
Ring-opening metathesis polymerization of a strained stilbene-based macrocyclic monomer†
Brock E.
Lynde
 ab,
Ruth L.
Maust
c,
Penghao
Li
c,
Daniel C.
Lee
d,
Ramesh
Jasti
*c and
Andrew J.
Boydston
*ab
ab,
Ruth L.
Maust
c,
Penghao
Li
c,
Daniel C.
Lee
d,
Ramesh
Jasti
*c and
Andrew J.
Boydston
*ab
aDepartment of Chemistry, University of Washington, Seattle, WA 98195, USA
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: aboydston@wisc.edu
cDepartment of Chemistry and Biochemistry, Materials Science Institute, and Knight Campus for Accelerating Scientific Impact, University of Oregon, Eugene, OR 97403, USA
dMolecular Engineering and Sciences Institute, University of Washington, Seattle, WA 98195, USA
First published on 21st November 2019
Abstract
We report the synthesis of a new class of strained macrocycle that performs well in ring-opening metathesis polymerization (ROMP). The polymerization displays chain growth characteristics with evidence of secondary metathesis in the form of chain transfer. The unique structure enables access to stilbene-based polymers that are traditionally prepared via uncontrolled polymerizations.
Introduction
Ring-opening metathesis polymerization (ROMP) has become an indispensable synthetic tool in modern polymer chemistry and materials science.1–6 The monomer landscape for ROMP is dominated largely by four motifs: norbornenes, cyclobutenes, cyclopropenes, and cyclooctenes (Fig. 1).2,7–15 These motifs share relatively high ring strain that provides a driving force for polymerization. From these frameworks, functional groups are typically introduced via side chains, whereas increasing the diversity of the backbone composition within ROMP is generally achieved by the polymerization of macrocycles (ring size >14 atoms). Most macrocyclic monomers however, have little or no ring strain.16 Therefore, a trade off exists between selection of monomers with high ring strain versus macrocyclic systems of greater diversity albeit without an enthalpic driving force.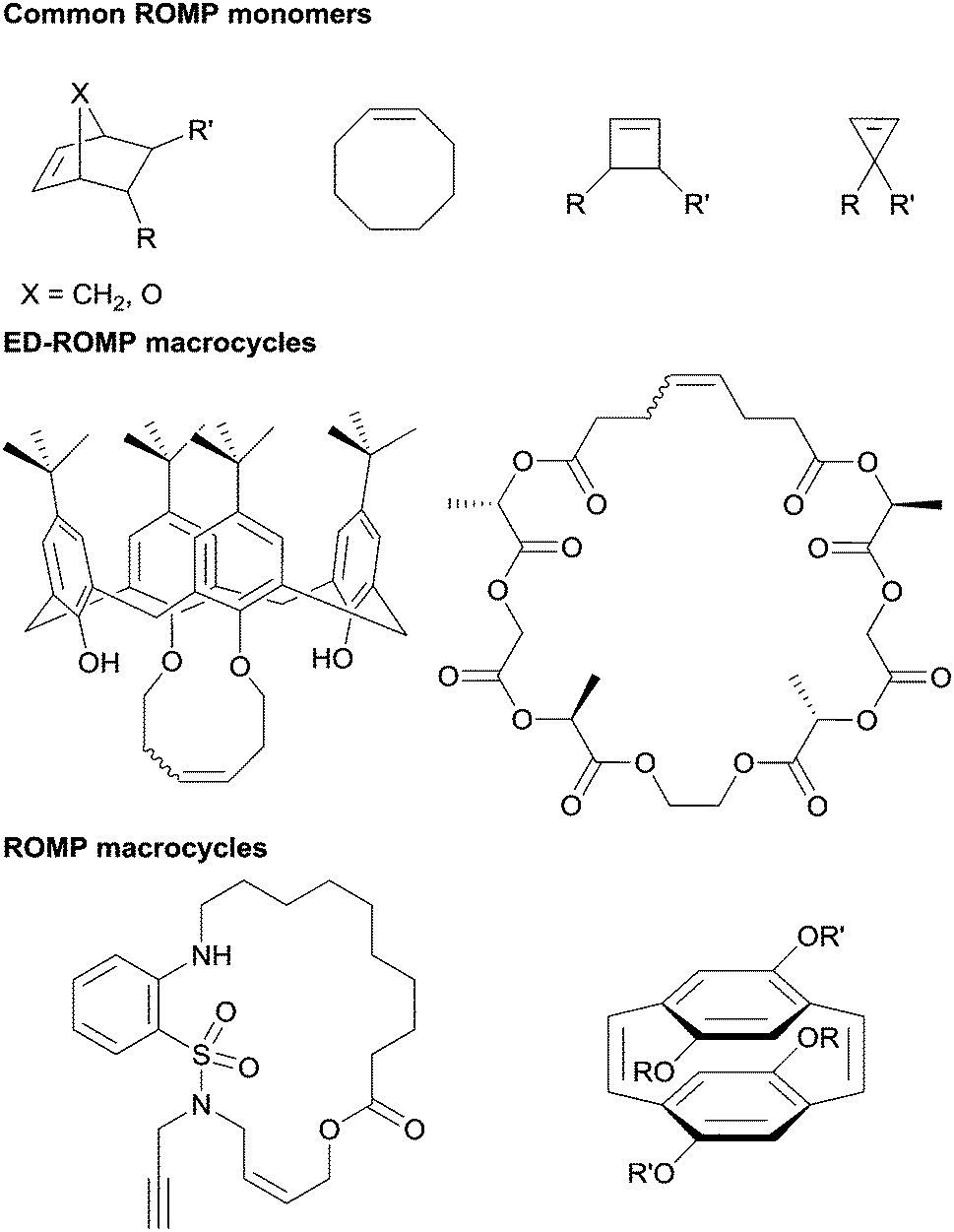 | ||
