DOI:
10.1039/C9QM00648F
(Research Article)
Mater. Chem. Front., 2020,
4, 899-904
A zinc-responsive fluorophore based on 5′-(p-hydroxyphenyl)-pyridylthiazole†
Received
23rd October 2019
, Accepted 18th January 2020
First published on 20th January 2020
Abstract
The synthesis and photophysical properties of an ion-sensitive fluorescent compound are described, featuring 5′-(p-hydroxyphenyl)pyridylthiazole (HPPT) as a highly emissive fluorophore. Density functional theory (DFT) calculations indicate that HPPT is capable of intramolecular charge transfer (ICT), with further polarization upon complexation with Zn(II). A 4′-picolyloxy–HPPT derivative was prepared and determined to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with Zn(NO3)2 in acetonitrile, with a Stokes shift of 137 nm (6323 cm−1) and a 67 nm bathochromic shift in emission relative to the neutral ligand, and a fluorescence quantum yield (ΦPL) of 92%. An X-ray crystal structure of the HPPT–Zn(II) complex confirmed a tridentate structure with a seven-membered chelate ring, and the picolyloxy unit rotated out of plane.
:1 complex with Zn(NO3)2 in acetonitrile, with a Stokes shift of 137 nm (6323 cm−1) and a 67 nm bathochromic shift in emission relative to the neutral ligand, and a fluorescence quantum yield (ΦPL) of 92%. An X-ray crystal structure of the HPPT–Zn(II) complex confirmed a tridentate structure with a seven-membered chelate ring, and the picolyloxy unit rotated out of plane.
Introduction
Chemically responsive fluorescent dyes are useful as probes for biological imaging and as components in the design of molecular logic gates.1–5 In many cases, molecular recognition events can be transduced by fluorescence using photoinduced electron transfer (PET) or intramolecular charge transfer (ICT) mechanisms.6,7 The latter has been broadly applied toward the design of ratiometric fluorescence sensing modalities that can quantify changes in the concentration of specific analytes.
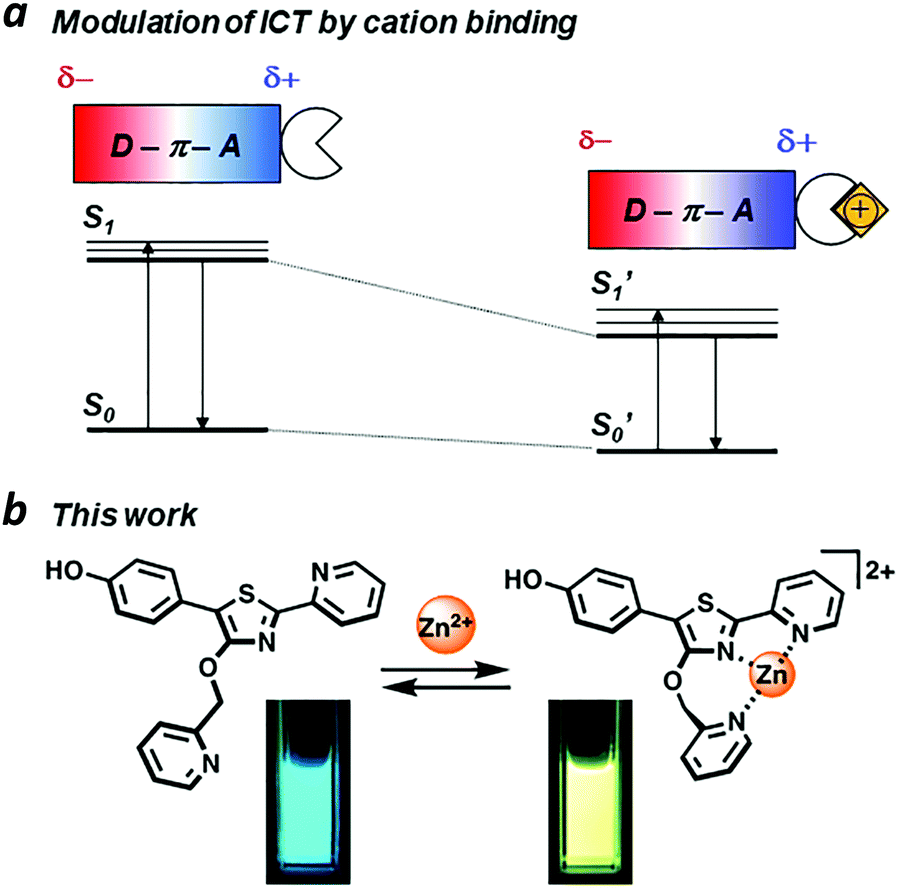
ICT is commonly observed in π-conjugated systems with electron-rich and electron-poor (donor and acceptor) units at opposite ends of the molecule. If the excited-state dipole is proximal to an ion binding site, then ICT efficiency can be modulated in a supramolecular fashion. For example, acceptor units that can bind metal ions may experience an increased electric polarization upon chelation, causing a bathochromic shift in photoemission (Fig. 1a). Ratiometric fluorescence sensing has been applied successfully toward the detection of transition-metal ions including Zn(II), an essential cofactor in many biological functions.8–10 The Zn(II) ion is compatible with excited-state processes, and can be incorporated into the design of fluorophores with large Stokes shifts.11–14
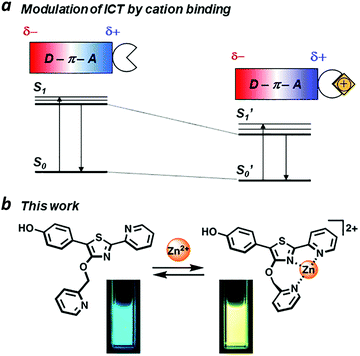 |
| | Fig. 1 (a) Intramolecular charge transfer (ICT) within a π-conjugated donor–acceptor system, modulated by ion binding; (b) a Zn(II)-binding fluorophore using 5′-(p-hydroxyphenyl)pyridylthiazole (HPPT) as an ICT platform. | |
Several Zn(II)-binding fluorophores are based on well-established dye scaffolds such as BODIPY,1,15 xanthene,3,16 and coumarin,3b,11 which all have high quantum yields (ΦPL) but produce small Stokes shifts upon metal ion binding. Fluorophores containing quinoline units have been shown to respond to Zn(II) with Stokes shifts of over 200 nm (8500 cm−1), but are limited by their low ΦPL.13 These earlier examples set the stage for developing new classes of ion-responsive fluorophores, featuring supramolecular binding motifs that have direct impact on their excited-state dipoles.
