
Bioinspired tough, conductive hydrogels with thermally reversible adhesiveness based on nanoclay confined NIPAM polymerization and a dopamine modified polypeptide†
Xiang
Di
,
Chen
Hang
,
Yue
Xu
,
Qiyue
Ma
,
Feifan
Li
,
Pingchuan
Sun
 and
Guolin
Wu
*
and
Guolin
Wu
*
Key Laboratory of Functional Polymer Materials, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: guolinwu@nankai.edu.cn
First published on 12th November 2019
Abstract
Hydrogels with excellent conductivity and flexibility have a wide range of applications in the biomedical field, such as in wearable devices, soft electronic skins, and biosensors. Inspired by marine mussels, a biocompatible hydrogel with controllable adhesiveness and a wearable strain-sensor were successfully prepared by polymerization of N-isopropylacrylamide (NIPAM) and a dopamine-modified polypeptide (PDAEA). The PNIPAM-clay-PDAEA hydrogel displayed repeatable and controllable adhesiveness due to the presence of free catechol groups in the PDAEA chains and the intrinsic thermo-sensitivity of the PNIPAM-based hydrogel. These materials also exhibited outstanding strain and pressure sensitive conductivity, which could be ascribed to a sufficient number of free moving ions in the hydrogel system. Test results showed that these conductive hydrogels possessed excellent biocompatibility, stable drug release behavior, and improved cell proliferation. Therefore, the as-prepared nanocomposite hydrogel has great potential applications as a wearable soft electronic skin and in tissue engineering.
1. Introduction
In the past few decades, stretchable, wearable and human-friendly electronic materials have gained significant attention in the biomedical field and smart wearable devices such as electronic skins, electrochemical sensors, and tissue engineering.1–6 However, conventional electronic devices are rigid, brittle, less flexible, costly, and non-biocompatible, which has limited their widespread applications. Compared to traditional electronic devices, conductive hydrogels, with high water contents and three-dimensional network structures, are similar to soft tissues, and have good biocompatibility and electronic properties. They can be used as an ideal ionic conductor.7–11 These hydrogels have been successfully fabricated by incorporating conductive nanofillers (graphene,12,13 metallic nanoparticles,4 carbon nanotube14,15) or conductive polymers (polypyrrole, polyaniline).16,17 For example, Zhang et al. introduced functionalized single-wall carbon nanotubes (FSWCNT) into biocompatible polyvinyl alcohol to obtain a hybrid hydrogel sensor, which could sense both gentle and fast human motions (including bending of fingers, walking, and pulse rate).18 Lu et al. developed a good biocompatible and conductive hydrogel using polydopamine nanoparticles and partly reduced graphene oxide (pGO), which displayed not only high stretchability and conductivity but also self-healability.11 Yu et al. synthesized a thermally responsive and conductive hydrogel with an interpenetrating binary network structure by addition of a conductive polymer (polypyrrole or polyaniline) and crosslinked with phytic acid in a poly(N-isopropylacrylamide) matrix.17 However, most of these conductive materials exhibit low stretchability and flexibility, and poor durability, which restricts their widespread applications where suitable strength is required.To overcome the shortcomings of poor mechanical properties, several strategies were developed to synthesize hydrogels with high strength by building multiscale network structures or incorporating effective nanomaterials such as nanocomposite hydrogels,19,20 double network hydrogels,21,22 and interpenetrating network hydrogels.23 For example, Bao et al. synthesized nanocomposite hydrogels (NC gels) by introducing metallic nanoparticles into an organic polymeric network, which exhibited electrical self-healing properties and good mechanical strength under ambient conditions due to a nanocomposite-reinforced mechanism.4 Gao et al. reported robust and flexible strain sensors based on dual physically cross-linked double network hydrogels, but they needed additional adhesive to adhere on external skin.24 Subsequently, Dong et al. proposed a mechanosensory electronic hydrogel through an interpenetrating polymer network (IPN) system, which displayed impressive stretchability, along with an ultra-stretchable pressure sensor.23 However, it also lacked another desirable feature of self-adhesiveness, by which it could adhere to the soft parts of the human body without any scotch tape, bandages or polyacrylate adhesives and promote the signal detection of wearable strain sensors under different deformation conditions. Therefore, it is necessary to combine both superior mechanical properties and self-adhesiveness into a conductive hydrogel.
Nature is always full of fascination and wisdom. Inspired by the adhesion mechanism of mussels, various strategies have been proposed to mimic this phenomenon.25–28 These materials usually contain 3,4-dihydroxy phenylalanine (DOPA) groups and derivatives found in adhesive proteins of mussels, such as polydopamine (PDA),29,30 and catechol-containing polymers.8,31–34 For instance, Liu et al. fabricated an epidermal strain sensor with the addition of polydopamine into polyvinyl alcohol hydrogel to monitor physiological signals, detect body movements and control robots.35 Wan et al. designed a tough and self-adhesive ionic gel with a great strain sensitivity by constructing synergistic multiple coordination bonds among tannic acid-coated cellulose nanocrystals (TA@CNCs), poly(acrylic acid) chains, and metal ions in a covalent polymer network.8
Herein, we developed a conductive hydrogel with excellent mechanical properties and thermo-reversible adhesion by combining a nanocomposite-reinforced matrix and dopamine modified polypeptide for the first time. Compared to the chemically crosslinked PNIPAM-based hydrogel, the PNIPAM-clay-PDAEA hydrogel exhibited excellent stretchability and elasticity due to the physically crosslinked network. The incorporation of polypetide endowed the material with excellent controllable adhesiveness and good biocompatibility. Besides, the ionic hydrogel could serve as a wearable strain sensor that could quickly and precisely detect electrical signal changes induced by different motions, such as stretching, compression deformation or subtle motions of the finger, wrist bending and blood pulse. Moreover, the in vitro cytotoxicity assay indicated that the as-prepared hydrogel possessed excellent cell affinity and biocompatibility. It is expected that this bioinspired material will provide an important alternative to construct deformable sensors, which have broad biomedical application areas, such as in wearable electronic skins, cell/drug delivery and tissue engineering.