| Fig. 1 Examples of both small molecule and macrocyclic monomers for ROMP and ED-ROMP. | ||
The lack of an enthalpic driving force for ROMP of macrocycles suggests that an entropic driving force must be present for polymerization to occur, and as such, these polymerizations are categorized as entropy-driven ring-opening metathesis polymerizations (ED-ROMPs).16–18 ED-ROMPs exist in a ring-chain equilibrium between macrocyclic oligomers and linear polymers, and the thermodynamic drive is provided by the increase in conformational entropy as the macrocyclic oligomers become linear polymer chains.16 Macrocyclic platforms for ED-ROMP have been used to synthesize polymers with many unique features including liquid crystalline polymers, poly(catenates), poly(calixarenes), as well as sequence-controlled polymers (Fig. 1).19–22 Disadvantages of using ED-ROMP include high molecular weight dispersity (Đ) for the resulting polymers, often the Đ for these polymerizations fall between 1.5–2.0, with notable exceptions.16,18,23,24
Approaches to address the challenges with ED-ROMP include designing macrocycles with high ring strain and engineering effectively irreversible reactions into the polymerization mechanism, the former being the more common of these approaches.25 For instance, Miao et al. utilized [2.2]paracyclophan-1-ene, a highly strained macrocycle, to synthesize a homopolymer as well as block and random co-polymers with norbornene initiated by a Schrock-type catalyst.26 Since this initial demonstration, several other cyclophanenes and cyclophanedienes have been used in ROMP to synthesize homo- and co-polymers.27–29 In addition, a series of donor–acceptor block co-polymers have been synthesized via ROMP of macrocycles based on arylenevinylenes.27,30,31 Inspired by this work, we set out to investigate methods to synthesize a new class of strained macrocycles capable of undergoing ROMP.
