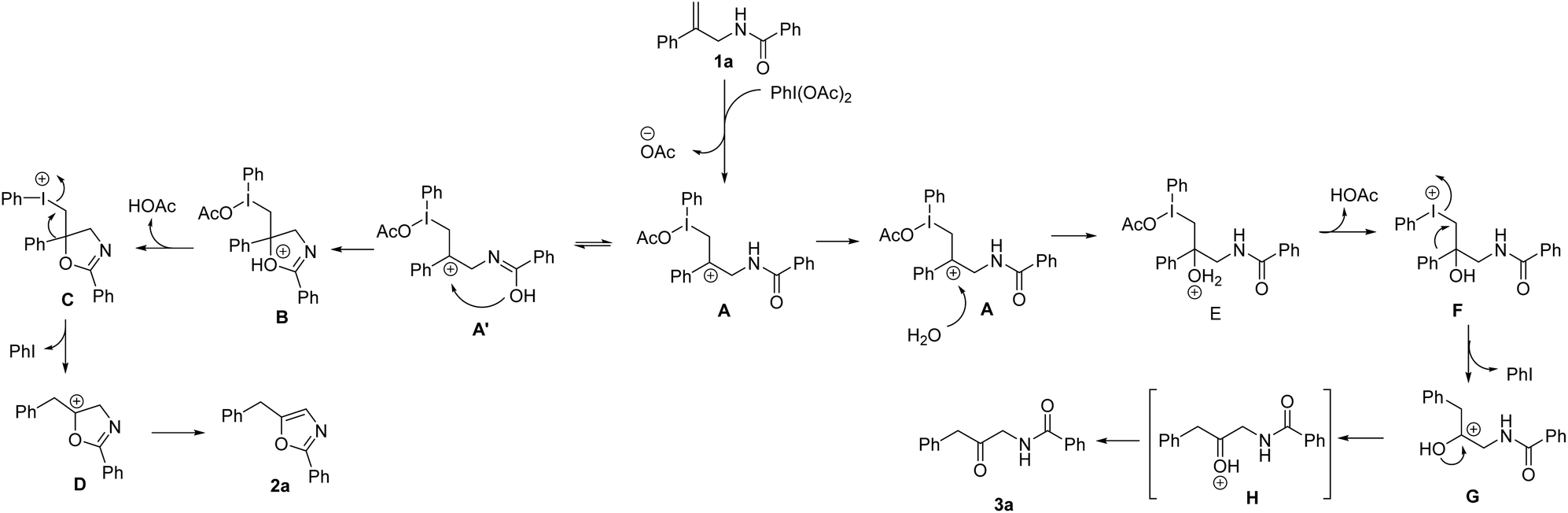
On the basis of our results and previous reports, we proposed a possible mechanistic rationale (Scheme 2). Initially, the reaction of allylic amide 1a with PhI(OAc)2 gives the iodinated intermediate A with the removal of acetate. For oxazole formation, the hydroxyl group attacks the carbocation of A′ and generates cyclic intermediate B, followed by elimination of acetic acid of B to form species C. The intermediate C might undergo migration of the phenyl group and removal of iodobenzene to give species D, which is further converted into the product 2a. On the other hand, when water is involved in the reaction system, the carbocation will be attacked by water to give intermediate E. The removal of acetic acid of E is followed by phenyl migration and iodobenzene elimination to give intermediate G. Subsequent deprotonation takes place to afford the desired β-keto amide product 3a.
 | ||
| Scheme 2 Mechanistic rationale. | ||
Conclusions
In summary, we have developed an efficient method for the synthesis of 2,5-disubstituted oxazoles and β-keto amides via an oxidative rearrangement of allylic amides. The transformation was realized by using PhI(OAc)2 as an oxidant under mild reaction conditions. A variety of functional groups could be tolerated well in the reaction and various products were obtained in moderate to excellent yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (NSFC 81602952), the Natural Science Foundation of Jiangsu Province (BK20160748), and the Project Program of Jiangsu Key Laboratory of Drug Design and Optimization, China Pharmaceutical University (2018KFKT-3) is acknowledged.Notes and references
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS; (b) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS; (c) F. V. Singh and T. Wirth, Chem. – Asian J., 2014, 9, 950–971 CrossRef CAS; (d) V. V. Zhdankin, Chem. Rev., 2008, 108, 5299 CrossRef CAS.
- F. V. Singh and T. Wirth, Synthesis, 2013, 45, 2499 CrossRef CAS.
