
Total syntheses of (−)-15-oxopuupehenol and (+)-puupehenone and formal syntheses of (−)-puupehenol and (+)-puupehedione†
Huayue
Song
,
Lin
Liu
,
Mingyang
Yang
,
Guangmiao
Wu
,
Peng
Chen
,
Xingang
Xie
 * and
Xuegong
She
* and
Xuegong
She
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou, 730000, P. R. China. E-mail: xiexg@lzu.edu.cn
First published on 15th November 2019
Abstract
A concise total synthesis of antitumor and antimalarial marine sponge metabolite (−)-15-oxopuupehenol has been accomplished in 8 steps (longest linear route) with an overall yield of 18% from R-(−)-carvone. Our synthesis involves a Suzuki carbonylative coupling reaction to assemble the compact tetra-substituted α,β-unsaturated aryl ketone and a KOH promoted intramolecular cyclization reaction to construct the unique chromanone core. The antituberculosis drug (+)-puupehenone is obtained via a three-step transformation from the synthetic (−)-15-oxopuupehenol. The formal syntheses of (−)-puupehenol and (+)-puupehedione can also be achieved from the same advanced intermediate 19.
Introduction
Quinone/hydroquinone sesquiterpenes based on drimane skeletons are mainly of marine origin and constitute a wide and diverse group of secondary metabolites of mixed biogenesis.1 Puupehenone, a typical example of such types of natural meroterpenoids, was firstly isolated together with its 21-chloro and bromo analogues (Fig. 1, 1–3) by Scheuer et al. in 1979.2a Puupehenones 1–3 contain an uncommon cis–anti–trans fused ring system which was formulated by the analysis of the X-ray data of their ozonolysis product.2g The unique rigid and compact configuration in space enables these types of natural chromane-sesquiterpenoids to display moderate activity against Gram-positive bacteria and against fungi. While the intriguing molecular skeleton and pharmaceutical potential of puupehenones prompted much research interest in this field, more than twenty congeners of puupehenone 1 were subsequently isolated in the following 40 years (Fig. 1, 4–10), exhibiting diverse and interesting bioactivities, such as antiviral, antimalarial, antibiotic, immunomodular and antitubercular activities.2,3 The important wide-spectrum bioactivities of these marine meroterpenoids together with their unusual fused ring system have also stimulated continuous interest in the synthetic community and served as a platform to develop and test new synthetic methods and strategies.4,5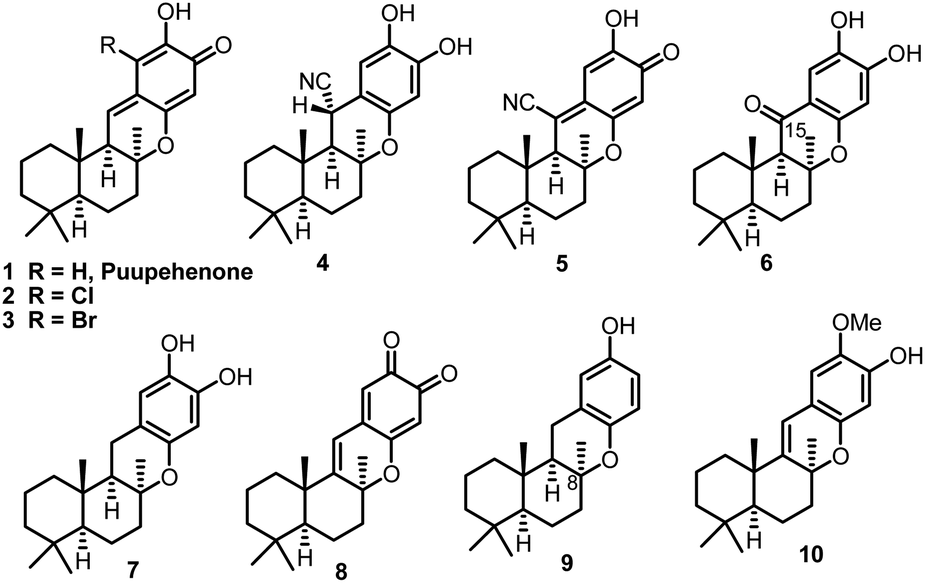 | ||
| Fig. 1 Puupehenone and its congeners. | ||
In 1995, (−)-15-oxopuupehenol (Fig. 1, 6), the only member containing a ketone group at C-15, was isolated by the Scheuer group from two Hawaiian sponges.6 Sharing a common tetracyclic skeleton, this congener, however, can be distinguished from other puupehenones by the unique presence of a carbonyl group at C-15.7 In a preliminary bioassay, (−)-15-oxopuupehenol showed significant and differential antitumor and antimalarial activities.6 So far, there have been two synthetic studies reported on 15-oxopuupehenol.8 Both of them firstly combined a drimane aldehyde unit with an aryl lithium reagent and assembled the C-15 ketone group by a lateral benzylic oxidation reaction. As for the key cyclization strategy, the Arjona and Plumet group utilized a CSA promoted cyclization reaction to access both enantiomers of 15-oxopuupehenol methylenedioxy derivatives (Scheme 1a).8a However, Alvarez-Manzaneda et al. later found that the above process occurred with a low degree of diastereoselectivity, with the undesired 8-epi-derivative being the major product.8b Thus, their approach for the first enantioselective total synthesis of (−)-15-oxopuupehenol was based on the palladium(II)-mediated oxa-Heck cyclization of a drimenylphenol prepared from (−)-sclareol (Scheme 1b). Recently, we reported an enantioselective total synthesis of (−)-8-epi-chromazonarol (9), a natural analogue of (−)-puupehenol (7), via a Stille carbonylative cross coupling reaction to combine the bicyclic unit and an aryl tin reagent. N2H4·H2O promoted highly diastereoselective oxa-Michael cyclization was utilized to construct the central chromane framework and assemble the unusual C-8 stereocenter.9 Although the completed synthesis was efficient and represented an expedient chemical synthetic solution to these types of chromane-meroterpenoids, we wondered whether the key coupling reaction could be replaced by a Suzuki carbonylative cross coupling reaction, avoiding the use of toxic tin reagents whose residue in the final product could hamper biological evaluations. In addition, we also want to capitalize on the developed strategy for a collective total synthesis of this class of secondary metabolites. With these ideas in mind, we continued our synthetic endeavours. Herein we report a concise total synthesis of (−)-15-oxopuupehenol and its transformation into (+)-puupehenone. Furthermore, also accomplished are the formal syntheses of (−)-puupehenol and (+)-puupehedione that can be achieved using the same advanced intermediate 19.