Results and discussion
We conceived of cis-stilbene-based macrocycle 1, which we predicted would possess a high degree of ring strain and would enable predefined control of the structure of the resulting polymer backbone (Fig. 2).32 By synthesizing a polymer through chain growth polymerization instead of intensive step growth condensation polymerization, we envisioned that we could readily obtain polymers with low Đ and controlled molecular weight. Additionally, the polymer resulting from macrocycle 1 would be similar in structure to many high-performance polymers, such as poly(phenylene)s, poly(phenylenevinylene)s, and poly(aryletherketone)s, that with a few exceptions have traditionally been synthesized through uncontrolled polymerizations.33–40 The potential to synthesize high-performance polymers through readily accessible chain growth polymerizations instead of step growth polymerizations could be an exciting advancement toward complex polymer structures that were previously unachievable.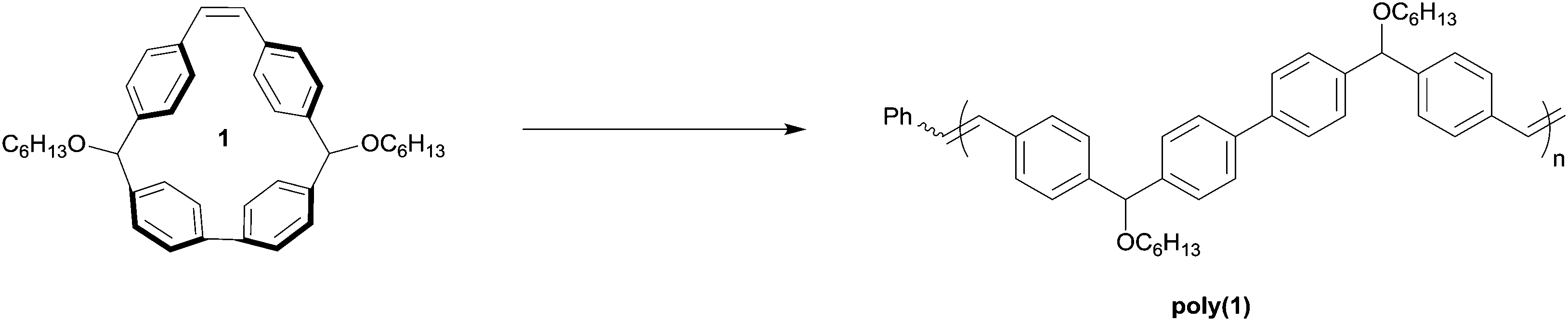 | ||
| Fig. 2 Proposed polymerization of macrocycle (1). | ||
Despite the broad utility of strained macrocycles for ROMP, there are few efficient synthetic routes to obtain macrocyclic monomers with enough ring strain to drive ring-opening polymerization. We therefore employed oxidative bisboronate homocoupling—a simple, scalable, and efficient strain-building reaction—for the preparation of macrocycle 1 (Fig. 3).41 First, we constructed curved diol intermediate 2 by double lithiation of 4,4′-dibromostilbene and subsequent nucleophilic addition to 4-bromobenzaldehyde. Deprotonation of the free alcohols with sodium hydride and treatment with 1-bromohexane yielded 3. Lithium-halogen exchange followed by treatment