2. Experimental
2.1. Materials
L-Aspartic acid and dopamine hydrochloride were obtained from Tianjin Lianxing Biotechnology Co. Ltd. Ethanolamine (EA), dimethylsulfoxide (DMSO) and phosphoric acid were purchased from Tianjin Heowns Biotechnology Co. Ltd. N-Isopropylacrylamide (NIPAM), potassium persulfate (KPS) and tetramethylethylene-diamine (TEMED) were obtained from TCI. Hectorite clay, “LAPONITE® XLG” [Mg5.34Li0.66Si8O20(OH)4]Na0.66, was procured from Rockwood Ltd. All other reagents were used without further purification.2.2. Characterization
2.3. Preparation of PNIPAM-clay-PDAEA hydrogels
Briefly, clay and sodium pyrophosphate were uniformly dispersed in water and sonicated in an ice water bath for 30 min. This was followed by the addition of the required PDAEA under stirring. Subsequently, the monomer (NIPAM) was added and degassed under vigorous stirring to remove oxygen. Finally, TEMED and KPS were added to the above solution and stirred for 5 min under N2 bubbling. The solution was quickly poured into a sealed glass mold provided with a 1 mm-thick spacer at ambient temperature for 6 h to obtain a PNIPAM-clay-PDAEA hydrogel. The constituents of different gels are shown in Table S1 (ESI†).2.4. Swelling ratio of the hydrogels
The swelling ratios of different hydrogels in deionized water were measured. Briefly, the weights of the samples (pure PNIPAM, PNIPAM-clay(7.5%), PNIPAM-clay(7.5%)-PDAEA(1%) hydrogels) at different time intervals were measured after wiping off the excess water, using filter paper. The swelling ratios were calculated according to the following equation:where, Wd was the dry weight of the lyophilized sample and Ws was the weight of the swollen sample at any time interval.
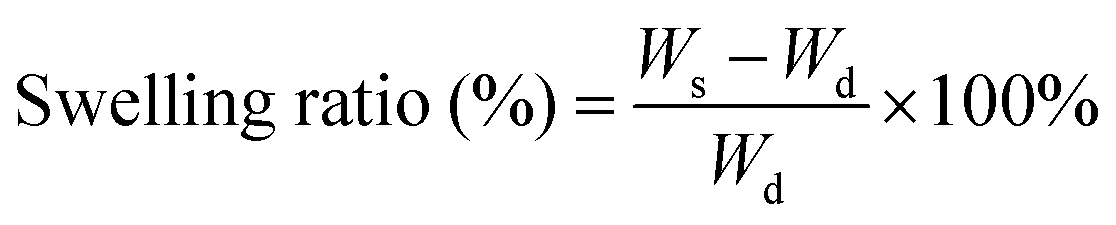
2.5. The volume phase transition temperature (VPTT) of the hydrogels
DSC measurements were performed to determine the VPTT of different hydrogels using a differential scanning calorimeter (DSC, Mettler-Toledo), using N2 purge gas and at cooling and heating rates of 2 °C min−1 in the temperature range of 20 °C to 55 °C. About 10 mg of each sample was encapsulated in a 40 μL aluminum pan for DSC measurements.2.6. Measurement of adhesive strength
Lap shear adhesion tests for different hydrogels were conducted to measure the adhesive strength. Glass, silicon sheet, and porcine skin were used as substrates. Repeatability and reversibility of adhesion on the glass surface in response to temperature were also evaluated for number of cycles.2.7. Mechanical properties of the hydrogels
The mechanical properties of different hydrogels were tested using a UTM tensile strength instrument (Shenzhen Suns Technology Stock Co. Ltd). For the tensile test, the dimensions of the hydrogels were 5.5 mm (diameter) × 60 mm (length) and the crosshead speed was 100 mm min−1 at room temperature. For a compression test, the hydrogel was cylindrical in shape with 13.2 mm (diameter) × 14 mm (height) dimensions. Tensile stress was evaluated from the ratio of real-time force to cross-section of the original hydrogel.2.8. The electrical conductivity measurement and conductive behavior of the hydrogels
The electrical conductivities (σ) of different hydrogels were determined by a four-probe method using a digital tester (Model 2400, Keithley), as reported in previous studies.37 Prior to the test, the hydrogels were cut into square shaped specimens with dimensions of 20 mm × 20 mm × 1 mm. The conductivities of the hydrogels were calculated according to the following equation:where, t is the thickness of the hydrogel and Rs is the sheet resistance of the hydrogel. Variations in real-time resistances of the hydrogels were detected and recorded in different states using a UT61E Digital Multimeter. Variation in electrical resistance of the hydrogel was defined by the following equation:
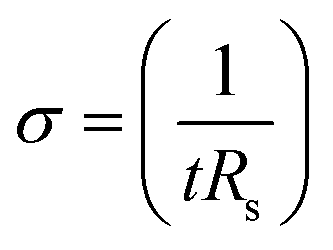
where, R and R0 are the real-time resistance and the primary resistance without strain, respectively.
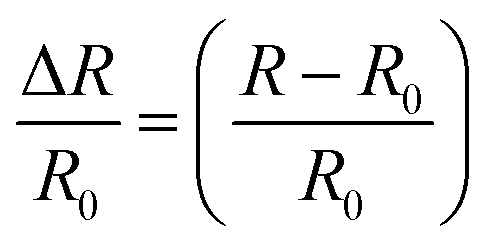
2.9. In vitro drug loading and release
Rhodamine B was chosen as the model drug for loading and release experiments. Lyophilized hydrogels attained equilibrium state in rhodamine B (0.3 mg mL−1) solution at ambient temperature. Then the hydrogel was immersed in buffer (PBS) and placed on a shaking bed at 37 °C (100 rpm) in the dark. At predetermined intervals, 3 mL of release medium was collected and replaced with 3 mL of fresh PBS. The release of rhodamine B from the hydrogel was quantified by UV-Vis spectrophotometry at 554 nm. The cumulative rhodamine B release was calculated as:where, M0 is the total mass of rhodamine B; Ve and V0 are the volumes of release media (3 mL) and the original volume (20 mL), respectively; Ci is the concentration of rhodamine B released from the sample and i is the displacement time. All tests were repeated three times.

2.10. Cytocompatibility evaluation and cell behavior on the hydrogel
The cytocompatibilities were determined from the viability of cells exposed to hydrogel extracts, using a quantitative methyl thiazolyl tetrazolium (MTT) assay. The suspension of NIH3T3 cells was cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and then seeded into 96-well culture plates with densities of 8 × 103 cells per 100 μL per well. Before cell culturing, sterilization was carried out by soaking the hydrogel in 75% ethanol for 2 h and washing three times with sterile PBS for 45 min. The hydrogel was submerged in DMEM (10 mg mL−1) for 24 h to obtain hydrogel extracts. Then the media were replaced with 100 μL per well hydrogel extracts and the cells without added hydrogel extracts were set as control groups. After incubation for 3 days, the hydrogel extracts were removed and replaced with 50 μL MTT solution (1 mg mL−1) for 2 h. Finally, 100 μL DMSO was added into each well to dissolve the crystals completely. The absorbance was measured by a microplate reader (Model 354, Finland) at 570 nm. The relative cell viability was calculated as:where, Abshydrogel and Abscontrol are the absorbance of the hydrogel extracts and control wells, respectively. The morphologies of the hydrogel surfaces cultured with fibroblasts were carefully studied by SEM. The proliferation of cells was observed by fluorescence microscopy (Olympus IX51) after staining with fluorescein dye.