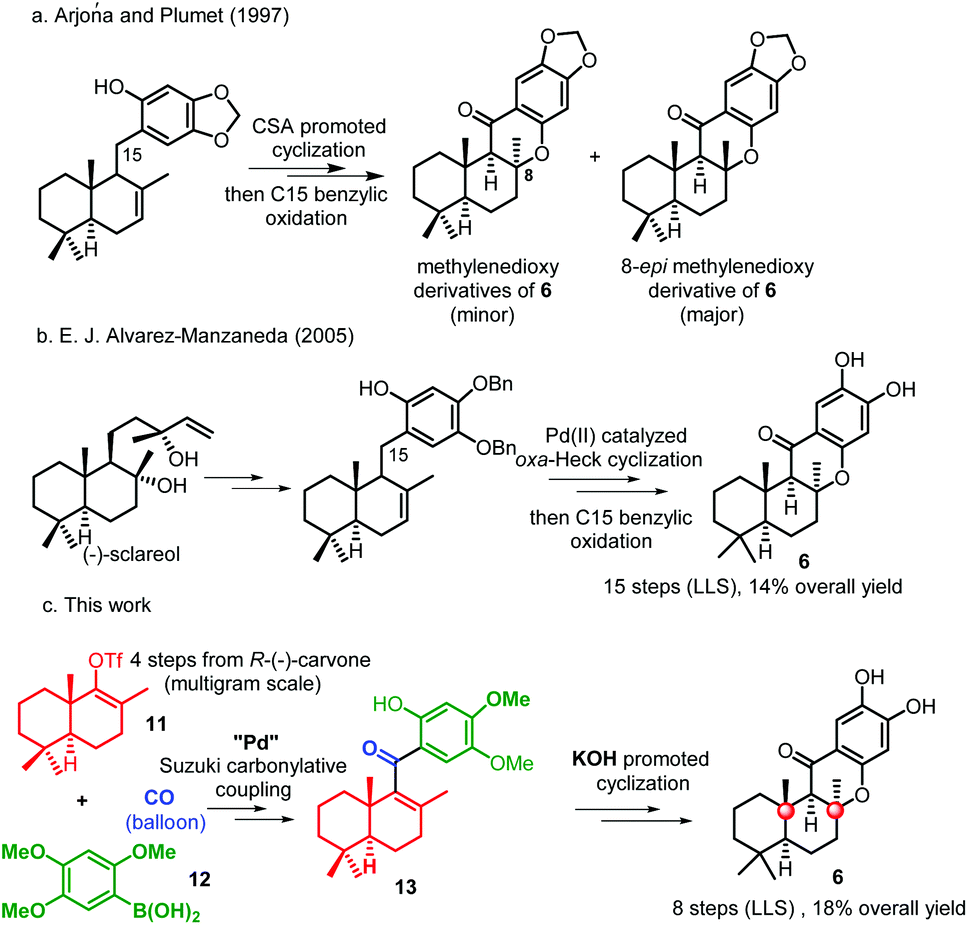 | ||
| Scheme 1 Total syntheses of (−)-15-oxopuupehenol 6. | ||
Results and discussion
Our synthesis commenced with the preparation of two essential coupling segments: bicyclic vinyl triflate 11 and aryl boronic acid 12 (Scheme 2). Compound 11 was quickly obtained in multigrams starting from the commercially available R-(−)-carvone via a modified 4-step synthetic route.9 The second coupling segment 12 was also easily prepared from the commercially available 1,2,4-trimethoxybenzene (17) on a multigram scale based on a two-step process reported by Aggarwal et al.10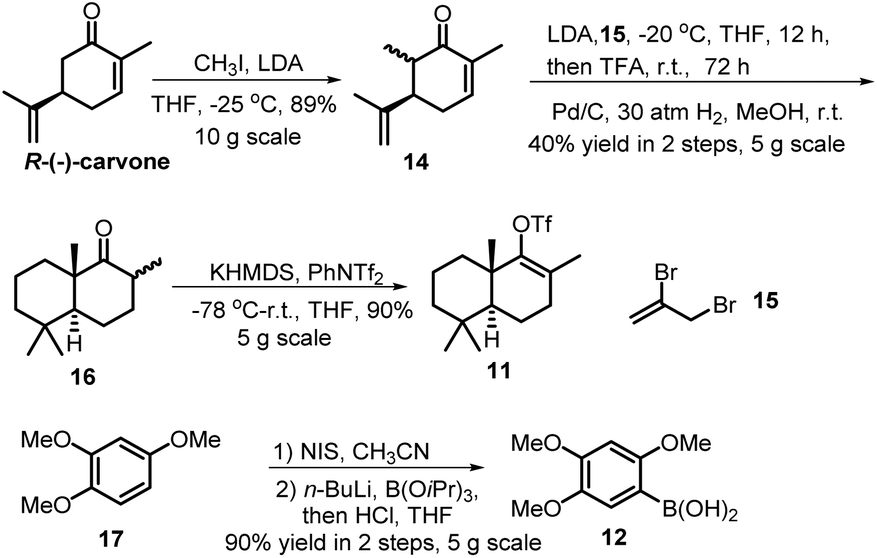 | ||
| Scheme 2 Preparation of aryl boronic acid 12 and bicyclic triflate 11. | ||
With sufficient amounts of two coupling segments in hand, we then studied their carbonylative union under Suzuki coupling reaction conditions which would give the required compact aryl vinyl ketone 18 (Scheme 3). To our delight, under a 1 atm CO atmosphere (balloon), using PdCl2/PPh3 as the catalyst system and K2CO3 as the essential base, the carbonylative coupling product 18 was obtained in 73% yield. This reaction could be smoothly scaled up to 2 g without a significant decrease in the yield. As only a few examples of the Suzuki carbonylative coupling reaction of vinyl triflates had been reported, several other aryl boronic acids were also prepared and tested in the coupling with bicyclic vinyl triflate 11 under the above conditions to roughly assess the generality of the reaction. As shown in Scheme 3, aryl boronic acids with strong electron donating methoxy substituents participated smoothly in the carbonylative coupling reactions to give the corresponding vinyl aryl ketones 18a–18e in 37%–76% isolated yields. However, aryl boronic acids with a strong electron withdrawing substituent such as a CF3 or Cl group did not react at all. Further screening of phenylboronic acids revealed the methoxy substituent on the aryl boronic acids to be essential for the success of the present carbonylative coupling reaction. The subsequent selective demethylation reaction proved to be a problem. The common demethylation reagents, such as BBr3, only led to the global demethylation of 18 even at −78 °C, and no reaction occurred when p-TsOH·H2O was utilized in CH2Cl2 at room temperature. To our delight, a softer Lewis acid AlCl3 later proved to be the reagent of choice. As shown in Scheme 4, the reaction of 18 with 3 equiv. of AlCl3 in refluxing CH3CN led to the selective removal of the methyl ether ortho to the C-15 carbonyl group, affording the corresponding demethylated product in 90% yield as a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of tautomers 13 and 13a (see the ESI† for details). Thus, the mixture was directly used in the subsequent Michael cyclization reaction. Treatment of compound 13 with excess N2H4·H2O in refluxing diethylene glycol (DEG), the conditions we previously used in the total synthesis of 8-epi-chromazonarol 9,9 smoothly gave the desired cyclization product 19 in 40% yield.