- (a) K. Miyamoto, Y. Sakai, S. Goda and M. Ochiai, Chem. Commun., 2012, 48, 982 RSC; (b) A. Yoshimura, K. R. Middleton, M. W. Luedtke, C. Zhu and V. V. Zhdankin, J. Org. Chem., 2012, 77, 11399 CrossRef CAS.
- (a) L. F. Silva, R. S. Vasconcelos and M. A. Nogueira, Org. Lett., 2008, 10, 1017 CrossRef CAS; (b) T. Abo, M. Sawaguchi, H. Senboku and S. Hara, Molecules, 2005, 10, 183 CrossRef CAS.
- (a) L. F. Silva, F. A. Siqueira, E. C. Pedrozo, F. Y. M. Vieira and A. C. Dorigutto, Org. Lett., 2007, 9, 1433 CrossRef CAS; (b) M. S. Yusubov, G. A. Zholobova, I. L. Filimonova and K. Chi, Russ. Chem. Bull. Int. Ed., 2004, 53, 1735 CrossRef CAS; (c) M. Kameyama, F. A. Siqueira, M. Gracia-Mijares, L. F. Silva and M. T. A. Silva, Molecules, 2011, 16, 9421 CrossRef CAS.
- (a) L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 2906 CrossRef CAS PubMed; (b) U. Farid, F. Malmedy, R. Claveau, L. Albers and T. Wirth, Angew. Chem., Int. Ed., 2013, 52, 7018 CrossRef CAS; (c) A. C. Boye, D. C. Meyer, K. Ingison, A. N. French and T. Wirth, Org. Lett., 2003, 5, 2157 CrossRef CAS.
- A. C. Boye, D. Meyer, C. K. Ingison, A. N. French and T. Wirth, Org. Lett., 2003, 5, 2157 CrossRef CAS.
- L. Liu, H. Lu, H. Wang, C. Yang, X. Zhang, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 2906 CrossRef CAS.
- L. Liu, L. Du, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2014, 16, 5772 CrossRef CAS.
- S. E. Wang, Q. He and R. Fan, Org. Lett., 2017, 19, 6478 CrossRef CAS.
- (a) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143 RSC; (b) Z. Jin, Nat. Prod. Rep., 2009, 26, 382 RSC.
- (a) H. An, S. Mai, Q. Xuan, Y. Zhou and Q. Song, J. Org. Chem., 2019, 84, 401–408 CrossRef CAS PubMed; (b) H. Liang, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, Chem. – Eur. J., 2013, 19, 9789 CrossRef CAS; (c) C. W. Cheung and S. L. Buchwald, J. Org. Chem., 2012, 77, 7526 CrossRef CAS; (d) A. S. K. Hashmi, M. C. B. Jaimes, A. M. Schuster and F. Rominger, J. Org. Chem., 2012, 77, 6394 CrossRef CAS; (e) E. Merkul and T. J. J. Müller, Chem. Commun., 2006, 4817 RSC; (f) P. Wipf, Y. Aoyama and T. E. Benedum, Org. Lett., 2004, 6, 3593 CrossRef CAS.
- (a) M. Heidrich, M. Bergmann, D. Müller-Borges and H. Plenio, Adv. Synth. Catal., 2018, 360, 3572 CrossRef CAS; (b) A. Lee, S. Oh and H. Kim, Org. Lett., 2018, 20, 3319 CrossRef CAS; (c) X.-L. Han, C.-J. Zhou, X.-G. Liu, S.-S. Zhang, H. Wang and Q. Li, Org. Lett., 2017, 19, 6108 CrossRef CAS; (d) T. Yagyu, Y. Takemoto, A. Yoshimura, V. V. Zhdankin and A. Saito, Org. Lett., 2017, 19, 2506 CrossRef CAS.
- (a) Y. Huang, L. Ni, F. Gan, Y. He, J. Xu, X. Wu and H. Yao, Tetrahedron, 2011, 67, 2066 CrossRef CAS; (b) X. Li, C. Li, B. Yin, P. Liu, J. Li, Z. Shi and C. Li, Chem. – Asian J., 2013, 8, 1408 CrossRef CAS.
- T. Ibata and K. Fukushima, Chem. Lett., 1992, 21, 2197 CrossRef.
- K. Xu, R. Yang, S. Yang, C. Jiang and Z. Ding, Org. Biomol. Chem., 2019, 17, 8977 RSC.
- Crystallographic data for 2g have been deposited at the Cambridge Crystallographic Data Centre with the deposition number CCDC 1959103.†.
- Crystallographic data for 3c have been deposited at the Cambridge Crystallographic Data Centre with the deposition number CCDC 1959105.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1959103 and 1959105. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo01298b |
| This journal is © the Partner Organisations 2020 |