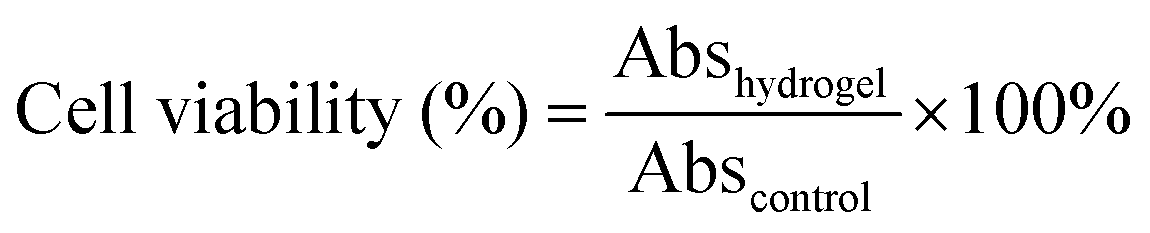
3. Results and discussion
Fig. 1 shows the process for fabrication of PNIPAM-clay-PDAEA hydrogels. Catechol-modified polypeptide (PDAEA) was prepared by ring-opening reaction of polysuccinimide with dopamine (Fig. S1, ESI†). NMR and FTIR spectra confirmed the successful preparation and the structure of PDAEA (Fig. S2 and S3, ESI†). The PNIPAM-clay-PDAEA hydrogels were prepared in the presence of an initiator and accelerator by in situ free radical polymerization as shown in Fig. 1a. The prepared hydrogel contained different types of functional groups, such as –OH, –CONH, –NH and –Si–OH, which formed a network of different hydrogen-bonds and improved the mechanical properties and self-healing ability of the hydrogel. Besides, the hydrogel exhibited excellent conductivity and cell affinity, and is biocompatible and safe to be applied for wound dressings, wearable electronic devices, smart actuators, and so on.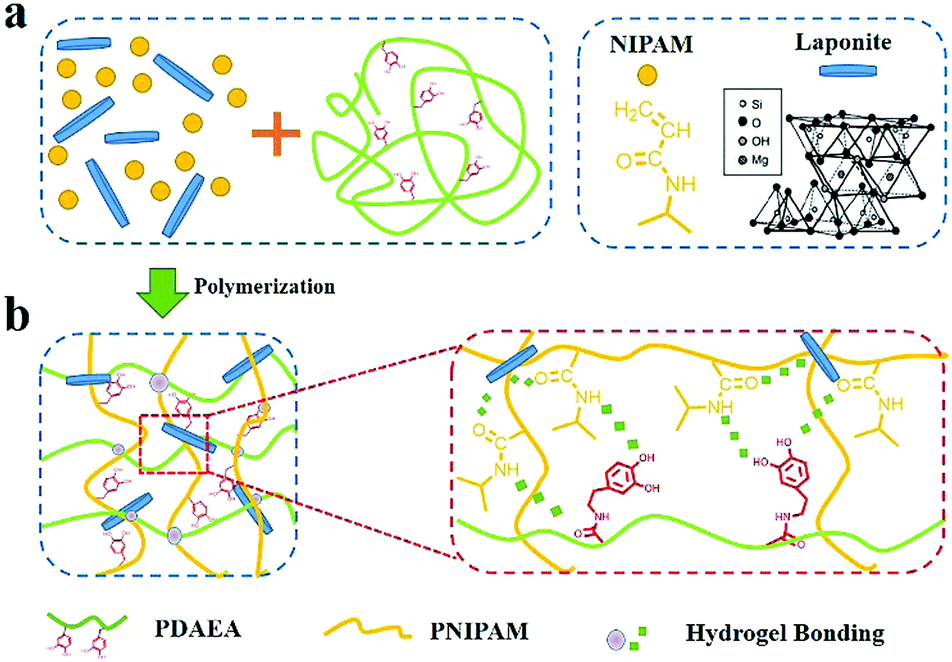 | ||
| Fig. 1 (a) Scheme depicting the fabrication of a PNIPAM-clay-PDAEA hydrogel. (b) Hydrogen bonding interactions between the clay, PDAEA chains, and PNIPAM chains. | ||
Compared to the chemically crosslinked PNIPAM-based hydrogel, the PNIPAM-clay-PDAEA hydrogel exhibited excellent stretchability and elasticity, owing to its fully physically crosslinked network. As illustrated in Fig. 2a and b, the prepared hydrogel could not only be stretched to seven times its original length without breaking, but was also able to withstand different deformations such as bending, twisted stretching and crossed stretching. In Fig. 2c, the cylindrical PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel was fast to recover from its deformation after removing the external force, indicating that the hydrogel has good elasticity and tough characteristics due to the reinforcement of the nanosheets. To further investigate the effect of PDAEA and clay composition on the mechanical properties of the hydrogels, a series of hydrogels with different PDAEA/NIPAM and clay/water weight ratios were prepared and tested for their tensile strengths at a stretch rate of 100 mm min−1 as shown in Fig. 2d. An increase in clay content increased the extension ratio and tensile strength of the hydrogel. It reached its maximum value with 7.5 wt% clay and 1 wt% of PDAEA/NIPAM (Fig. S4, ESI†). It should be noted that an excessive number of PDAEA chains hindered the polymerization of NIPAM monomers and resulted in a decrease of mechanical strength. This was ascribed to the catechol groups of PDAEA, which deactivated some of the initiator molecules. The tensile and compressive strengths of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel were about 73.3 kPa and 308 kPa (Fig. 2e), respectively, higher than those of the other nano-enhanced adhesive hydrogels.38 Therefore, due to the combined effect of clay and PDAEA on mechanical strength, the PNIPAM-clay-PDAEA hydrogel with 7.5 wt% of clay and 1 wt% of PDAEA was chosen for the subsequent studies.