:1 mixture of tautomers 13 and 13a (see the ESI† for details). Thus, the mixture was directly used in the subsequent Michael cyclization reaction. Treatment of compound 13 with excess N2H4·H2O in refluxing diethylene glycol (DEG), the conditions we previously used in the total synthesis of 8-epi-chromazonarol 9,9 smoothly gave the desired cyclization product 19 in 40% yield.
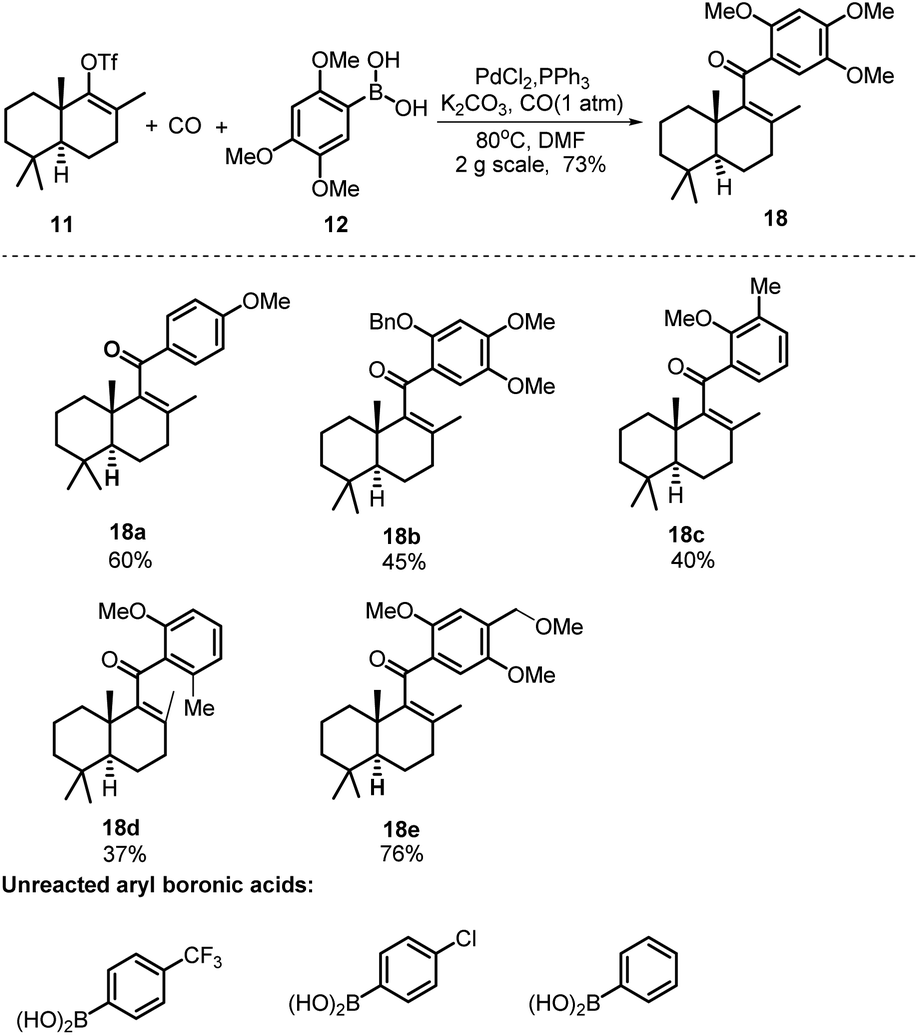 | ||
| Scheme 3 Synthesis of vinyl aryl ketone 18via a Suzuki carbonylative coupling reaction. | ||
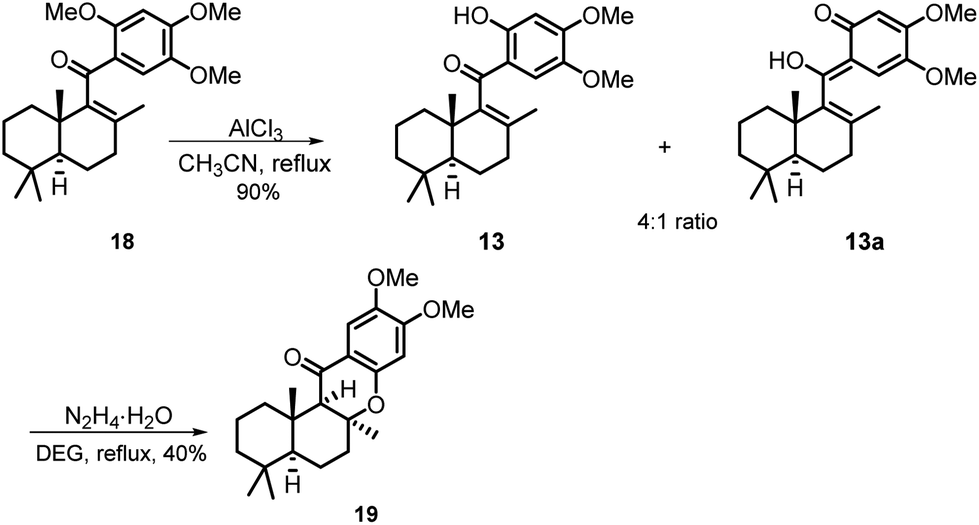 | ||
| Scheme 4 Synthesis of tetracyclic chromanone 19 using N2H4·H2O as the promoter. | ||
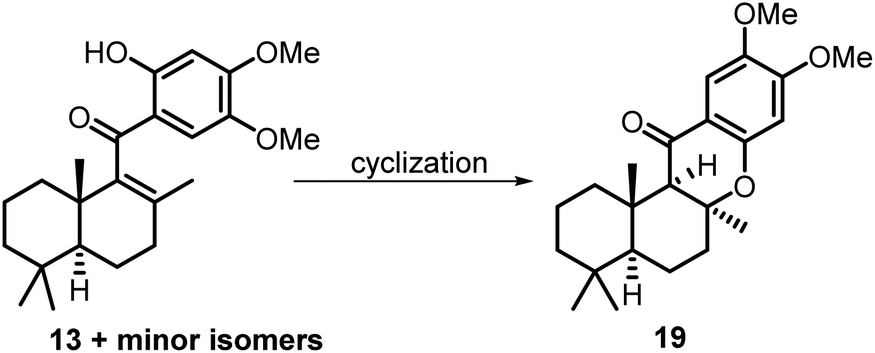
Although the N2H4·H2O promoted intramolecular Michael cyclization reaction was still efficient, we pondered whether the yield of the reaction could be improved by using a different base as the promoter. Thus, a further optimization study of the cyclization reaction was performed, which mainly focused on screening the additive bases (Table 1). To our delight, KOH soon proved to be the most efficient promoter, and the desired cyclization product 19 was obtained in 86% yield (Table 1, entries 1–5). Further screening of the amounts of the base disclosed that 5 equivalents of KOH were essential for this cyclization reaction (Table 1, entries 6 and 7). Finally, the solvent screening studies indicated that the combination of KOH and DEG was essential for the success of the cyclization reaction (Table 1, entries 8–10).