We have identified 5′-phenyl-2′-(2-pyridyl)thiazole (PPT) as a promising scaffold for the rational design of ratiometric fluorescent probes, using the ICT approach (Fig. 1b). The bidentate cleft of the pyridylthiazole unit can be fashioned into a receptor for transition-metal ions, providing a useful mechanism for lowering the energy of its acceptor state.17,18 Earlier studies on the chemistry and photophysical properties of 2′-(2-pyridyl)thiazoles by Yan19 and Beckert20 have shown these compounds to be capable of supporting high ΦPL and large Stokes shift due to structural changes in their excited states, with considerable tolerance for chemical modification. Their studies provide an excellent starting point for designing PPT derivatives whose fluorescence outputs can be tuned by well-defined supramolecular interactions. In this paper, we introduce 5′-(p-hydroxyphenyl)pyridylthiazole (HPPT) as a highly emissive ICT-type fluorophore, and a Zn(II)-binding HPPT derivative that exhibits a 67 nm bathochromic shift in emission upon complexation, with 92% ΦPL and a Stokes shift of 137 nm (6323 cm−1).
Results and discussion
DFT calculations
To determine the feasibility of generating excited-state ICT within the PPT scaffold, we first conducted density functional theory (DFT) calculations to evaluate the effects of p-substitution on the phenyl ring of the PPT unit, with and without Zn(II) complexation.21 HPPT was selected as an ICT-type fluorophore due to the strong polarization of its molecular orbital (MO) states by the p-hydroxyl group, relative to that of unsubstituted PPT (Fig. 2 and ESI†). Coordination between the pyridylthiazole unit and Zn(NO3)2 further increased polarization of the LUMO coefficients as expected, with the decrease in energy band gap (ΔEg) being most pronounced for HPPT. We also performed DFT calculations on pyridylthiazole derivatives bearing a picolyloxy group (4′-O-Pic-PPT 3 and 4′-O-Pic-HPPT 4), a well-known ancillary ligand for stabilizing Zn(II) binding, and confirmed that its presence had a negligible effect on the spatial distribution of MO coefficients (see ESI†).
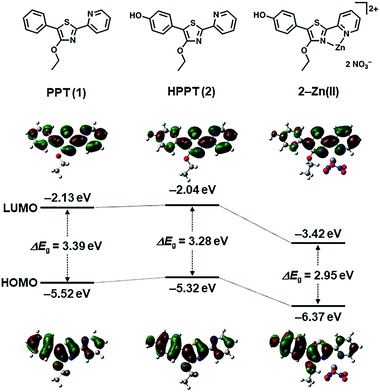 |
| | Fig. 2 DFT calculations of energy bands and spatial distributions of molecular orbital coefficients (HOMO and LUMO) for 4′-O-Et-PPT (1), 4′-O-Et-HPPT (2), and 2–Zn(II). See ESI† for additional DFT calculations. | |