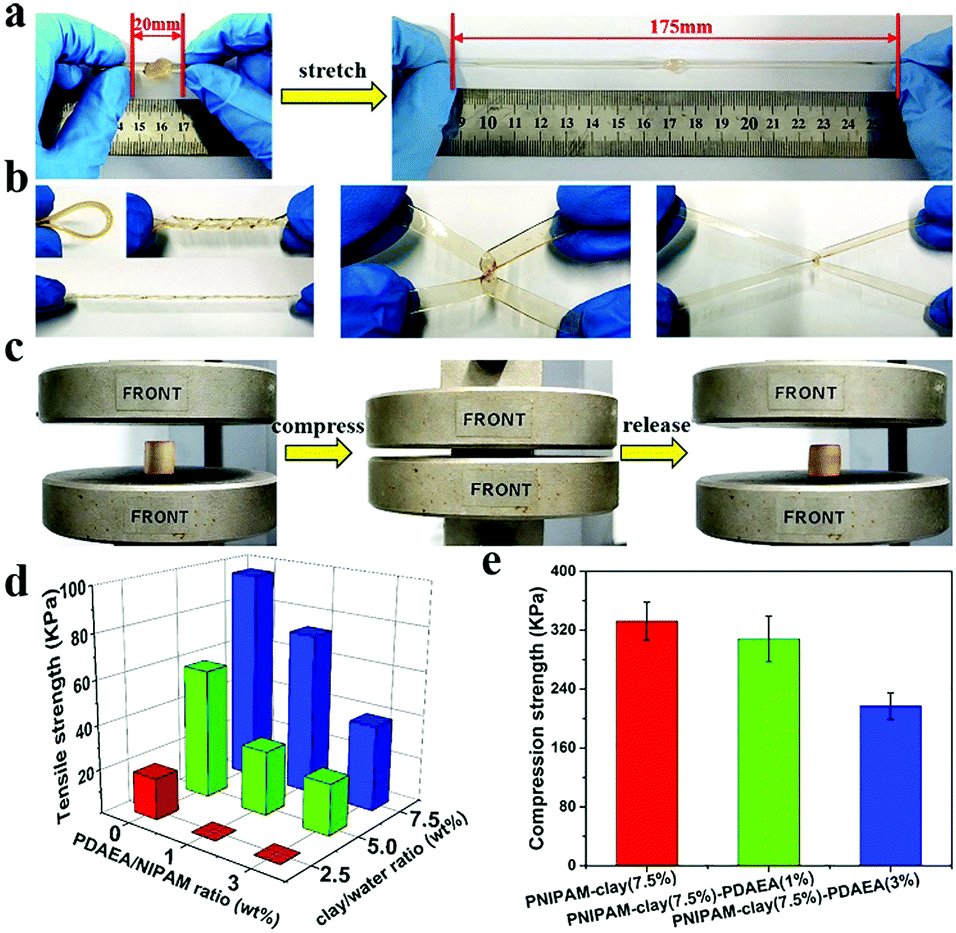 | ||
| Fig. 2 Mechanical properties of the PNIPAM-clay-PDAEA hydrogel. (a) The PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel could be easily stretched and (b) twisted and knotted. (c) The recoverability of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel under compression of 85% strain. (d) The tensile and (e) compression strengths of PNIPAM-clay hydrogels with different contents of PDAEA and clay. | ||
Fig. 3 shows the adhesive properties of the PNIPAM-clay-PDAEA hydrogel. As illustrated in Fig. 3a, the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel could adhere to different substrates such as glass, silicone, plastic, paper, aluminum, and copper, due to the catechol groups and multiple physical interactions between the contact surfaces and hydrogels, which include hydrogen bonding, π–π stacking, hydrophobic interactions, or a synergetic effect of these interactions. Fig. 3b shows that the hydrogel could adhere to the author's hand even when stretched and did not fall off even when the hand was shaken rapidly (Video S1, ESI†). Besides, the adhesive hydrogel could be easily peeled off without any residue, irritation, or inflammatory response. To quantify the strength with which the hydrogel adhered to different surfaces, lap shear strength was tested and the results are shown in Fig. 3d. The adhesive strength with the surface of the silicone sheet was highest amongst all the substrates. This could be attributed to the synergistic effects of π–π stacking and hydrogen bonding. The adhesion to the glass surface was more than that of porcine skin because of the interactions between the hydrogel and the hydroxyl groups of the smoother glass surface. It should be noted that the PNIPAM-clay hydrogel with a large number of PDAEA chains could have caused a decrease in adhesive strength, due to inhibition of NIPAM polymerization by the catechol groups, which led to cracking of the hydrogel during the lap shear test. Therefore, the PNIPAM-clay-PDAEA hydrogel with 1 wt% of PDAEA was chosen for the reversible and repeatable adhesion test.
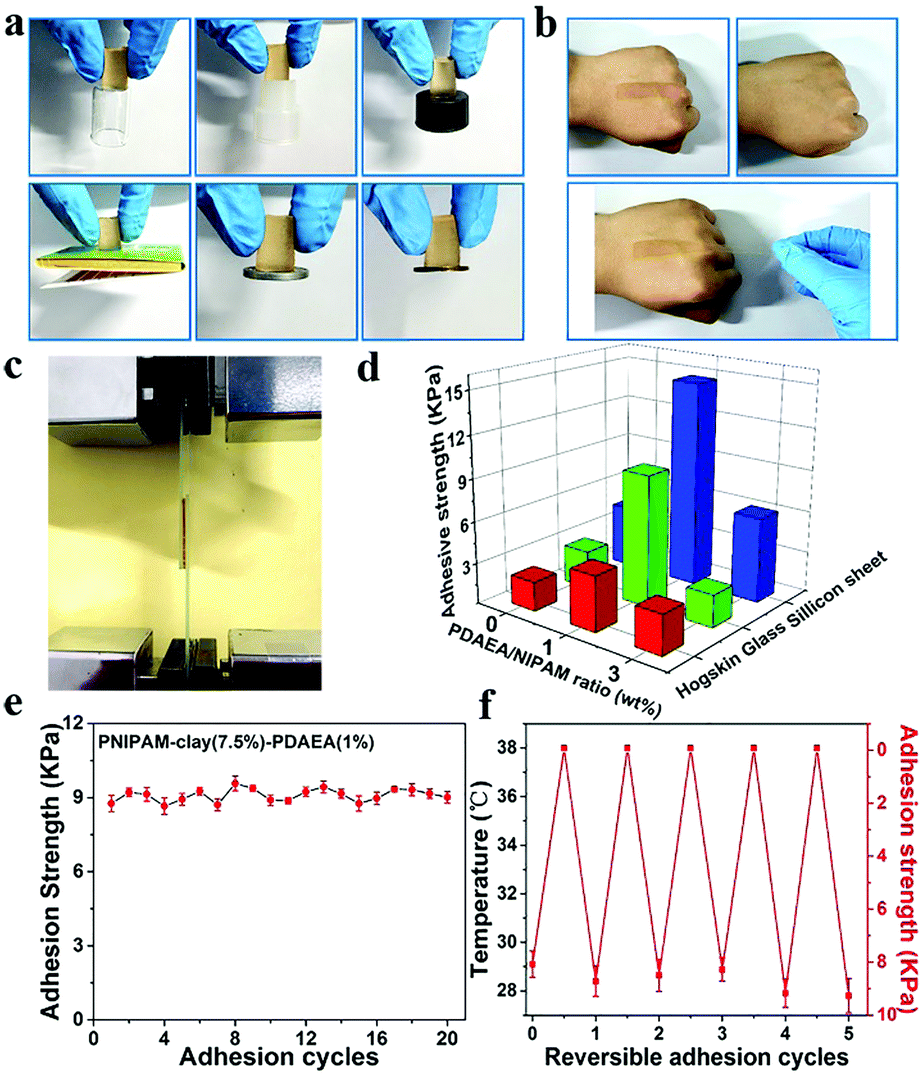 | ||
| Fig. 3 Adhesive properties of the PNIPAM-clay-PDAEA hydrogel. (a) Adhesion of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel on different substrates: glass, silicone, plastic, paper, aluminum and copper. (b) Tissue adhesiveness and toughness of the hydrogel on the author's hand. (c) Tests for adhesive strength of the hydrogel on a glass slide. (d) Adhesive strengths of hydrogels on different substrates. (e) Repeatable and (f) thermally reversible adhesion of the hydrogel to glass. | ||
In addition to good adhesive properties, the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel also displayed thermal reversibility and repeatable adhesion. The cyclic peel-off tests on glass for 20 cycles were performed to demonstrate this. In one cycle, the hydrogel was allowed to adhere to the glass surface and then peeled off using a tensile load before the next cycle. As illustrated in Fig. 3e, the adhesive strength of the hydrogel was approximately 9.5 kPa with almost no loss in adhesion strength during all 20 cycles. To investigate the thermally reversible adhesiveness of the PNIPAM-based hydrogel, typical volume phase transition temperature (VPTT) profiles of different hydrogels were obtained by DSC, as shown in Fig. S5a (ESI†). The results showed that the addition of PDAEA chains increased the VPTT to 35.5 °C, which could be ascribed to the hydrogen bonding between PDAEA chains and the hydrogel network. Therefore, the relationship between adhesiveness and temperature was established by a thermally reversible adhesion test on a glass surface, as shown in Fig. S5b (ESI†). The PNIPAM chain in the hydrogel system changed from hydrophilic to hydrophobic as the temperature was higher than the VPTT. Moreover, some