with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane yielded bisboronate 4. Finally, 4 was subjected to mild Pd-catalysed oxidative homocoupling conditions to yield final macrocyclic monomer 1 on a multigram scale. The key cyclization reaction is 50% yielding with the remaining mass balance primarily attributed to oligomeric byproducts. In principle, other sized macrocycles could form as well, but we did not observe these products to any appreciable extent.
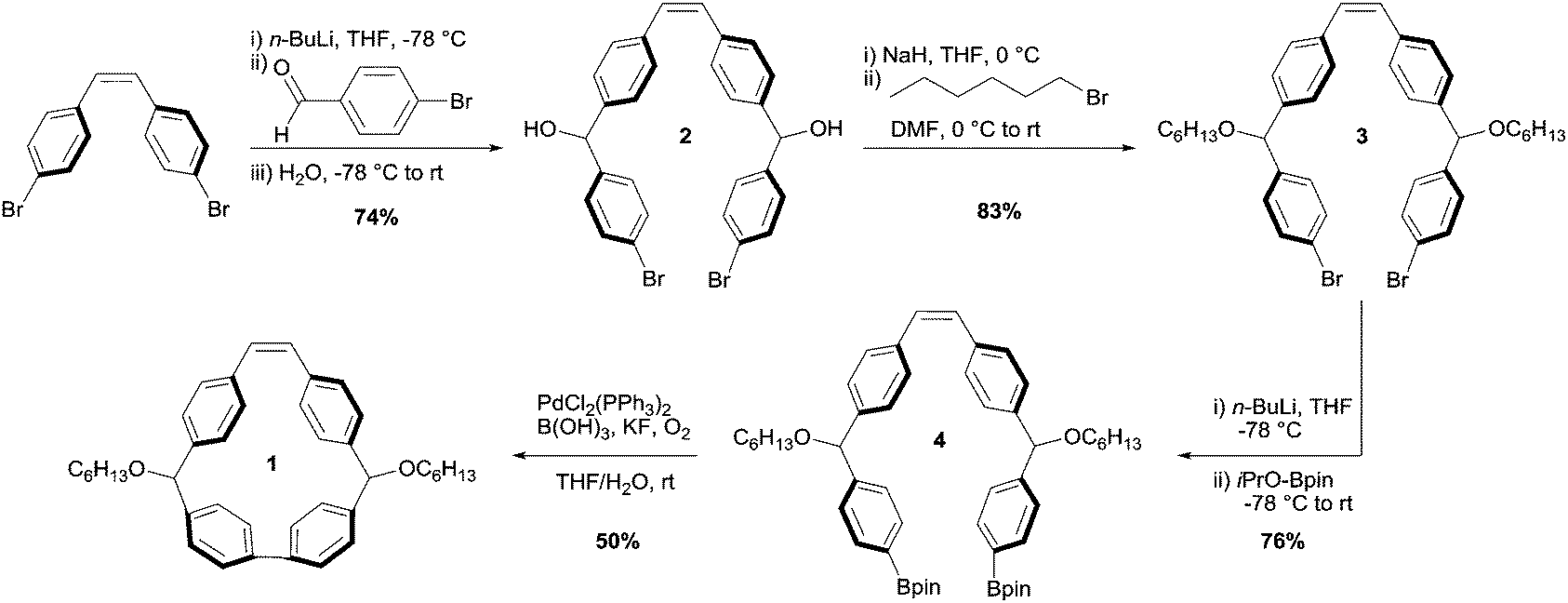 | ||
| Fig. 3 Synthetic scheme for 1. | ||
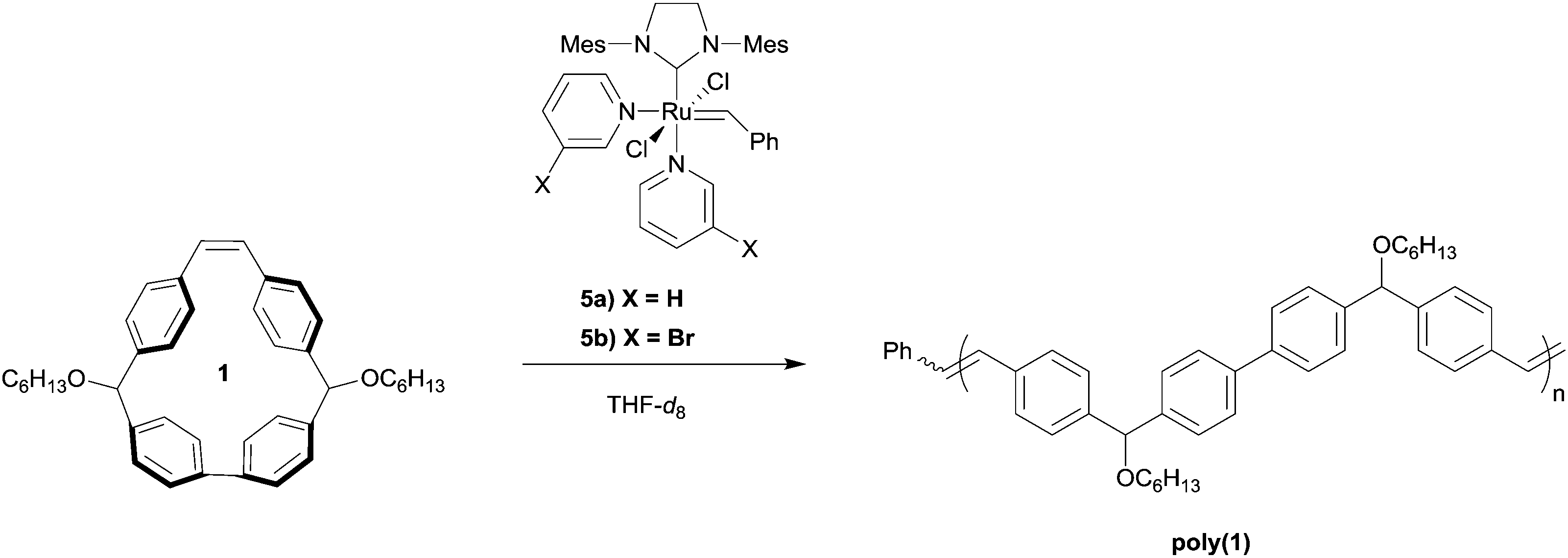
With monomer 1 in hand, we investigated the polymerization of 1 using the third generation Grubbs catalysts in tetrahydrofuran-d8 ([1]0 = 1 M) with an initial monomer to initiator ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entries 1 and 2). With each initiator, conversion reached >99% within 12 h at 60 °C, as determined by 1H NMR spectroscopy. From these experiments, we found that the molecular weight distribution of poly(1) was monomodal with a Mw = 107 kDa and Đ of 1.7, based upon SEC analysis using multi-angle laser light scattering and refractive index detection. The structure of poly(1) was confirmed by 1H NMR spectroscopy and matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF/MS). In the case of poly(1) only a single vinylic signal at δ = 7.24 ppm was observed, while no other vinylic signals were present above the detection limit for 1H NMR spectroscopy. This observation is consistent with the backbone of poly(1) being primarily trans-stilbene isomers (Fig. S11, ESI†).42–44 MALDI-TOF/MS then was used to better understand the structural speciation within samples of poly(1). The repeat unit for poly(1) has an experimental mass of 558.9 amu, which is consistent with the predicted molecular weight of 1 (Fig. 4). Notably, we did not see evidence of cyclic polymer structures from any of the analyses.