Synthesis
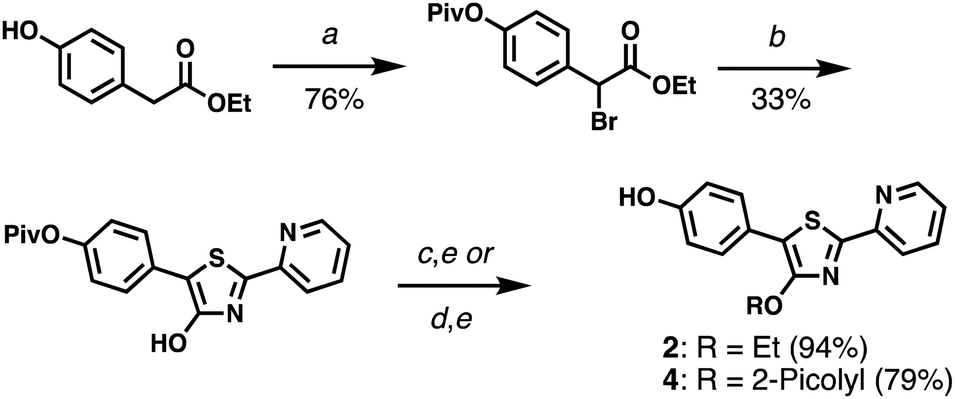
PPT derivatives with a 4′-hydroxyl on the central thiazole ring were prepared using Hantzsch conditions,22 similar to those described by Beckert and coworkers (Scheme 1).23,24 In the case of HPPT, ethyl p-hydroxyphenylacetate was converted to a pivaloyl ester then brominated at the methylene position, and condensed with 2-pyridylthioamide by microwave heating to yield the corresponding 4′-hydroxythiazole in 25% yield over three steps. O-Alkylation with ethyl iodide or picolyl bromide followed by methanolysis produced 4′-O-Et-HPPT (2) or 4′-O-Pic-HPPT (4) respectively. PPT derivatives lacking the p-hydroxyl group, namely 4′-O-Et-PPT (1) and 4′-O-Pic-PPT (3), were prepared according to literature procedure and alkylated as described above.24
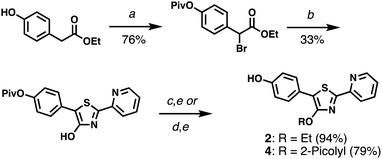 |
| | Scheme 1 Synthesis of HPPT derivatives 2 and 4. (a) (i) PivCl, DMAP, pyridine; (ii) NBS, AIBN, TFT, 80 °C. (b) 2-Pyridylthioamide, EtOH, 90 °C (μW). (c) EtI, K2CO3, acetone. (d) 2-PicBr·HBr, K2CO3, acetone. (e) K2CO3, MeOH. See ESI† for reagent abbreviations. | |
X-ray crystal structure determination
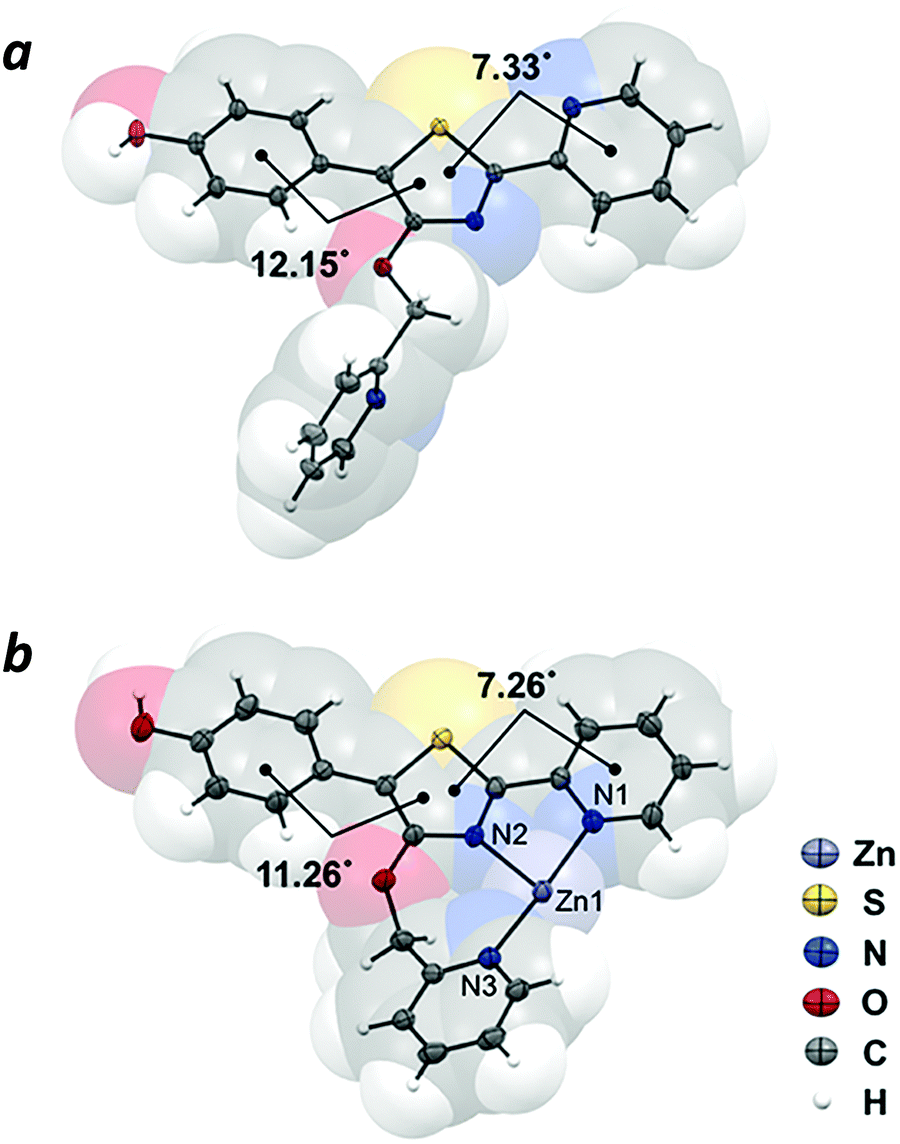
The crystal structure of compound 4 revealed the π-conjugated chromophore to be nearly planar (phenyl-thiazolyl dihedral angle = 12.15°; pyridyl-thiazolyl dihedral angle = 7.33°), with the nitrogen atoms adopting a transoid geometry to minimize steric interactions between rings. In contrast, the 4′-O-picolyl group is nearly perpendicular with respect to the thiazole ring (Fig. 3a). The crystal structure of complex 4–Zn(II), which was prepared by mixing Zn(NO3)2 with 4 in equimolar ratio, confirmed the tridentate nature of Zn coordination by the 4′-O-picolyl unit and the two pyridylthiazole nitrogens in cisoid geometry (Fig. 3b). The bite angles for N1–Zn–N2 and N2–Zn–N3 are 77.8° and 100.2°, respectively. Notably, there are no significant differences in the distances between Zn–N bonds (Zn–N1 bond length = 2.121 Å; Zn–N2 = 2.125 Å; Zn–N3 = 2.110 Å), despite the fact that the pyridine ring in the picolyloxy arm is rotated out of plane by 26.4° relative to the thiazole. The seven-membered chelate ring is uncommon,25 but may be a useful binding motif in the design of other coordination complexes.