volume of water was extruded from the hydrogel due to volume shrinkage, which further prevented the hydrogen bonding interactions between the gel and glass surface. This caused a decrease in adhesiveness until it was finally lost. Furthermore, the adhesiveness was recovered to its original state after cooling down to room temperature and there was almost no loss of this property for over 5 cycles. (Video S2, ESI†). These results suggest that the hydrogel displayed excellent repeatable and thermally reversible adhesiveness, and has potential applications such as in wound dressings and wearable strain sensors.
To study the relationship between macroscopic properties and microstructure, the self-healing behavior and internal microstructure of the hydrogel were investigated and explained (Fig. 4). As shown in Fig. 4a, the fractured hydrogel samples with different colors were contacted with the cut interfaces and were left to heal at ambient temperature without any external stimuli and healing agents. After only 5 min of healing time, the self-healed hydrogel was able to carry its own weight. After contacting for 6 h, the diffusion of rhodamine B from one-half of the hydrogel to the other could be observed directly and the cutting at the interface disappeared. Besides, the self-healing hydrogel could even withstand stretching. The self-healing ability of the hydrogel under ambient conditions was attributed to the nanostructure of the hydrogel system and the different physical interactions between the contacting surfaces. In Fig. 4b, the fracture morphology at a cross-section of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel showed a honeycomb-like densely microporous structure. The magnified region showed a uniform porous network promoting the formation of a large number of hydrogen bonds and the transport of ions in the network system. In contrast, the hydrogel without PDAEA chains had a smooth, uneven surface and nonuniform porous structure, due to which it lacked self-healing ability and adhesiveness. The average pore diameter of PNIPAM-clay(7.5%)-PDAEA(1%) and PNIPAM-clay(7.5%) hydrogel was 20 μm and 15 μm, respectively (Fig. S8, ESI†). The pore diameter of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel was bigger than that of the PNIPAM-clay(7.5%) sample, due to the addition of hydrophilic polymeric chains (PDAEA), which was consistent with the swelling behavior of the hydrogels (Fig. S7, ESI†).
 | ||
| Fig. 4 (a) The self-healing behavior of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel. (b) SEM images of cross-sectional morphologies of PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel and (c) PNIPAM-clay(7.5%) hydrogel (one stained with rhodamine B). | ||
To explore the viscoelastic characteristics of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel, oscillatory rheology testing was carried out as shown in Fig. 5. The storage modulus (G′), and loss modulus (G′′) of the prepared hydrogel displayed frequency and temperature dependence. The value of G′ was found to be consistently higher than that of G′′ over the entire frequency range (10−3–10 Hz), indicating the stable and elastic nature of the hydrogel. Further, a small loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) suggested that the hydrogel had excellent elastic recovery properties. Besides, the G′ value of the hydrogel was maximum at 37 °C due to volume shrinkage and structure packing of the hydrogel above the volume phase transition temperature, almost consistent with the results of VPTT of DSC analyses (Fig. S5, ESI†).
δ) suggested that the hydrogel had excellent elastic recovery properties. Besides, the G′ value of the hydrogel was maximum at 37 °C due to volume shrinkage and structure packing of the hydrogel above the volume phase transition temperature, almost consistent with the results of VPTT of DSC analyses (Fig. S5, ESI†).