:1 (Table 1, entries 1 and 2). With each initiator, conversion reached >99% within 12 h at 60 °C, as determined by 1H NMR spectroscopy. From these experiments, we found that the molecular weight distribution of poly(1) was monomodal with a Mw = 107 kDa and Đ of 1.7, based upon SEC analysis using multi-angle laser light scattering and refractive index detection. The structure of poly(1) was confirmed by 1H NMR spectroscopy and matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF/MS). In the case of poly(1) only a single vinylic signal at δ = 7.24 ppm was observed, while no other vinylic signals were present above the detection limit for 1H NMR spectroscopy. This observation is consistent with the backbone of poly(1) being primarily trans-stilbene isomers (Fig. S11, ESI†).42–44 MALDI-TOF/MS then was used to better understand the structural speciation within samples of poly(1). The repeat unit for poly(1) has an experimental mass of 558.9 amu, which is consistent with the predicted molecular weight of 1 (Fig. 4). Notably, we did not see evidence of cyclic polymer structures from any of the analyses.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | [1]:[5] |
Conc. (M) | Temp. (°C) | Conv. (%) | M n, theo (kg mol−1) | M n (kg mol−1) | M w (kg mol−1) | Đ | k (s−1) |
| a Initiator 5b. b Initiator 5a. c Average of 3 experiments. d Error represents the standard deviation. | |||||||||
| 1a | 100:1 |
1 | 60 | >99 | 55.9 | 62.1 | 107.0 | 1.7 | — |
| 2b | 100:1 |
1 | 60 | 99 | 55.9 | 55.4 | 102.2 | 1.7 | — |
| 3a | 75:1 |
0.1 | 40 | >99 | 42.4 | 53.4 | 79.7 | 1.5 | — |
| 4a,c | 35:1 |
1 | 20 | 45 | 8.6 | 8.0 | 8.7 | 1.1 | 9.97 × 10−6 ± 0.02 |
| 5a,c | 35:1 |
1 | 30 | >99 | 20.1 | 18.3 | 27.8 | 1.5 | 5.1 × 10−5 ± 0.4 |
| 6a,c | 35:1 |
1 | 40 | >99 | 18.4 | 23.9 | 36.0 | 1.5 | 2.2 × 10−4 ± 0.3 |
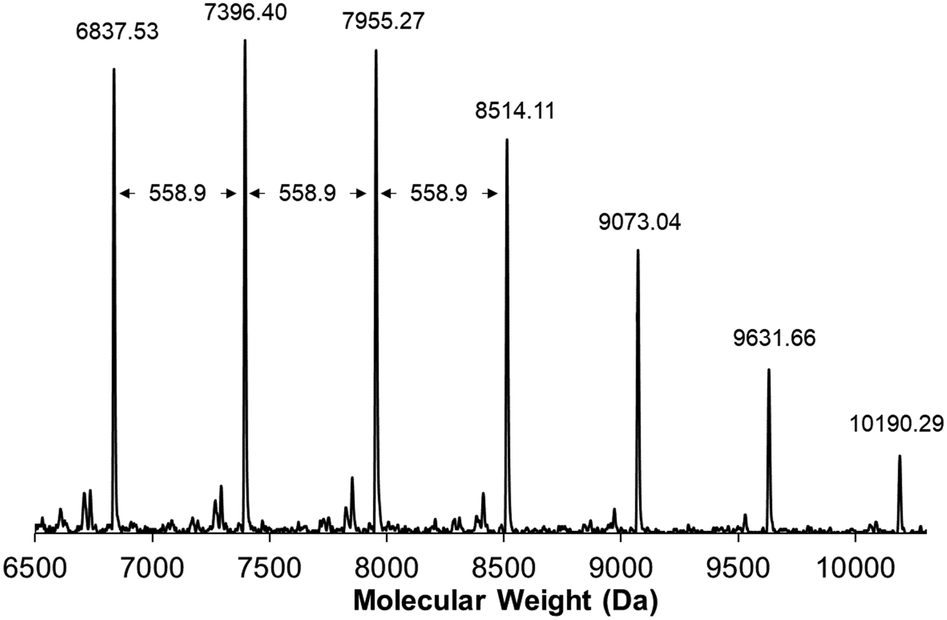 | ||
| Fig. 4 MALDI-TOF/MS spectrum for poly(1). The molecular weight between repeat units was measured to be 558.9 amu, consistent with the molecular weight of the monomer. | ||
We also evaluated the thermal properties of poly(1) using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The decomposition temperature (Td) for poly(1) was found to be 281 °C under N2, determined by the onset of weight loss using TGA (Fig. S13, ESI†). A close comparison to poly(1) would be poly(phenylenevinylene)s, which have Td > 300 °C.43 A glass transition temperature (Tg) for poly(1) was found to be 94 °C determined by DSC, and no other thermal transitions were observed (Fig. S14, ESI†).