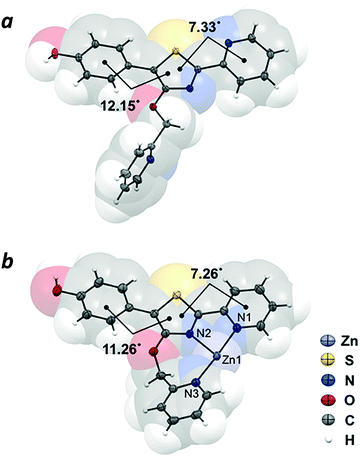 |
| | Fig. 3 X-ray crystal structures of (a) 4′-O-Pic-HPPT (4) and (b) 4–Zn(II). Nitrate counterions are omitted for clarity. Thermal ellipsoids are shown at 50% probability. | |
Photophysical properties
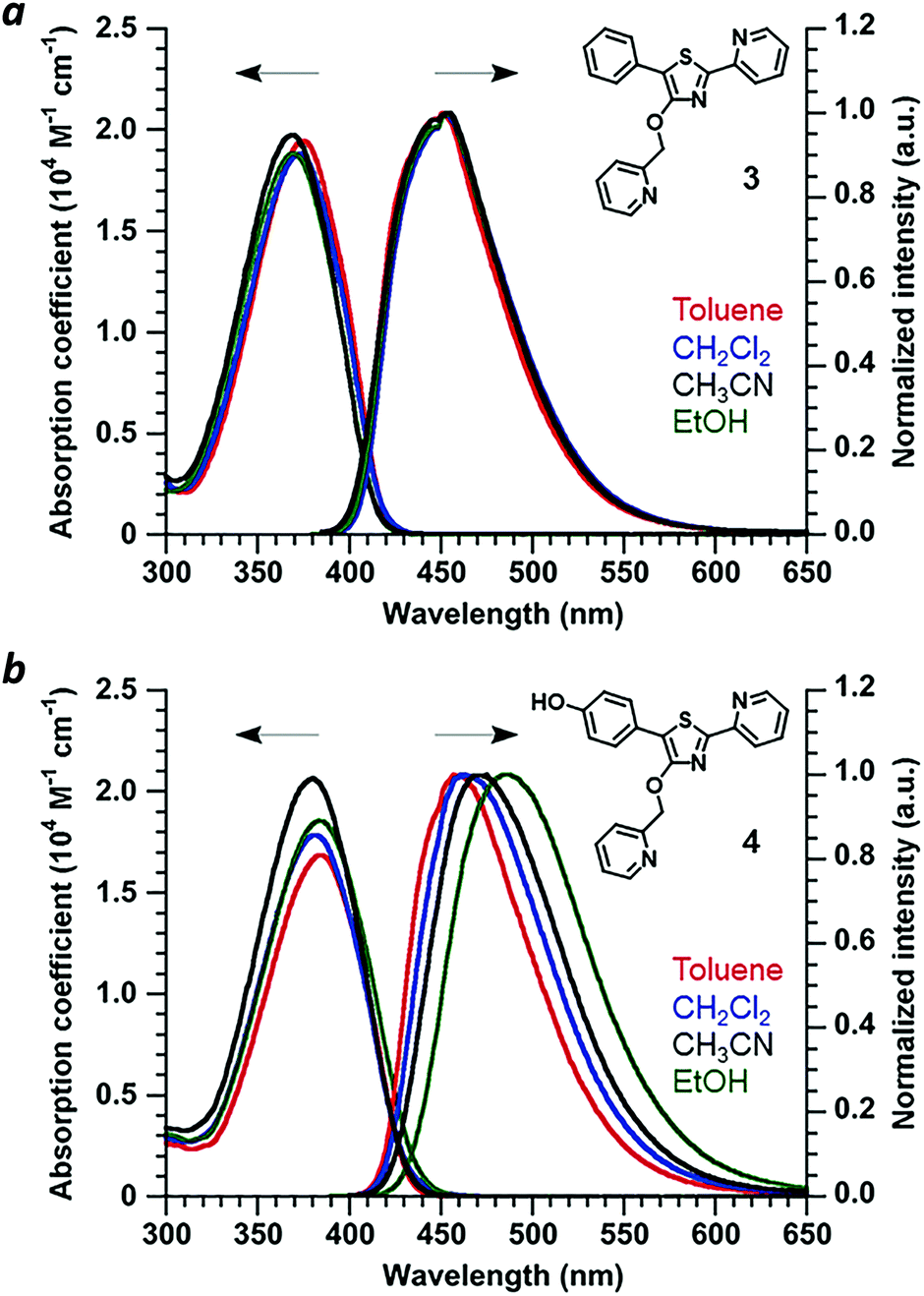
Absorbance and photoluminescence (PL) spectra of 4′-O-Pic-PPT 3 and 4′-O-Pic-HPPT 4 were acquired in solvents of varying polarity to determine the extent of electric polarization within HPPT (Fig. 4). HPPT 4 has a strong absorption band near 380 nm (εM = 2 × 104 M−1 cm−1, CH3CN) corresponding with its π–π* transition, and is redshifted by 11 nm relative to that of PPT 3 (λmax = 369 nm). 4 also exhibits solvatochromism in its PL spectra, with a shift in peak emission (λem) from 461 nm in toluene (εd 2.38) to 487 nm in ethanol (εd 24.5), indicating increased stabilization of its excited-state dipole by a polar environment (see ESI†).26 Similar phenomena were observed with 4′-O-ethyl derivative 2, validating HPPT as a donor–acceptor ICT system (Table 1 and ESI†). The ΦPL for all PPT derivatives in acetonitrile are well over 90%, and correlate well with previous studies.20,23 Optical band gaps (Eg) were estimated from the absorption edge to be 3.0 eV for 1 and 3 and 2.9 eV for 2 and 4, indicating minimal perturbation of the π-conjugated system by the 4′-O-alkyl unit.
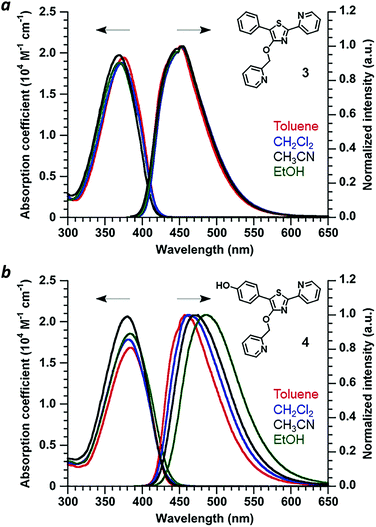 |
| | Fig. 4 (a) Optical absorption and normalized PL spectra of 4′-O-Pic-PPT 3 and (b) 4′-O-Pic-HPPT 4. Spectra recorded at 30 μM and 20 °C. | |
Table 1 Photophysical properties of PPT derivatives (in CH3CN)
| Compound |
Abs (λmax) (nm) |
ε
M × 104 (M−1 cm−1) |
λ
em
(nm) |
ν
St (cm−1/nm) |
Φ
PL
(%) |
|
λ
ex = 370 nm for 1 and 3, 380 nm for 2 and 4, 383 nm for 3–Zn(II), and 402 nm for 4–Zn(II).