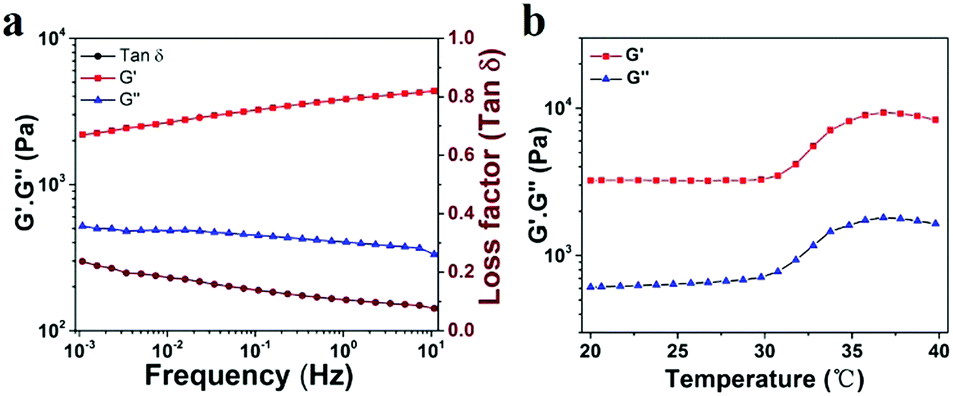 | ||
| Fig. 5 (a) Frequency-sweep curves of rheological behavior of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel. (b) Thermal rheological behavior of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel. | ||
Given that the large number of ions could freely move in the hydrogel, the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel exhibited excellent ionic conductivity. The conductivity of the hydrogel was about 0.2 S m−1 and increased with the increase of clay content. It showed reasonably good electrical conductivity at 25 °C (Fig. S6, ESI†).7 An increase in the number of PDAEA chains also improved the conductivity. This signified that the uniform porous network system facilitated free movement of ions (Fig. 4b). To demonstrate the strain and pressure sensing ability of the fabricated hydrogel in wearable electronic devices, a test was conducted by using the hydrogel in a complete circuit connecting a light-emitting diode (LED) light. The brightness of the LED bulb gradually decreased when it was elongated from 0% to 200%. Importantly, the blue-colored LED exhibited a rapid response to the changes of strain, indicating that the hydrogel possessed high strain sensitivity (Video S3, ESI†). The changes in relative resistance vs strain are shown in Fig. 6b. It was obvious that the relative resistance displayed a step-like trend with a step-by-step stretching cycle. The inset image showed that the real-time resistance changed on applying 50%, 100%, and 200% stretching strains on the gel. The changes in LED brightness were consistent with the state of compression. Similarly, Fig. 6d showed that the relative resistance decreased with increase in degree of compression, which further indicated that the hydrogel also possessed excellent pressure sensing ability (Video S4, ESI†). The inset image showed changes in real-time resistance on applying 0%, 30%, and 50% compression strains on the gel.
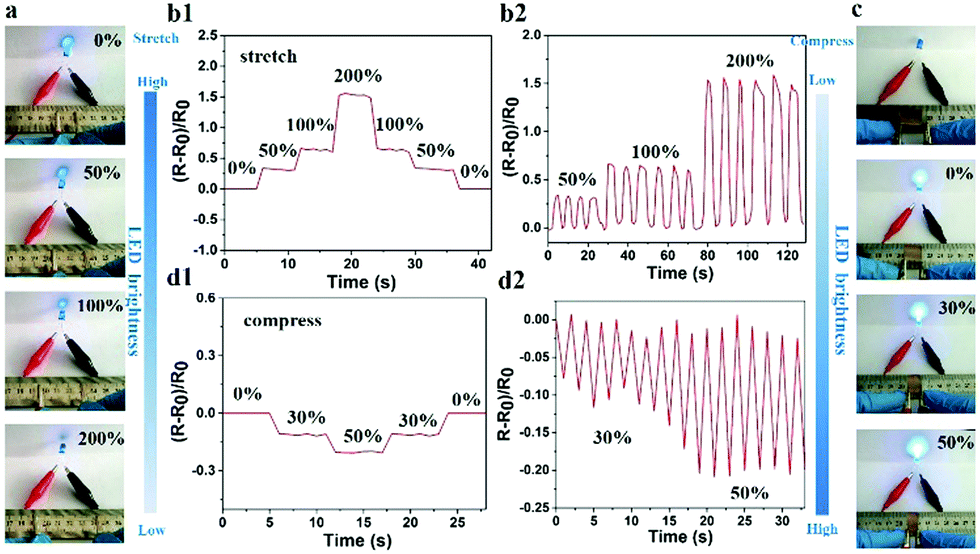 | ||
| Fig. 6 Demonstration of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel as a wearable sensor with strain-sensitive conductivity. (a) Photographs showing changes in LED brightness and (b1) relative resistance response when stretched to different extents and (b2) real-time resistance change in applying 50%, 100%, and 200% stretching strains on the gel. (c) Photographs showing changes in LED brightness and (d1) relative resistance variations under different compression states and (d2) real-time resistance change in applying 30% and 50% compression strains on the gel. | ||
The prepared hydrogel could not only detect macroscopic deformation but could also monitor slight physiological changes. As shown in Fig. 7a, the hydrogel sensor adhered to the wrist of the author was used to monitor the slight pressure differences in wrist pulse. Changes in real-time relative resistance curve showed that the regular pulse rate was 66 beats min−1 in relaxation. After exercise the pulse frequency was 92 beats min−1 with irregular amplitude, indicating the high strain sensitivity of the hydrogel. Hence, the hydrogel can serve as a human-motion sensor to detect movements of the finger and wrist (Fig. 7b and c). The relative resistance of the hydrogel sensor increased with increase in bending angle. This was related to the elongation of the hydrogel-based sensor, which led to an increase of resistance. In addition to sensing human motions, the prepared hydrogel could also be applied as a soft touch pen for a smart phone (Fig. 7d, e and Video S5, ESI†), where it could be used to write smoothly on the screen without any scratches (Video S6, ESI†). This made it ideal for use in wearable electronic sensors and flexible input devices.