Given the ring strain and addition of an enthalpic driving force for the ROMP of 1, we expected the polymerization to demonstrate chain growth characteristics, and a high degree of molecular weight control. To better understand the polymerization mechanism of poly(1), monomer conversion was monitored by 1H NMR spectroscopy using the benzylic ether hydrogen of the monomer and polymer at δ = 5.39 and 5.48 ppm, respectively (Fig. S15, ESI†). The polymerization displayed first order kinetics with respect to consumption of 1 (Fig. S16, ESI†) and a linear correlation between Mn and conversion (Fig. 5). Collectively, these results are consistent with a chain growth polymerization mechanism that does not exhibit slow initiation or early irreversible termination. Rate constants (k) for propagation where measured at 20, 30, and 40 °C (Table 1 entries 4–6), in all experiments the polymerization was stopped after 16 hours. An activation energy was determined to be 28.2 kcal mol−1 (Fig. S17, ESI†). We next turned our attention toward understanding the underlying reason for the high Đ.
 | ||
| Fig. 5 M n vs. conversion plot for the polymerization of 1 follows a linear progression consistent with a chain growth mechanism, Đ for each time point in parenthesis. | ||
Chain transfer has been observed during ROMP, typically facilitating an equilibration of chain lengths via intermolecular cross metathesis reactions. We investigated the occurrence of chain transfer by combining two different molecular weight polymers (Mn = 71.5 kDa and 15.2 kDa) in the presence of 5b in THF. After 4 hours, the resulting polymer had an intermediate molecular weight (Mn = 24 kDa) that was consistent with the weighted average of the feed polymers (Fig. 6a) and a higher Đ of 2.0. Chain transfer with trans-stilbene was also found to be efficient under our polymerization conditions. Specifically, 1 equivalent of trans-stilbene (relative to repeat unit) was combined with a sample of poly(1) (Mn = 41.5 kDa, Đ = 1.6) and 5b in THF. After 3.5 hours, the molecular weight of poly(1) decreased (Mn = 16.9 kDa) (Fig. 6b). These results are consistent with chain transfer occurring during the polymerization of 1. It should be noted in the latter experiment no change in Đ was observed contrary to what is expected, this result could be due to a change in column resolution between the molecular weights.
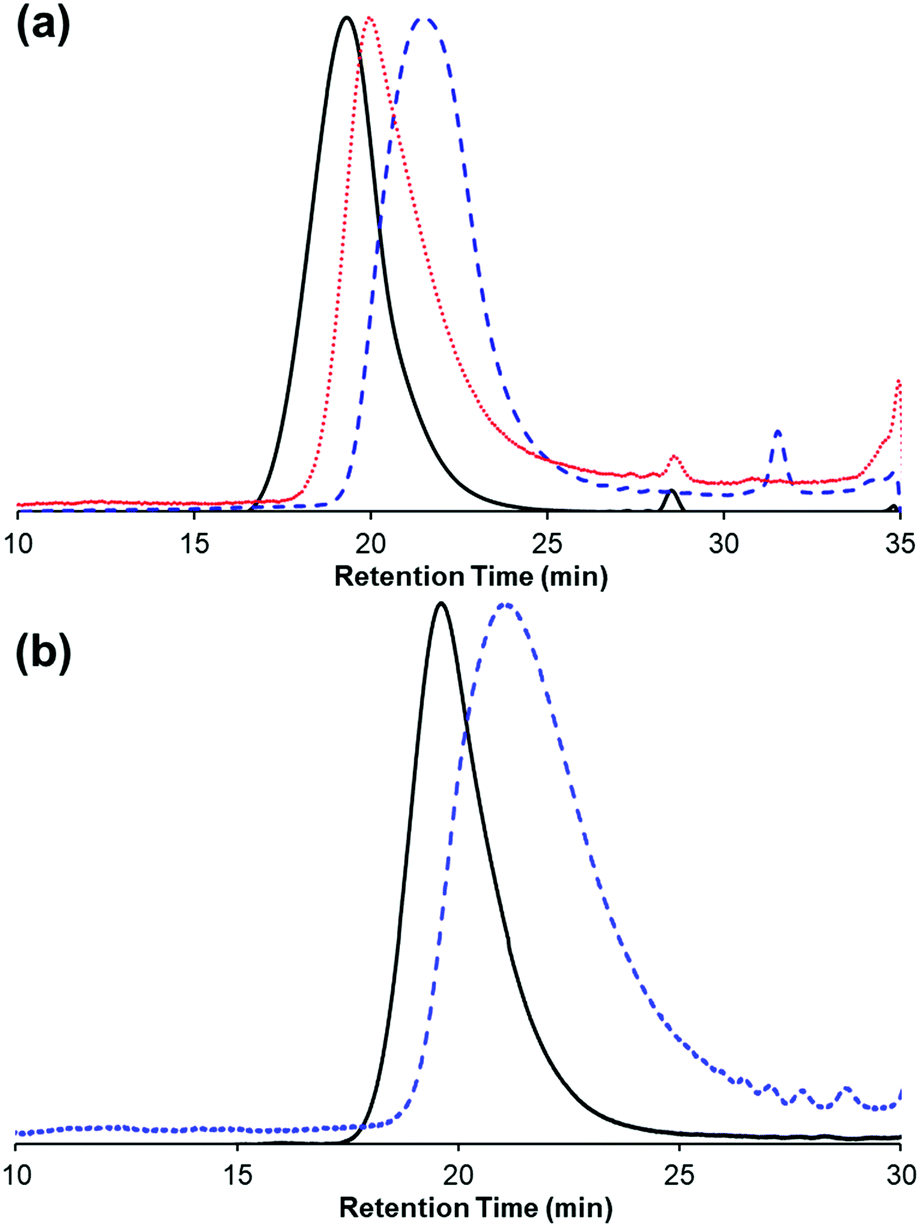 | ||
| Fig. 6 (a) GPC traces for polymer–polymer chain transfer experiment for the two starting polymers (black solid, Mn = 71 kDa; blue dash, Mn = 15.2 kDa) and the final polymer (red dot, Mn = 24 kDa). (b) GPC traces for polymer–stilbene chain transfer experiment for the starting polymer (black solid, Mn: 41.5 kDa) and the final polymer (blue dash, Mn: 16.9 kDa). | ||
Taken together, our results suggested to us that, likely due to the ring strain of 1, the polymerization proceeds through a chain growth mechanism and is not an entropy-driven polymerization. During the course of the polymerization of 1 the Đ of poly(1) increased from 1.0 to 1.5 further corroborating the presence of chain transfer during the polymerization (Fig. 5).