Φ
PL determined using an integrating sphere.
|
|
1 (4′-O-Et-PPT) |
371 |
1.90 |
454 |
4928/83 |
96 |
|
2 (4′-O-Et-HPPT) |
383 |
1.77 |
476 |
5101/93 |
96 |
|
3 (4′-O-Pic-PPT) |
369 |
1.97 |
454 |
5074/85 |
94 |
|
4 (4′-O-Pic-HPPT) |
380 |
2.07 |
472 |
5129/92 |
92 |
|
3–Zn(II)
|
383 |
1.82 |
476 |
5101/93 |
87 |
|
4–Zn(II)
|
402 |
1.85 |
539 |
6323/137 |
92 |
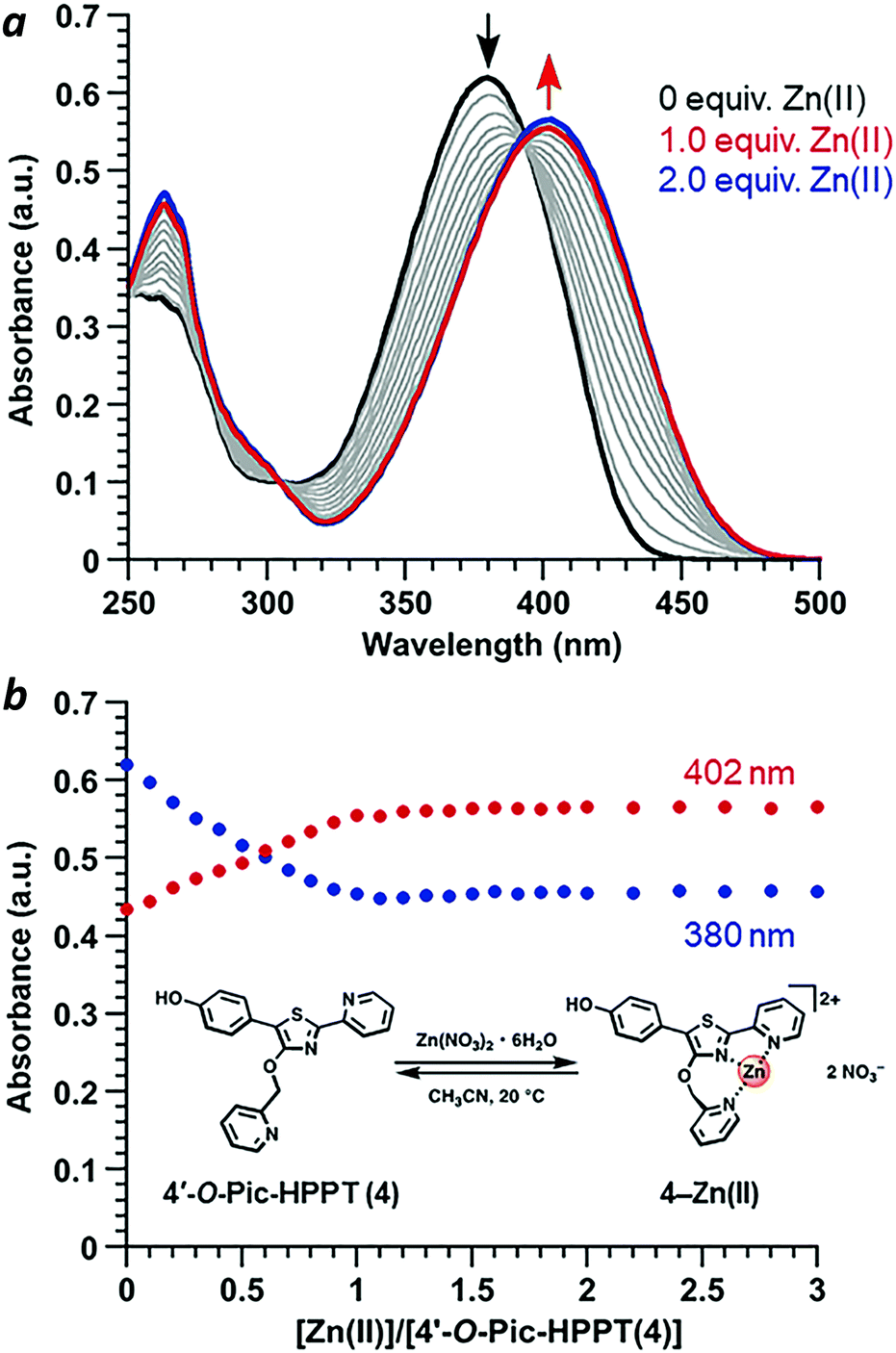
Coordination of Zn(II) to 4′-O-Pic-HPPT 4 led to striking changes in emission colour with retention of high ΦPL (Table 1). The λem of 4 shifted by 67 nm toward the red upon Zn(II) binding, from 472 to 539 nm. A 22 nm redshift in absorbance (Δλmax) from 380 to 402 nm was also observed, with well-defined isosbestic points at 305 and 393 nm (Fig. 5a). Stoichiometric titration of Zn(NO3)2 into a CH3CN solution of 4 produced linear changes in both absorption and emission peak intensities, with no further change after one molar equivalent (Fig. 5b). The association constant (Ka) for Zn2+ binding by 4 was determined to be 9.6 × 107 M−1 (see ESI†), higher than that reported for some similar compounds in CH3CN.18 In comparison, coordination of Zn(II) to 4′-O-Pic-PPT 3 induced much smaller redshifts in emission (Δλem = 22 nm) and absorbance (Δλmax = 14 nm), as well as a slight loss in ΦPL and a lower Ka value (see ESI†). No redshifts were observed for 4′-O-ethyl derivatives 1 or 2, meaning that the pyridyl nitrogen in the 4′-O-picolyl group was necessary for Zn(II) binding, and could adopt an appropriate geometry for stable complex formation. Lastly, exchanging counterions from nitrate to perchlorate or triflate had little effect on the photophysical properties of 4 (see ESI†).