|
|
||
|---|---|---|
| Entry | Base | Yields (%) |
| a All reactions were performed on a 0.1 mmol scale, using 10 equiv. base in 10 mL DEG at 125 °C for 24 h unless otherwise mentioned. b KOH (5 equiv.) was used. c KOH (2 equiv.) was used. d Toluene was used as the solvent. e Ethylene glycol was used as the solvent. f Without base. | ||
| 1 | KOH | 86 |
| 2 | K2CO3 | 15 |
| 3 | NaOH | 83 |
| 4 | DBU | 40 |
| 5 | n-U4NOH | 50 |
| 6b | KOH | 86 |
| 7c | KOH | 36 |
| 8d | KOH | — |
| 9e | KOH | — |
| 10f | — | — |
When the cyclization reaction was performed on a 600 mg scale under the optimal conditions, a less polar minor product was obtained in 6% yield along with compound 19, which was determined to be 8-epi-19 by a comprehensive spectra data analysis. A control experiment using 19 as the starting material under the identical cyclization reaction conditions was carried out to explore the interconversion possibility of 19 and 8-epi-19. Compound 19 remained unaltered and no 8-epi-19 was produced after 24 h which indicates that the cyclization reaction is not reversible.
At this stage, our proposal for the reaction mechanism of the above base promoted cyclization is shown in Scheme 5. When the mixture of 13 and its tautomer 13a is treated with a base, an oxyanion (I or I′) is generated which adopts two possible ultimate conformations (II or II′vsIII or III′). Conformer II or II′ is more favored due to the conformation constraint of the boat-like B ring and it undergoes anionic cyclization from the β-face followed by a protonation reaction to give the desired 19 as the major product. Although 8-epi-19 is a more stable cyclization product based on DFT calculation (34.95 kJ mol−1 lower than that of 19, see the ESI† for details), it can only be obtained from the cyclization reaction of a disfavored conformer III or III′ and thus it serves as a minor product.
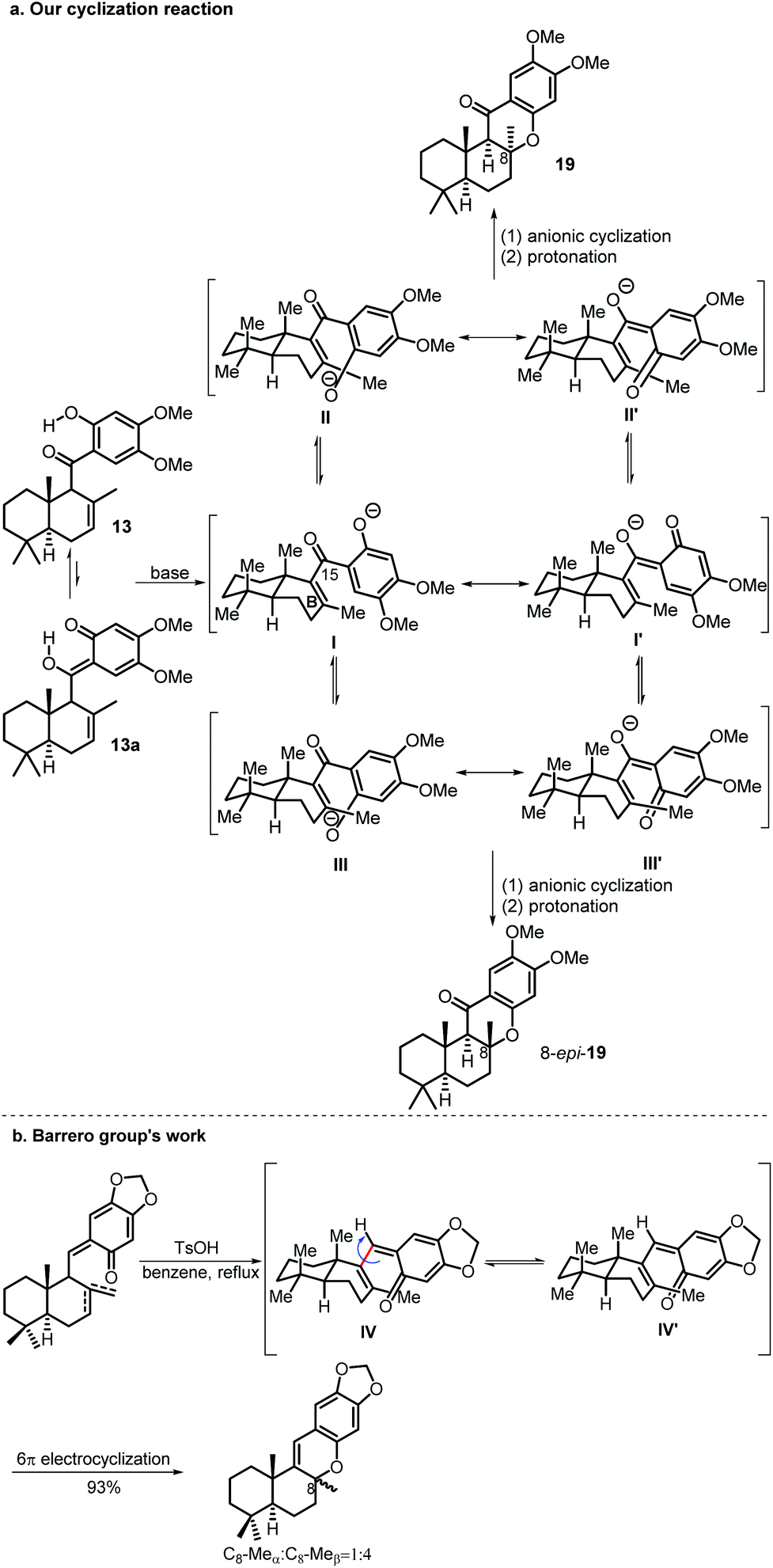 | ||
| Scheme 5 Possible mechanisms of the KOH promoted cyclization reaction. | ||
Several previous syntheses of these kinds of natural products using 6π electrocyclization gave the 8-epi-cyclization product as the major product.2c,3d,5b,11 For example, the Barrero group reported that a 1:4 mixture (C8-Meαvs. C8-Meβ) was obtained from conformers IV and IV′ (Scheme 5b). The main difference between our cyclization precursor and previous similar 6π electrocyclization precursors used in the syntheses of these kinds of natural products is the oxygen atom attached to C-15. In all the previous cases, a smaller hydrogen atom is attached at the same position instead, which favors the interconversion of conformers IV and IV′, so the more stable 8-epi-cyclization product is obtained as the major product.