Conclusions
In summary, we have reported the synthesis and subsequent polymerization of a new class of strained macrocycle. The polymerization of 1 demonstrated first order kinetics and a linear correlation between Mn and conversion, consistent with chain growth polymerization. The resulting linear polymer obtained through this method had a Tg of 94 °C and a Td of 281 °C. Further work is being done to use variations of 1 to make more complex polymeric materials that cannot be achieved using traditional small molecule-based poly(olefins). In this way, we hope to be able to control and modify the thermal and physical properties of these modular polymers. Ultimately, ROMP of highly strained macrocyclic monomers provides an exciting avenue to create and develop new polymeric materials from efficient synthetic methods.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. J. B. acknowledges partial financial support from the Yamamoto Family, the Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin – Madison with funding from the Wisconsin Alumni Research Foundation, the National Science Foundation (DMR-1452726), and the Camille and Henry Dreyfus Foundation. R. L. M. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1309047. Additional support for R. L. M., P. L., and R. J. was provided by NSF CHE-1800586. Mass spectrometry support was provided by NSF CHE-1625529. We would also like to acknowledge the UW School of Pharmacy, Mass Spec Center.Notes and references
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- H. Unsal, S. Onbulak, F. Calik, M. Er-Rafik, M. Schmutz, A. Sanyal and J. Rzayev, Macromolecules, 2017, 50, 1342–1352 CrossRef CAS.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y. Xia, A. C. Durrell, D. A. Tirrell and R. H. Grubbs, Macromolecules, 2010, 43, 10326–10335 CrossRef CAS PubMed.
- Y. Hu, X. Li, A. W. Lang, Y. Zhang and S. R. Nutt, Polym. Degrad. Stab., 2016, 124, 35–42 CrossRef CAS.
- Y. Wang, L. Zhang, J. Sun, J. B. Bao, Z. Wang and L. Ni, Ind. Eng. Chem. Res., 2017, 56, 4750–4757 CrossRef CAS.
- H. Martinez, N. Ren, M. E. Matta and M. A. Hillmyer, Polym. Chem., 2014, 5, 3507–3532 RSC.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Science, 2017, 357, 475–479 CrossRef CAS.
- B. R. Elling, J. K. Su and Y. Xia, Chem. Commun., 2016, 52, 9097–9100 RSC.
- W. H. Binder, S. Kurzhals, B. Pulamagatta, U. Decker, G. M. Pawar, D. Wang, C. Kühnel and M. R. Buchmeiser, Macromolecules, 2008, 41, 8405–8412 CrossRef CAS.
- R. Singh, C. Czekelius and R. R. Schrock, Macromolecules, 2006, 39, 1316–1317 CrossRef CAS.
- Z. Wu and R. H. Grubbs, Macromolecules, 1994, 27, 6700–6703 CrossRef CAS.
- N. T. Lin, Y. Z. Ke, K. Satyanarayana, S. L. Huang, Y. K. Lan, H. C. Yang and T. Y. Luh, Macromolecules, 2013, 46, 7173–7179 CrossRef CAS.
- F. Leroux, S. Pascual, V. Montembault and L. Fontaine, Macromolecules, 2015, 48, 3843–3852 CrossRef CAS.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS.