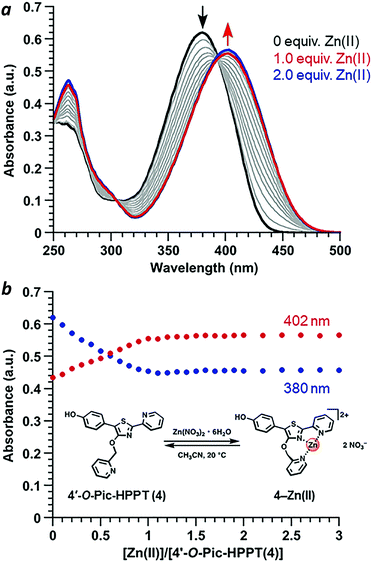 |
| | Fig. 5 (a) Spectral changes in the absorbance of 4 (30 μM) upon titration of Zn(NO3)2·6H2O (3 mM in CH3CN). (b) Stoichiometry of 4–Zn(II) complex formation in CH3CN using the molar ratio method, based on absorbance maxima. | |
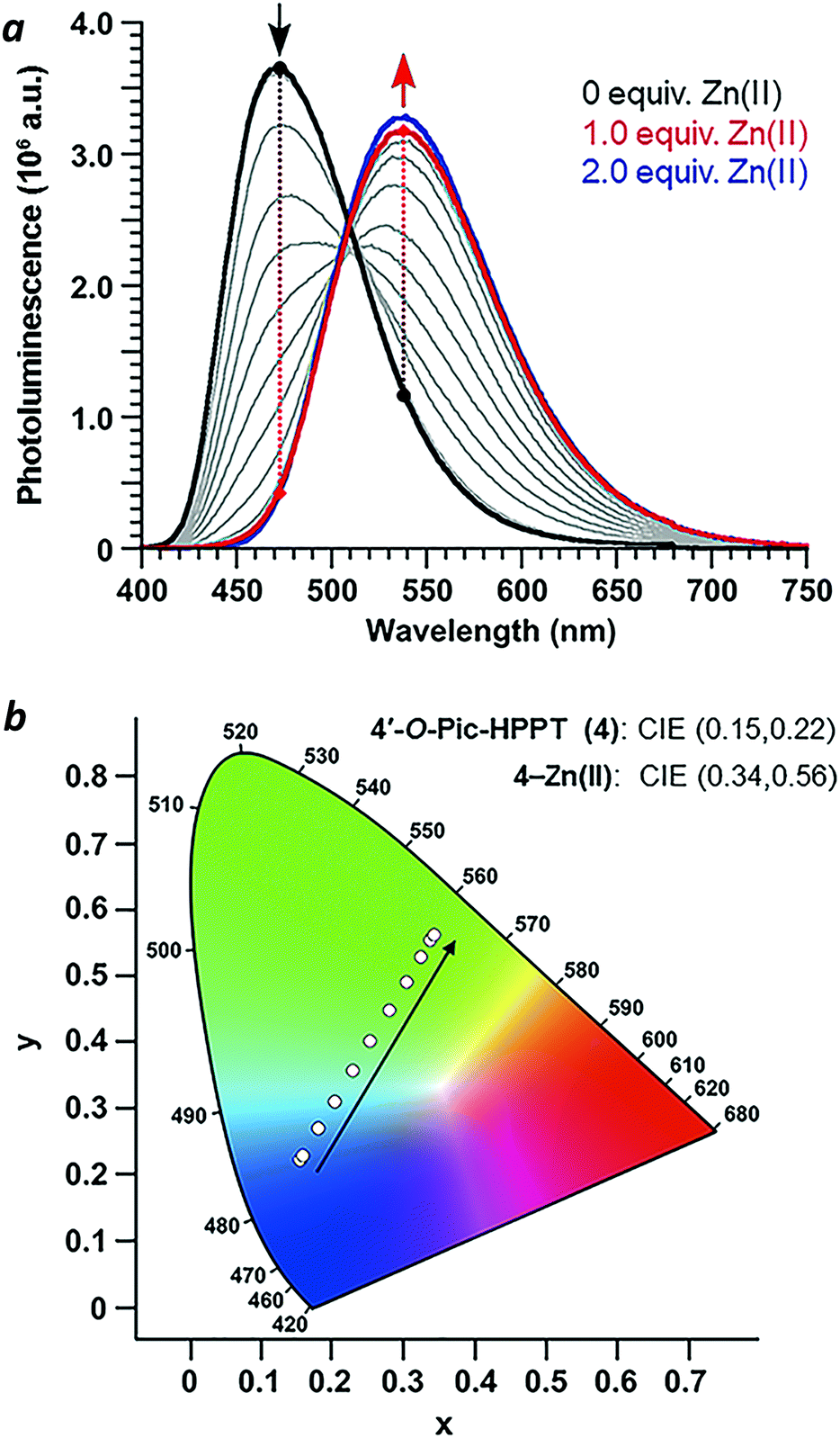
With regard to visual perception, HPPT derivatives produce photons that are easily detected by the human eye. The photoluminescence of 4 (λex/λem = 393/472 nm) corresponds with chromaticity coordinates (0.15,0.22) in Commission Internationale de L'Eclairage (CIE) 1931 colour space (Fig. 6).27 Formation of the 4–Zn(II) complex results in a peak emission at 539 nm, which shifts the CIE coordinates to (0.34,0.56) within the green region. To translate these CIE coordinates into an optical readout of Zn(II) binding, the ratio of emission intensities at 539 and 472 nm (F539/F472) increases from 0.12 in the absence of Zn(II) to 2.77 in the presence of equimolar Zn(II). This ratiometric increase is greater than 20-fold, and implies that 4 can function as a single-excitation dual-emission probe for Zn(II) at wavelengths appropriate for unaided visual analysis. For comparison, the CIE coordinates for PL emission from PPT derivative 3 (λex/λem = 377/454 nm) shift in response to Zn(II) from CIE (0.15,0.10) to CIE (0.16,0.25) (λem for 3–Zn(II) = 476 nm). The ratio of emission intensities (F476/F454) changes from 0.72 in the absence of Zn(II) to 1.38 in the presence of equimolar Zn(II), corresponding to less than a 2-fold increase. Moreover, 4–Zn(II) has a Stokes shift of 137 nm (νSt = 6323 cm−1), whereas that of 3–Zn(II) is 93 nm or 5101 cm−1 (Table 1 and ESI†). The larger Stokes shift reduces the overlap between excitation and emission bands, and minimizes self-quenching.