As shown in Scheme 6, the demethylation reaction of tetracyclic chromanone 19 by BBr3 was smoothly performed to afford (−)-15-oxopuupehenol 6 in 99% yield. The spectroscopic data of our synthetic 6 are in good agreement with those of the natural (−)-15-oxopuupehenol. The relative stereochemistry of our synthetic 6 was further confirmed by a NOE experiment.12 Since a direct reduction of (−)-15-oxopuupehenol 6 with the common carbonyl reducing reagents, such as DIBAL-H, NaBH4, LiAlH4, PdCl2/Et3SiH or Pd/C, H2 (1 atm or 20 atm), was not successful, its two free phenolic groups were firstly masked as the corresponding MOM ethers. Then, reduction of the C-15 ketone group of the obtained MOM ether 20 with LiBH4 followed by p-TsOH·H2O promoted a one pot MOM ether removal/dehydration elimination/isomeric rearrangement reaction sequence affording (+)-puupehenone 1 in 56% overall yield (brsm). When tetracyclic chromanone 19 was firstly reduced by LiAlH4 to the corresponding benzylic alcohol, prior to further reduction with PdCl2/Et3SiH, tetracyclic chromane 21 was obtained in 89% yield, which had been previously used in the total synthesis of (−)-puupehenol (7) by Alvarez-Manzaneda and Barranco's group.13a On the other hand, when compound 19 was sequentially treated with LiAlH4 and TFA, tetracyclic chromene 22 was obtained instead. According to the report of Armstrong and Sepúlveda et al.,13b simultaneous demethylation–oxidation of chromene 22 would give (+)-puupehedione (8). Thus, the formal syntheses of (−)-puupehenol (7) and (+)-puupehedione (8) could also be achieved with the same advanced intermediate 19.
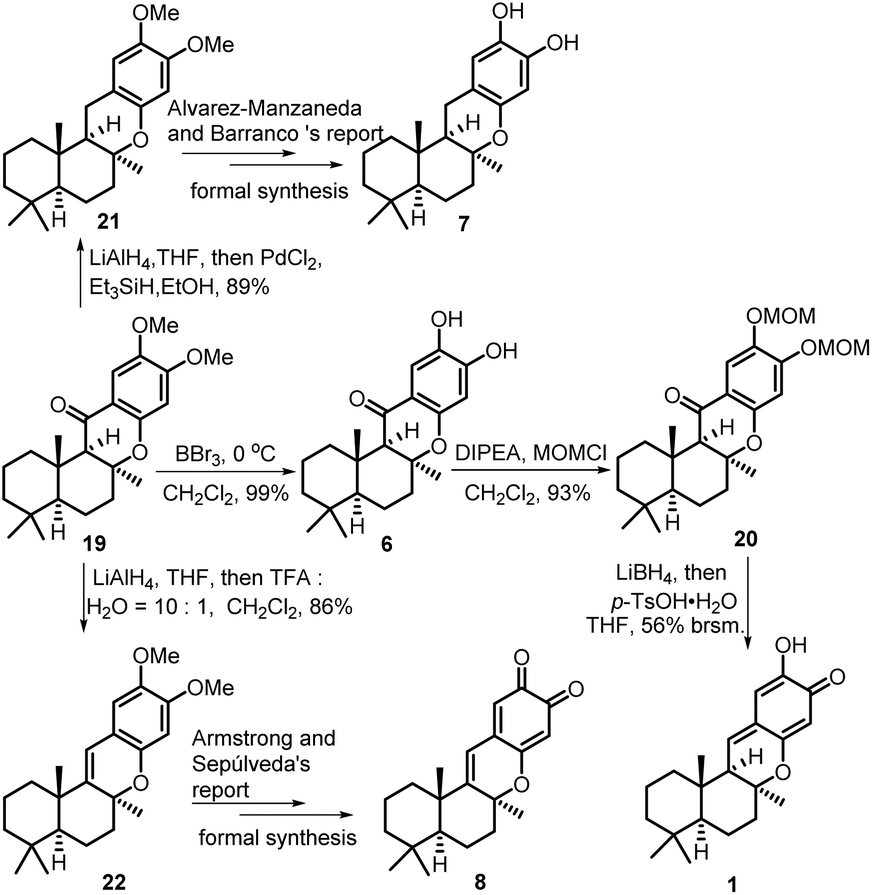 | ||
| Scheme 6 Total synthesis of (−)-15-oxopuupehenol 6, its derivative transformation to (+)-puupehenone 1 and the formal syntheses of (−)-puupehenol 7 and (+)-puupehedione 8. | ||
Conclusions
In conclusion, a concise total synthesis of (−)-15-oxopuupehenol 6 has been achieved from the readily available R-(−)-carvone in only 8 steps (longest linear route) with an overall yield of 18%. The key features of the present synthesis are the use of a Suzuki carbonylative reaction to assemble the compact vinyl aryl ketone 18 and the KOH-assisted highly diastereoselective cyclization that constructed the unique chromanone skeleton with its unusual cis–anti (BC fused ring system) stereochemistry. (+)-Puupehenone 1 has been prepared in three steps from 6. Subjection of the key tetracyclic chromanone 19 to different reduction conditions also led to the formal syntheses of (−)-puupehenol 7 and (+)-puupehedione 8. All of these indicate the great potential of the developed strategy in the synthesis of a diverse array of chromane-containing meroterpenoids which will provide a solid foundation for their further SAR studies.Experimental
General procedure for the synthesis of α,β-unsaturated aryl ketones 18 and 18a–18e