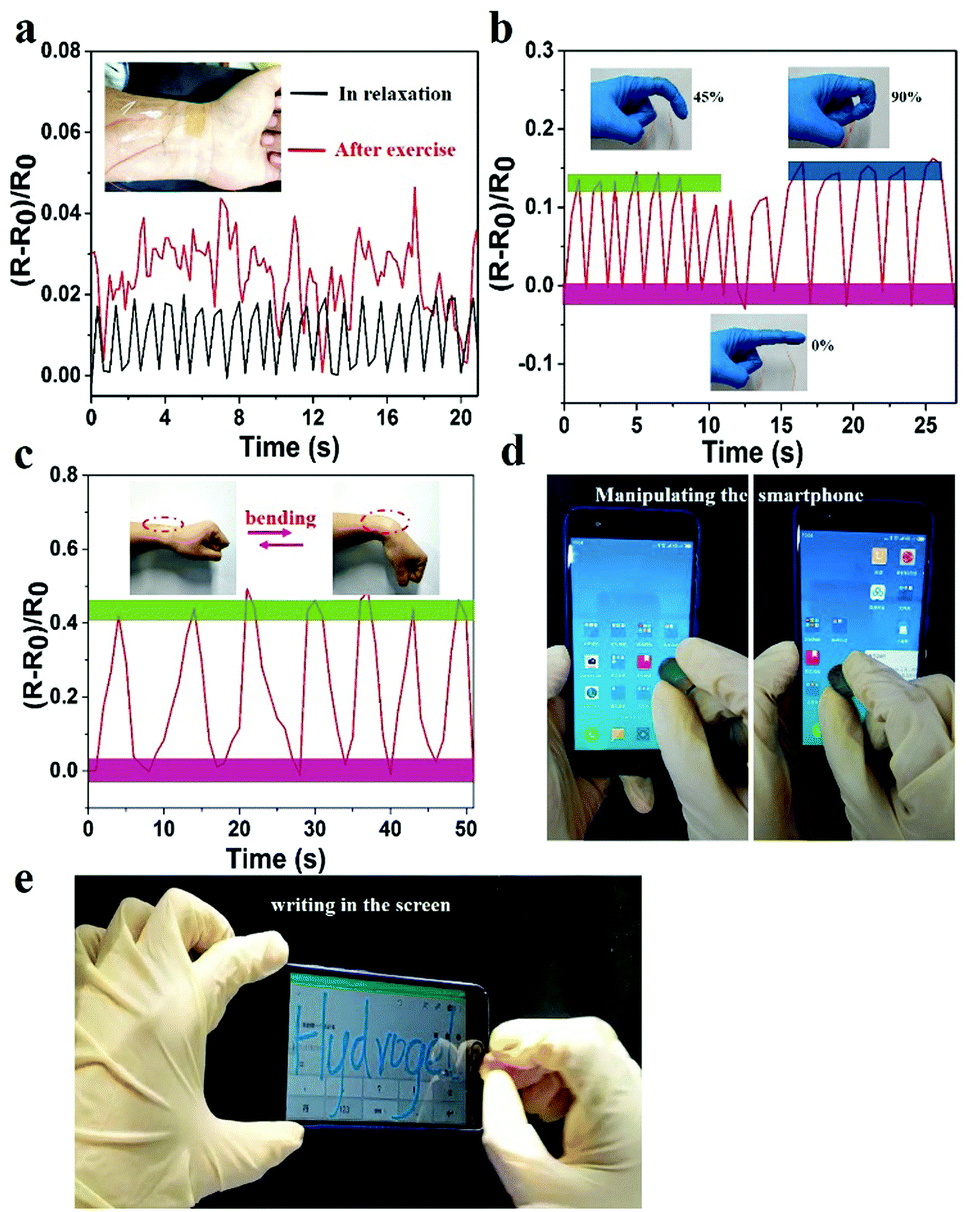 | ||
| Fig. 7 The relative resistance change versus time for real-time monitoring of different human motions. (a) Real-time resistance change signal of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel sensor as a wearable pulse monitor worn by the author under relaxation and exercise conditions. (b) The relative resistance changes versus time when the finger and (c) the wrist bend at different angles. (The bending angle of the finger was 0°, 45°, and 90°.) (d) The hydrogel as a smart pen to manipulate a smart phone and (e) to write on the screen. | ||
To evaluate the cytocompatibility of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel, NIH3T3 cells were chosen for studying their proliferation on the hydrogel surface and the live cells were stained green with fluorescein dye according to the literature.39,40 As shown in Fig. 8a, the intensity of green fluorescence gradually brightened with increase in incubation time, indicating that the hydrogel was conducive for proliferation and growth of cells. Compared to the smooth surface of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel, a large number of cells formed a confluent monolayer on the hydrogel with many extended filopodia after 3 days of culturing. Fusiform morphology of cells further showed that the cells were in a healthy growing state (Fig. 8c and d). SEM and fluorescence images unambiguously demonstrated that the hydrogel possessed excellent cell affinity and promoted cellular adhesion and proliferation. The relative cell viability of NIH3T3 on the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel was assessed by a quantitative MTT assay. As shown in Fig. 8e, the viability of cells was more than 80% during the entire period of culturing and increased with time. These results indicated that the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel was non-toxic to the cells and possessed excellent cell affinity, which is significantly important for smart wearable devices, soft electronic skins and so on.
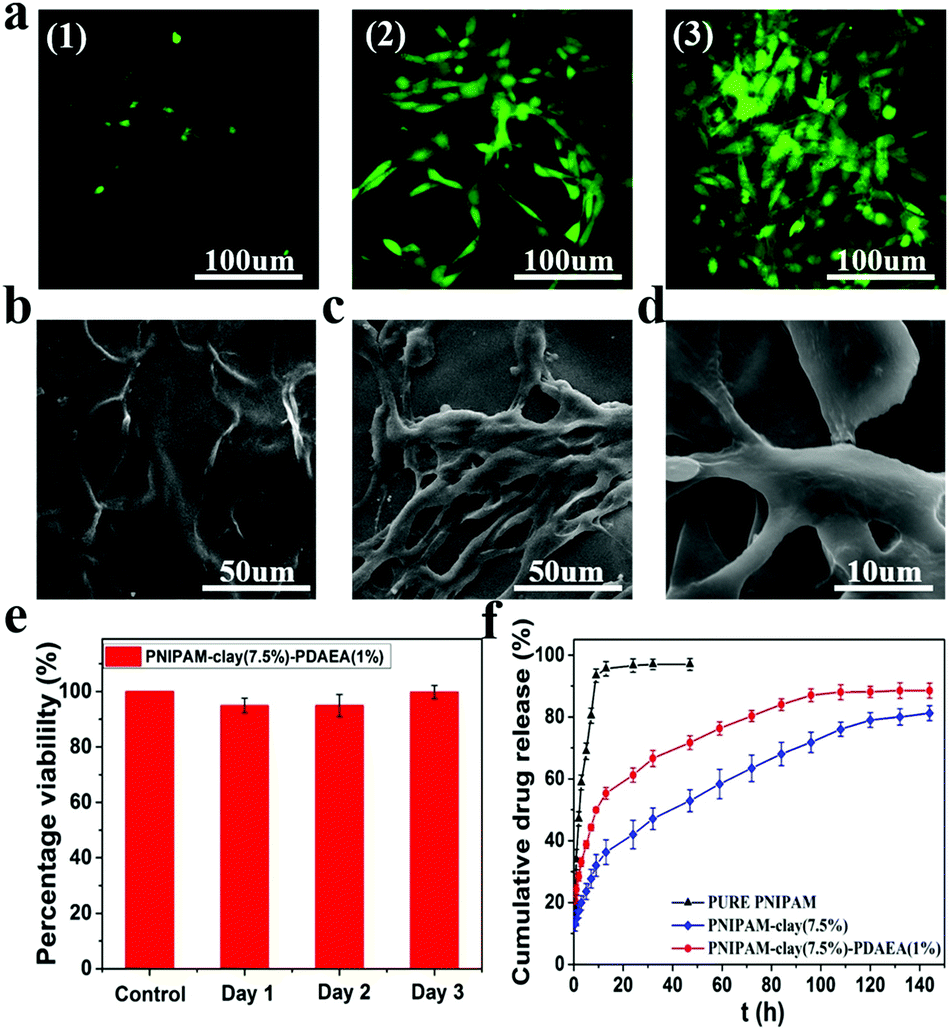 | ||
| Fig. 8 Cell behaviors on PNIPAM-clay-PDAEA hydrogels. (a) Fluorescence images of fibroblasts incubated on the surface of PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel after 1, 2, and 3 days of culture. (b) SEM images of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel without fibroblasts and (c) with fibroblasts after 3 days culture. (d) The magnified morphology of cells on the hydrogel. (e) In vitro cytocompatibility of fibroblasts, as measured via MTT assay. (f) The release profiles of rhodamine B from different hydrogels. | ||
Fig. 8f shows the drug releases profile from different hydrogels. The results showed that the release rate of the model drug from the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel was faster than that from PNIPAM-clay(7.5%) and it was more stable and durable than the pure PNIPAM hydrogel. This was ascribed to the honeycomb-like uniform porous structure of the PNIPAM-clay(7.5%)-PDAEA(1%) hydrogel. The increase in a weight swelling ratio (Fig. S7, ESI†) indicated that the addition of hydrophilic polymeric chains (PDAEA) could make the release behavior more effective and controllable, which further broadened the scope of its potential applications in biomedical fields such as wound dressing and controllable drug release.