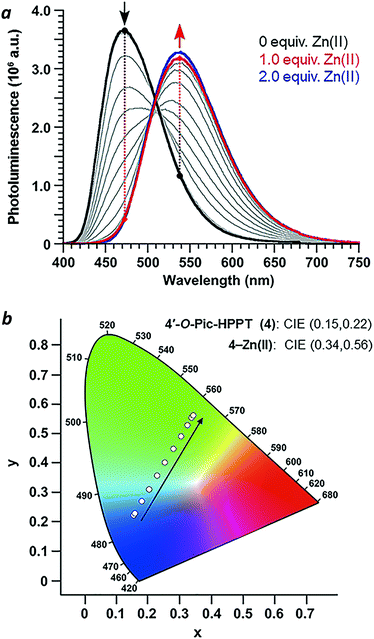 |
| | Fig. 6 (a) Spectral changes in the photoluminescence of 4 (30 μM in CH3CN) upon titration of Zn(NO3)2·6H2O (3 mM in CH3CN) and excitation at the isosbestic point (λex = 393 nm). (b) CIE chromaticity diagram of 4 showing the progression in emitted light with Zn(NO3)2 addition. | |
Conclusions
5′-(p-Hydroxyphenyl)pyridylthiazole is a new ICT-type fluoro-phore that can be configured for a strongly emissive response to Zn(II) ions. The HPPT platform is easy to synthesize and amenable to derivatization, particularly at the 4′ position. The 4′-O-picolyl arm stabilizes the coordination of Zn(II), as shown by x-ray crystallography and by the absence of a Zn-induced shift in emission for 4′-O-ethyl HPPT 2 in solution. The p-hydroxyphenyl unit in Pic-HPPT 4 is responsible for a 67 nm shift in emission upon Zn(II) binding, compared with a 22 nm shift in emission for PPT 3. The 4–Zn(II) complex has a relatively large Stokes shift of 137 nm or 6323 cm−1, which bodes well for optical sensing and imaging applications. Investigations on the metal-ion binding properties of HPPT derivatives in aqueous solutions are in progress, and will be reported in due course.
Experimental
Chemical reagents and supplies obtained from commercial vendors were used as supplied unless otherwise noted. N-Bromo-succinimide (NBS) was recrystallized from water as a white crystalline solid. Microwave reactions were performed using a CEM Discovery microwave reactor. 1H and 13C NMR spectra were collected at room temperature on a Varian INOVA 300, Bruker AV400 HD, or AV800 HD spectrometer. NMR samples were prepared in chloroform-d, acetone-d6, or dimethyl sulfoxide-d6, with chemical shifts (δ) reported in ppm relative to residual solvent resonances as internal standards. Electrospray ionization mass spectra were obtained using an Advion Expression compact spectrometer. Full details on the synthesis and characterization of 1–4 and their intermediates are described in the ESI.†
Absorption spectra were acquired on a Varian Cary50 Bio UV-Vis spectrophotometer using a 1-cm quartz cuvette. Photoluminescence spectra and absolute quantum yields (ΦPL) were measured using an Edinburgh Instruments FLS 980 spectrometer with an integrating sphere accessory. Chromaticity (CIE) coordinates were analysed with F980 software. Quantum chemical calculations were performed using the hybrid density functional theory (DFT) functional (Gaussian 16).21 Geometries of PPT derivatives were optimized at the B3LYP level using the 6-31+G(d,p) basis set for the ground state. Geometries of PPT–Zn(II) complexes were optimized at the B3LYP/Gen level using the 6-31+G(d,p) basis set for H, C, N atoms and LANL2DZ basis set for the Zn(II) atom in the gas phase. Crystallographic data reported in this paper are tabulated in the ESI† and archived at the Cambridge Crystallographic Data Centre under reference numbers CCDC 1960137 and 1960138.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the ACS Petroleum Research Fund (58453-ND4). Y. W. gratefully acknowledges a fellowship provided by the Japan Society for the Promotion of Science (JSPS). Funding for the single crystal X-ray diffractometer was provided by the National Science Foundation (CHE-1625543). We thank Ericka (Kistler) Brandau, Benjamin Washer, Patricia Bishop, and Hartmut Hedderich for technical support.
References
-
(a) A. Loudet and K. Burgess, BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed;
(b) J. Han and K. Burgess, Fluorescent Indicators for Intracellular pH, Chem. Rev., 2010, 110, 2709 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, New Strategies for Fluorescent Probe Design in Medical Diagnostic Imaging, Chem. Rev., 2010, 110, 2620 CrossRef CAS PubMed.
-
(a) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Fluorescent Chemosensors Based on Spiroring-Opening of Xanthenes and Related Derivatives, Chem. Rev., 2012, 112, 1910 CrossRef CAS PubMed;
(b) D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Coumarin-Based Small-Molecule Fluorescent Chemosensors, Chem. Rev., 2019, 119, 10403 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Reaction-based small-molecule fluorescent probes for chemoselective bioimaging, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed.
- X. Li, X. Gao, W. Shi and H. Ma, Design Strategies for Water-Soluble Small Molecular Chromogenic and Fluorogenic Probes, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed.
-
(a) R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire and K. R. A. S. Sandanayake, Molecular fluorescent signalling with ‘fluor–spacer–receptor’ systems: approaches to sensing and switching devices via supramolecular photophysics, Chem. Soc. Rev., 1992, 21, 187 RSC;
(b) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Signaling Recognition Events with Fluorescent Sensors and Switches, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- B. Valeur and I. Leray, Design principles of fluorescent molecular sensors for cation recognition, Coord. Chem. Rev., 2000, 205, 3 CrossRef CAS.
- Z. Xu, J. Yoon and D. R. Spring, Fluorescent chemosensors for Zn2+, Chem. Soc. Rev., 2010, 39, 1996 RSC.