In a dried vial with 12 (1.4 g, 6.6 mmol, 1.1 equiv.), 11 (2.04 g, 6 mmol, 1 equiv.), PdCl2 (308 mg, 1.8 mmol, 0.3 equiv.), PPh3 (943 mg, 3.6 mmol, 0.6 equiv.) and K2CO3 (2.48 g, 18 mmol, 3 equiv.) was added freshly distilled DMF (60 mL). The general procedure was followed starting from 11 and 12. Then it was evacuated under vacuum and refilled with carbon monoxide (balloon) three times. The resulting mixture was then stirred at room temperature for 2 h. Then it was heated to 80 °C. After stirring for 12 h, the mixture was then cooled to 23 °C and filtered to remove the solid using Celatom and eluted with EtOAc (200 mL). Then the organic phase was washed with water (30 mL) and brine. The resulting aqueous layer was extracted with EtOAc (60 mL × 3). The combined organic phase was dried over anhydrous Na2SO4, filtered, concentrated, and purified by column chromatography on silica gel to afford the corresponding α,β-unsaturated aryl ketones 18a–18e and 18.:20) as the eluent to afford the mixture of compound 13 and its unknown minor isomer as a white solid (1.51 g, 90%). To a stirred solution of phenol 13 and its minor isomer (558 mg, 1.5 mmol, 1 equiv.) in dry diethylene glycol (15 mL) was added KOH (420 mg, 7.5 mmol, 5 equiv.). The resulting mixture was then heated to 120 °C. After stirring for 12 h, the reaction mixture was quenched with saturated NH4Claq. (5 mL). The mixture was washed with 2 N HCl, and then extracted with EtOAc (30 mL × 3). The combined organic phases were washed with saturated brine, dried over anhydrous Na2SO4, filtered, and concentrated to give a crude residue which was purified by column chromatography on silica gel and eluted with EtOAc/petroleum (1:30) to afford ketone 19 as the major product (white solid, 480 mg, 86%). M.p.: 197–203 °C (CH2Cl2). [α]20D = −54.0, (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 7.26 (s, 1H), 6.38 (s, 1H), 3.90 (s, 3H), 3.88 (s, 3H), 2.23–2.19 (m, 1H), 1.91 (s, 1H), 1.72–1.65 (m, 3H), 1.61–1.48 (m, 3H), 1.44–1.40 (m, 3H), 1.26 (s, 4H), 0.93 (s, 3H), 0.85 (s, 6H). 13C NMR (101 MHz, CDCl3) δ = 193.2, 156.3, 155.7, 143.8, 114.5, 106.2, 100.3, 80.6, 64.3, 56.1, 56.1, 54.2, 41.6, 40.1, 40.0, 38.4, 33.8, 33.4, 26.6, 22.0, 18.3, 18.1, 15.1. HRMS (ESI): m/z [M + H]+ calcd for C23H33O4: 373.2379, found: 373.2377.
8-epi-19 was obtained in 6% yield together with 19 when the cyclization reaction was performed at a 600 mg scale. M.p.: 199–206 °C (CH2Cl2). 1H NMR (400 MHz, CDCl3) δ = 7.21 (s, 1H), 6.35 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 2.75 (d, J = 12.8 Hz, 1H), 2.67 (s, 1H), 2.06 (m, 1H), 1.92 (m, 1H), 1.80–1.68 (m, 2H), 1.47–1.42 (m, 2H), 1.40 (s, 4H), 1.31–1.26 (m, 2H), 1.13 (s, 3H), 1.01–0.96 (m, 2H), 0.90 (s, 3H), 0.85 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 192.9, 155.3, 154.5, 143.6, 114.2, 106.7, 100.0, 83.4, 64.5, 56.1, 56.1, 55.5, 41.8, 40.8, 39.8, 37.3, 33.6, 33.3, 21.6, 21.1, 19.3, 18.4, 15.7. HRMS (ESI): m/z [M + H]+ calcd for C23H33O4: 373.2379, found: 373.2383.
:5) to afford compound 6 as a colorless glass (191 mg, 99%). [α]22D = −102.0, (c = 0.5, CH3OH). 1H NMR (400 MHz, CDCl3) δ = 7.55 (s, 1H), 6.43 (s, 1H), 2.21 (d, J = 14.2 Hz, 1H), 1.91 (s, 1H), 1.72–1.69 (m, 3H), 1.57–1.49 (m, 2H), 1.46–1.39 (m, 3H), 1.23 (s, 3H), 1.21–1.13 (m, 2H), 0.91 (s, 3H), 0.88 (d, J = 1.4 Hz, 1H), 0.85 (s, 3H), 0.84 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 195.6, 157.1, 153.5, 138.5, 114.8, 110.8, 103.7, 80.4, 64.5, 54.2, 41.6, 40.1, 39.9, 38.7, 33.8, 33.4, 26.6, 22.0, 18.4, 18.16, 15.2. HRMS (ESI): m/z [M + H]+ calcd for C21H29O4: 345.2066, found: 345.2060.