- P. Hodge, Chem. Rev., 2014, 114, 2278–2312 CrossRef CAS PubMed.
- C. Y. Tastard, P. Hodge, A. Ben-Haida and M. Dobinson, React. Funct. Polym., 2006, 66, 93–107 CrossRef CAS.
- Z. Xue and M. F. Mayer, Soft Matter, 2009, 5, 4600–4611 RSC.
- J. H. Swisher, J. A. Nowalk and T. Y. Meyer, Polym. Chem., 2019, 10, 244–252 RSC.
- J. Berrocal, L. M. Pitet, M. M. L. Nieuwenhuizen, L. Mandolini, E. W. Meijer and S. Di Stefano, Macromolecules, 2015, 48, 1358–1363 CrossRef CAS.
- L.-L. Deng, L.-X. Guo, B.-P. Lin, X.-Q. Zhang, Y. Sun and H. Yang, Polym. Chem., 2016, 7, 5265–5272 RSC.
- Y. Yang and T. M. Swager, Macromolecules, 2007, 40, 7437–7440 CrossRef CAS.
- A. L. Short, C. Fang, J. A. Nowalk, R. M. Weiss, P. Liu and T. Y. Meyer, ACS Macro Lett., 2018, 7, 858–862 CrossRef CAS.
- R. M. Weiss, A. L. Short and T. Y. Meyer, ACS Macro Lett., 2015, 4, 1039–1043 CrossRef CAS.
- W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2015, 137, 8038–8041 CrossRef CAS PubMed.
- Y.-J. Miao and G. C. Bazan, Macromolecules, 1994, 27, 1063–1064 CrossRef CAS.
- E. Elacqua and M. Gregor, Angew. Chem., Int. Ed., 2019, 58, 9527–9532 CrossRef CAS.
- C.-Y. Yu, J. W. Kingsley, D. G. Lidzey and M. L. Turner, Macromol. Rapid Commun., 2009, 30, 1889–1892 CrossRef CAS PubMed.
- A. M. Spring, C.-Y. Yu, M. Horie and M. L. Turner, Chem. Commun., 2009, 2676–2678 RSC.
- V. Komanduri, D. J. Tate, R. Marcial-Hernandez, D. R. Kumar and M. L. Turner, Macromolecules, 2019, 52, 7137–7144 CrossRef CAS.
- S.-W. Chang and M. Horie, Chem. Commun., 2015, 51, 9113–9116 RSC.
- Ring strain calculated to be 28.9 kcal mol−1. See ESI† for strain energy analysis.
- A. K. Schönbein, M. Wagner, P. W. M. Blom and J. J. Michels, Macromolecules, 2017, 50, 4952–4961 CrossRef.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, Macromolecules, 1988, 21, 294–304 CrossRef CAS.
- A. Abdulkarim, K. P. Strunk, R. Bäuerle, S. Beck, H. Makowska, T. Marszalek, A. Pucci, C. Melzer, D. Jänsch, J. Freudenberg, U. H. F. Bunz and K. Müllen, Macromolecules, 2019, 52, 4458–4463 CrossRef CAS.
- T. E. E. Attwood, P. C. Dawson, J. L. Freeman, L. R. J. Hoy, J. B. Rose and P. A. Staniland, Polymer, 1981, 22, 1096–1103 CrossRef CAS.
- F. Papadimitrakopoulos, K. Konstadinidis, T. M. Miller, R. Opila, E. A. Chandross and M. E. Galvin, Chem. Mater., 1994, 6, 1563–1568 CrossRef CAS.
- F. Louwet, D. Vanderzande, J. Gelan and J. Mullens, Macromolecules, 1995, 28, 1330–1331 CrossRef CAS.
- T. Junkers, J. Vandenbergh, P. Adriaensens, L. Lutsen and D. Vanderzande, Polym. Chem., 2012, 3, 275–285 RSC.
- E. R. Darzi, B. M. White, L. K. Loventhal, L. N. Zakharov and R. Jasti, J. Am. Chem. Soc., 2017, 139, 3106–3114 CrossRef CAS PubMed.
- Y. Xu, W. L. Xu, M. D. Smith and L. S. Shimizu, RSC Adv., 2014, 4, 1675–1682 RSC.
- D. Maker, C. Maier, K. Brodner, U. Bunz, D. Mäker, C. Maier, K. Brödner and U. Bunz, ACS Macro Lett., 2014, 3, 415–418 CrossRef.
- R. Adhikary, C. A. Barnes, R. L. Trampel, S. J. Wallace, T. W. Kee and J. W. Petrich, J. Phys. Chem. B, 2011, 115, 10707–10714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00604d |
| This journal is © the Partner Organisations 2020 |