-
(a) E. M. Nolan and S. J. Lippard, Small-Molecule Fluorescent Sensors for Investigating Zinc Metalloneurochemistry, Acc. Chem. Res., 2009, 42, 193 CrossRef CAS PubMed;
(b) E. Tomat and S. J. Lippard, Imaging mobile zinc in biology, Curr. Opin. Chem. Biol., 2010, 14, 225 CrossRef CAS PubMed.
- K. P. Carter, A. M. Young and A. E. Palmer, Fluorescent Sensors for Measuring Metal Ions in Living Systems, Chem. Rev., 2014, 114, 4564 CrossRef CAS PubMed.
- N. C. Lim, J. V. Schuster, M. C. Porto, M. A. Tanudra, L. Yao, H. C. Freake and C. Brückner, Coumarin-Based Chemosensors for Zinc(II): Toward the Determination of the Design Algorithm for CHEF-Type and Ratiometric Probes, Inorg. Chem., 2005, 44, 2018 CrossRef CAS PubMed.
- K. Komatsu, Y. Urano, H. Kojima and T. Nagano, Development of an Iminocoumarin-Based Zinc Sensor Suitable for Ratiometric Fluorescence Imaging of Neuronal Zinc, J. Am. Chem. Soc., 2007, 129, 13447 CrossRef CAS PubMed.
-
(a) L. Xue, H.-H. Wang, X.-J. Wang and H. Jiang, Modulating Affinities of Di-2-picolylamine (DPA)-Substituted Quinoline Sensors for Zinc Ions by Varying Pendant Ligands, Inorg. Chem., 2008, 47, 4310 CrossRef CAS PubMed;
(b) L. Xue, C. Liu and H. Jiang, A ratiometric fluorescent sensor with a large Stokes shift for imaging zinc ions in living cells, Chem. Commun., 2009, 1061 RSC.
- D. Bourassa, C. M. Elitt, A. M. McCallum, S. Sumalekshmy, R. L. McRae, M. T. Morgan, N. Siegel, J. W. Perry, P. A. Rosenberg and C. J. Fahrni, Chromis-1, a Ratiometric
Fluorescent Probe Optimized for Two-Photon Microscopy Reveals Dynamic Changes in Labile Zn(II) in Differentiating Oligodendrocytes, ACS Sens., 2018, 3, 458 CrossRef CAS PubMed.
- Y. Wu, X. Peng, B. Guo, J. Fan, Z. Zhang, J. Wang, A. Cui and Y. Gao, Boron dipyrromethene fluorophore based fluorescence sensor for the selective imaging of Zn(II) in living cells, Org. Biomol. Chem., 2005, 3, 1387 RSC.
- S. C. Burdette, G. K. Walkup, B. Spingler, R. Y. Tsien and S. J. Lippard, Fluorescent Sensors for Zn2+ Based on a Fluorescein Platform: Synthesis, Properties and Intracellular Distribution, J. Am. Chem. Soc., 2001, 123, 7831 CrossRef CAS PubMed.
-
(a) J. C. Jeffery, C. R. Rice, L. P. Harding, C. J. Baylies and T. Riis-Johannessen, Ligand Reprogramming in Dinuclear Helicate Complexes: A Consequence of Allosteric or Electrostatic Effects?, Chem. – Eur. J., 2007, 13, 5256 CrossRef CAS PubMed;
(b) S. Bullock, L. J. Gillie, L. P. Harding, C. R. Rice, T. Riis-Jojammessem and M. Whitehead, Isomeric pyridyl-thiazole donor units for metal ion recognition in bi- and tri-metallic helicates, Chem. Commun., 2009, 4856 RSC.
- M. H. Zheng, X. Hu, M. Y. Yang and J. Y. Jin, Ratiometically Fluorescent Sensing of Zn(II) Based on Dual-Emission of 2-Pyridylthiazole Derivatives, J. Fluoresc., 2015, 25, 1831 CrossRef CAS PubMed.
- M.-H. Zheng, J.-Y. Jin, W. Sun and C.-H. Yan, A new series of fluorescent 5-methoxy-2-pyridylthiazoles with a pH-sensitive dual-emission, New J. Chem., 2006, 30, 1192 RSC.
- U.-W. Grummt, D. Weiss, E. Birckner and R. Beckert, Pyridylthiazoles: Highly Luminescent Heterocyclic Compounds, J. Phys. Chem. A, 2007, 111, 1104 CrossRef CAS PubMed.
-
M. J. Frisch, et al., Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Hantzsch, Untersuchungen über Azole, Justus Liebigs Ann. Chem., 1889, 250, 257 CrossRef.
-
(a) E. Täuscher, D. Weiß, R. Beckert and H. Görls, Synthesis and Characterization of New 4-Hydroxy-1,3-thiazoles, Synthesis, 2010, 1603 Search PubMed;
(b) S. Wolfram, H. Würfel, S. H. Habenicht, C. Lembke, P. Richter, E. Birckner, R. Beckert and G. Pohnert, A small azide-modified thiazole-based reporter molecule for fluorescence and mass spectrometric detection, Beilstein J. Org. Chem., 2014, 10, 2470 CrossRef PubMed.
- M. Kaufmann, M. L. Hupfer, T. Sachse, F. Herrmann-Westendorf, D. Weiß, B. Dietzek, R. Beckert and M. Presselt, Introducing double polar heads to highly fluorescent thiazoles: Influence on supramolecular structures and photonic properties, J. Colloid Interface Sci., 2018, 526, 410 CrossRef CAS PubMed.
- D. Aguila, E. Escribano, S. Speed, D. Talancon, L. Yerman and S. Alvarez, Calibrating the coordination chemistry tool chest: metrics of bi- and tridentate ligands, Dalton Trans., 2009, 6610 RSC.
-
P. Suppan and N. Ghoneim, Solvatochromism, Royal Society of Chemistry, Cambridge, 1997 Search PubMed.
- T. Smith and J. Guild, The C.I.E. colorimetric standards and their use, Trans. Opt. Soc., 1931, 33, 73–134 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional DFT calculations and photophysical studies, details on synthesis and X-ray crystallography, and NMR spectra. CCDC 1960137 and 1960138. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00648f |
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Wanna
Sungnoi
a,
Wanna
Sungnoi