:20) to afford the corresponding compound 20 as a white solid (402 mg, 93%). M.p.: 120–123 °C (CH2Cl2). [α]24D = −39.0, (c = 1.0, CHCl3). 1H NMR (600 MHz, CDCl3) δ = 7.21 (s, 1H), 6.35 (s, 1H), 3.88 (s, 3H), 3.86 (s, 3H), 2.75 (d, J = 12.8 Hz, 1H), 2.67 (s, 1H), 2.06 (d, J = 12.5 Hz, 1H), 1.92 (t, J = 11.3 Hz, 1H), 1.78 (d, J = 13.4 Hz, 1H), 1.70 (dd, J = 27.1, 13.5 Hz, 1H), 1.47–1.36 (m, 7H), 1.31–1.25 (m, 2H), 1.20–1.18 (m, 1H), 1.15 (d, J = 13.7 Hz, 4H), 1.00–0.97 (m, 2H), 0.91 (s, 3H), 0.85 (s, 3H). 13C NMR (151 MHz, CDCl3) δ = 192.9, 155.4, 154.7, 143.6, 114.2, 106.8, 100.1, 83.4, 83.4, 64.6, 56.1, 56.1, 55.6, 41.9, 40.9, 39.8, 37.4, 33.6, 33.4, 31.6, 21.6, 21.1, 19.4, 18.4, 15.7. HRMS (ESI): m/z [M + H]+ calcd for C25H37O6: 433.2590, found: 433.2585.
:5) to afford the corresponding alcohol (74 mg) and 20 (36 mg) was recovered. The obtained alcohol (36 mg, 0.08 mmol) was dissolved in a solution of THF/H2O (0.8/0.08 mL) and cooled to 0 °C. p-TsOH·H2O (63 mg, 0.32 mmol, 4.0 equiv.) was slowly added and the mixture was stirred at 40 °C for 2 h. After evaporation under vacuum, the resulting residue was purified by column chromatography on silica gel with EtOAc/petroleum (1:5) as the eluent affording (+)-puupehenone 1 (16 mg, 56% based on the recycled starting material) as a yellow oil. [α]25D = 278.0, (c = 0.5, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.85 (s, 1H), 6.65 (d, J = 6.9 Hz, 1H), 6.20 (s, 1H), 5.86 (d, J = 1.0 Hz, 1H), 2.17 (m, 1H), 2.04 (d, J = 6.9 Hz, 1H), 1.68 (dd, J = 12.4, 1.4 Hz, 1H), 1.59–1.53 (m, 3H), 1.51–1.39 (m, 3H), 1.23 (s, 3H), 1.21–1.13 (m, 2H), 0.97–0.93 (m, 1H), 0.91 (s, 3H), 0.84 (s, 3H), 0.82 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 182.0, 162.8, 147.4, 140.4, 129.3, 106, 105.0, 78.8, 54.8, 53.9, 41.6, 40.7, 40.0, 39.2, 33.7, 33.3, 28.0, 21.9, 18.4, 18.1, 15.0. HRMS (ESI): m/z [M + H]+ calcd for C21H29O3: 329.2117, found: 329.2111.
:5) as the eluent affording the corresponding benzylic alcohol as a colorless oil (122 mg). To a solution of the obtained benzylic alcohol (91 mg, 0.24 mmol, 1 equiv.) and triethylsilane (0.23 mL, 1.44 mmol, 6 equiv.) in ethanol (1.2 mL) was added a catalytic amount of palladium(II) chloride (10 mmol%). Then the mixture was heated to 40 °C and stirred under an argon atmosphere overnight. After evaporation under vacuum, the resulting residue was purified by column chromatography on silica gel with EtOAc/petroleum (1:50) as the eluent affording 21 (80 mg, two steps 89%) as a white solid. M.p.: 139–143 °C (CH2Cl2). [α]21D = −41.0 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ = 6.48 (s, 1H), 6.29 (s, 1H), 3.77 (d, J = 1.0 Hz, 6H), 2.80 (dd, J = 17.6, 8.1 Hz, 1H), 2.63 (d, J = 17.6 Hz, 1H), 2.07 (dd, J = 10.5, 2.4 Hz, 1H), 1.81 (d, J = 12.3 Hz, 1H), 1.62–1.24 (m, 9H), 1.13 (s, 3H), 1.11–1.07 (m, 1H), 0.86 (s, 3H), 0.78 (s, 3H), 0.68 (s, 3H). 13C NMR (101 MHz, CDCl3) δ = 148.2, 147.7, 142.6, 112.9, 111.3, 101.1, 75.3, 56.3, 55.7, 55.3, 49.6, 41.9, 40.7, 40.1, 38.3, 33.7, 33.2, 27.2, 22.3, 21.9, 18.5, 18.3, 14.2. HRMS (ESI): m/z [M + H]+ calcd for C23H35O3: 359.2586, found: 359.2581.
:5) as the eluent afforded the corresponding benzylic alcohol as a colorless oil (122 mg). The obtained benzylic alcohol (25 mg, 0.067 mmol, 1 equiv.) was dissolved in a solution of CH2Cl2 (0.7 mL) and cooled to 0 °C. H2O (0.012 mL, 0.67 mmol, 10 equiv.) and CF3COOH (0.03 mL, 0.4 mmol, 6 equiv.) were slowly added, warmed to room temperature and kept under stirring overnight. After evaporation under vacuum, the resulting residue was purified by column chromatography on silica gel with EtOAc/petroleum (1:50) as the eluent affording 22 (22 mg, two steps, 86%) as a white solid. M.p.: 139–143 °C (CH2Cl2). [α]23D = 78.0 (c = 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ = 6.58 (s, 1H), 6.45 (s, 1H), 6.10 (s, 1H), 3.83 (s, 6H), 2.20–1.99 (m, 3H), 1.95–1.78 (m, 2H), 1.75–1.40 (m, 6H), 1.36 (s, 3H), 1.21 (s, 3H), 0.95 (s, 3H), 0.87 (s, 3H). 13C NMR (75 MHz, CDCl3) δ = 149.7, 148.7, 145.6, 143.2, 116.1, 113.9, 109.5, 100.7, 77.2, 56.4, 55.9, 43.9, 42.0, 39.1, 38.6, 33.7, 32.6, 31.0, 25.5, 25.0, 21.2, 19.0, 17.3. HRMS (ESI): m/z [M + H]+ calcd for C23H33O3: 357.2430, found: 357.2424.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We wish to thank the NSFC (21472079 and 21572088) for their generous financial support. We thank the referees for helpful suggestions on the possible cyclization mechanism and Prof. Panpan Zhou at Lanzhou University for his help with the calculation of the relative energy of the cyclization products.References
- I. S. Marcos, A. Conde, R. F. Moro, P. Basabe, D. Diez and J. G. Urones, Mini-Rev. Org. Chem., 2010, 7, 230 CrossRef CAS.
- (a) B. N. Ravi, H. P. Perzanowski, R. A. Ross, T. R. Erdman, P. J. Scheuer, J. Finer and J. Clardy, Pure Appl. Chem., 1979, 51, 1893 CAS; (b) M. T. Hamann, P. J. Scheuer and M. Kelly-Borges, J. Org. Chem., 1993, 58, 6565 CrossRef CAS; (c) D. H. Hua, X. Huang, Y. Chen, S. K. Battina, M. Tamura, S. K. Noh, S. I. Koo, I. Namatame, H. Tomoda, E. M. Perchellet and J. P. Perchellet, J. Org. Chem., 2004, 69, 6065 CrossRef CAS PubMed; (d) M. T. Hamann and P. J. Scheuer, Tetrahedron Lett., 1991, 32, 5671 CrossRef CAS; (e) A. F. Barrero, E. J. Alvarez-Manzaneda, M. M. Herrador, M. V. Valdivia and R. Chahboun, Tetrahedron Lett., 1998, 39, 2425 CrossRef CAS; (f) K.-I. Takao, T. Sasaki, T. Kozaki, Y. Yaganisawa, K.-I. Tadano, A. Kawashima and H. Shinonaga, Org. Lett., 2001, 3, 4291 CrossRef CAS PubMed; (g) S. Urban and R. J. Capon, J. Nat. Prod., 1996, 59, 900 CrossRef CAS.
- (a) I. C. Piña, M. L. Sanders and P. Crews, J. Nat. Prod., 2003, 66, 2 CrossRef PubMed; (b) E. Sayed, K. A. Bartyzel, P. Shen, X. Perry, T. L. Zjawiony, J. K. Hamann and M. T, Tetrahedron, 2000, 56, 949–953 CrossRef; (c) M. L. Ciavatta, M. P. Lopez Gresa, M. Gavagnin, V. Romero, D. Melck, E. Manzo, Y. Guo, R. Van Soest and G. Cimino, Tetrahedron, 2007, 63, 1380 CrossRef CAS; (d) A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun, M. Cortés and V. Armstrong, Tetrahedron, 1999, 55, 15181 CrossRef CAS.
- Selected reported synthetic efforts towards puupehenone and its analogues: (a) R. M. Kamble and M. M. V. Ramana, Helv. Chim. Acta, 2011, 94, 261 CrossRef CAS; (b) R. G. Pritchard, H. M. Sheldrake, I. S. Taylor and T. W. Wallace, Tetrahedron Lett., 2008, 49, 4156 CrossRef CAS; (c) A. F. Barrero, J. F. Q. Moral, M. M. Herrador, P. Arteaga, M. Cortés, J. Benitesa and A. Rosellón, Tetrahedron, 2006, 62, 6012 CrossRef CAS; (d) B. S. Crombie, C. Smith, C. Z. Varnavas and T. W. Wallace, J. Chem. Soc., Perkin Trans. 1, 2001, 206 RSC.
- Reported total synthesis of puupehenone and its analogues: (a) A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun, M. Cortés and V. Armstrong, Tetrahedron, 1999, 55, 15181 CrossRef CAS; (b) S. Maiti, S. Sengupta, C. Giri, B. Achari and A. K. Banerjee, Tetrahedron Lett., 2001, 42, 2389 CrossRef CAS; (c) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432 CrossRef CAS PubMed; (d) H. Ishibashi, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 11122 CrossRef CAS PubMed; (e) S. Quideau, M. Lebon and A.-M. Lamidey, Org. Lett., 2002, 4, 3975 CrossRef CAS; (f) H. Wang, H. Li, J. Wang and Y. Wu, Green Chem., 2017, 19, 2140 RSC; (g) H. Wang, H. Li, X. Nan, Y. Luo and Y. Wu, J. Org. Chem., 2017, 82, 12914 CrossRef CAS; (h) A. F. Barrero, E. J. Alvarez-Manzaneda and R. Chahboun, Tetrahedron Lett., 1997, 38, 2325 CrossRef CAS.
- S. S. Nasu, B. K. S. Yeung, M. T. Hamman, P. J. Scheuer, M. Kelly-Borgues and K. Goins, J. Org. Chem., 1995, 60, 7290 CrossRef CAS.
- Although 15-oxopuupehenoic acid was reported to contain the C-15 ketone group as 15-oxopuupehenol, a recent total synthesis by Enrique Alvarez-Manzaneda et al. indicated that the originally assigned structure might be wrong. (a) S. J. Robinson, E. K. Hoobler, M. Riener, S. T. Loveridge, K. Tenney, F. A. Valeriote, T. R. Holman and P. Crews, J. Nat. Prod., 2009, 72, 1857 CrossRef CAS; (b) E. Boulifa, A. Fernández, E. Alvarez, R. Alvarez-Manzaneda, A. I. Mansour, R. Chahboun and E. J. Alvarez-Manzaneda, Org. Chem., 2014, 79, 10689 CrossRef CAS.
- (a) O. Arjona, M. Garranzo, J. Maluego, E. Maroto, J. Plumet and B. Sáez, Tetrahedron Lett., 1997, 38, 7249 CrossRef CAS; (b) E. J. Alvarez-Manzaneda, R. Chahboun, B. I. Pérez, E. Cabrera, E. Alvarez and R. Alvarez-Manzaneda, Org. Lett., 2005, 7, 1447 CrossRef.
- L. Liu, H. Song, P. Chen, Z. Yuan, S. Feng, W. Zhang, B. Fang, X. Xie and X. She, Org. Chem. Front., 2018, 5, 3013 RSC.
- A. Noble, S. Roesner and V. K. Aggarwal, Angew. Chem., Int. Ed., 2016, 55, 15920 CrossRef CAS.
- H. Wang, H. Li, Z. Zhang and Y. Wu, Eur. J. Org. Chem., 2018, 915 CrossRef CAS.
- See the ESI† for details.
- (a) E. J. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, A. Haidour, J. M. Ramos, R. Alvarez-Manzaneda, R. Tapia, H. Es-Samti, A. Fernández and I. Barranco, Eur. J. Org. Chem., 2009, 1139 CrossRef CAS; (b) V. Armstrong, A. F. Barrero, E. J. Alvarez-Manzaneda, M. Cortés and B. Sepúlveda, J. Nat. Prod., 2003, 66, 1382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo01027k |
| This journal is © the Partner Organisations 2020 |