4. Conclusions
In summary, a strain-sensitive, conductive, self-healing, and biocompatible hydrogel with controllable adhesiveness was designed and developed by combining the adhesive mechanism of mussels and temperature-sensitivity of PNIPAM-based materials. The hydrogels possessed high stretchability and self-healing properties without the requirement of any external stimuli or healing agents. The ample number of hydrogen bonding interactions and the presence of a sufficient number of catechol groups endowed the material with durable and repeatable adhesiveness. Importantly, the uniform porous network structure inside the PNIPAM-clay-PDAEA hydrogel could act as a pathway for ionic transport, had enhanced strain sensitivity as a strain sensor, and detected the changes in relative resistance induced by human motions, including fast deformations, gentle finger and wrist bending movements, and blood pulse rate. Moreover, the as-prepared hydrogel also exhibited good biocompatibility, cell affinity, and sustained drug release behavior. To sum up, this prepared nanocomposite hydrogel showed the possibility of fabricating stretchable, self-healing, biocompatible, strain-sensitive, and conductive soft materials with controllable adhesiveness, which have great potential applications in tissue engineering, and as electronic skins and wearable devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Key Program of National Natural Science Foundation of China (21534005).References
- T. Yang, W. Wang, H. Zhang, X. Li, J. Shi, Y. He, Q. Zheng, Z. Li and H. Zhu, ACS Nano, 2015, 9, 10867–10875 CrossRef CAS.
- J. Lee, S. Kim, J. Lee, D. Yang, B. C. Park, S. Ryu and I. Park, Nanoscale, 2014, 6, 11932–11939 RSC.
- J. Li, L. Geng, G. Wang, H. Chu and H. Wei, Chem. Mater., 2017, 29, 8932–8952 CrossRef CAS.
- B. C. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS.
- Z. Wang, H. Zhou, W. Chen, Q. Li, B. Yan, X. Jin, A. Ma, H. Liu and W. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 14045–14054 CrossRef CAS.
- S. Wei, G. Qu, G. Luo, Y. Huang, H. Zhang, X. Zhou, L. Wang, Z. Liu and T. Kong, ACS Appl. Mater. Interfaces, 2018, 10, 11204–11212 CrossRef CAS.
- X. Zhang, N. Sheng, L. Wang, Y. Tan, C. Liu, Y. Xia, Z. Nie and K. Sui, Mater. Horiz., 2019, 6, 326–333 RSC.
- C. Shao, M. Wang, L. Meng, H. Chang, B. Wang, F. Xu, J. Yang and P. Wan, Chem. Mater., 2018, 30, 3110–3121 CrossRef CAS.
- Y. J. Liu, W. T. Cao, M. G. Ma and P. Wan, ACS Appl. Mater. Interfaces, 2017, 9, 25559–25570 CrossRef CAS.
- Y. Zhu, S. Liu, X. Shi, D. Han and F. Liang, Mater. Chem. Front., 2018, 2, 2212–2219 RSC.
- L. Han, X. Lu, M. Wang, D. Gan, W. Deng, K. Wang, L. Fang, K. Liu, C. W. Chan, Y. Tang, L. T. Weng and H. Yuan, Small, 2017, 13, 1601916 CrossRef.
- Y. Wang, F. Huang, X. Chen, X. Wang, W. Zhang, J. Peng, J. Li and M. Zhai, Chem. Mater., 2018, 30, 4289–4297 CrossRef CAS.
- Y. R. Jeong, H. Park, S. W. Jin, S. Y. Hong, S.-S. Lee and J. S. Ha, Adv. Funct. Mater., 2015, 25, 4228–4236 CrossRef CAS.
- X. Zhang, C. L. Pint, M. H. Lee, B. E. Schubert, A. Jamshidi, K. Takei, H. Ko, A. Gillies, R. Bardhan, J. J. Urban, M. Wu, R. Fearing and A. Javey, Nano Lett., 2011, 11, 3239–3244 CrossRef CAS.
- S.-J. Park, J. Kim, M. Chu and M. Khine, Adv. Mater. Technol., 2018, 3, 1700158 CrossRef.
- Y. Liu, K. Xu, Q. Chang, M. A. Darabi, B. Lin, W. Zhong and M. Xing, Adv. Mater., 2016, 28, 7758–7767 CrossRef CAS.
- Y. Shi, C. Ma, L. Peng and G. Yu, Adv. Funct. Mater., 2015, 25, 1219–1225 CrossRef CAS.
- M. Liao, P. Wan, J. Wen, M. Gong, X. Wu, Y. Wang, R. Shi and L. Zhang, Adv. Funct. Mater., 2017, 27, 1703852 CrossRef.
- M. K. Jaiswal, J. R. Xavier, J. K. Carrow, P. Desai, D. Alge and A. K. Gaharwar, ACS Nano, 2016, 10, 246–256 CrossRef CAS.
- C. Yao, Z. Liu, C. Yang, W. Wang, X. Ju, R. Xie and L. Chu, ACS Appl. Mater. Interfaces, 2016, 8, 21721–21730 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, K. Sato, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Adv. Mater., 2015, 27, 2722–2727 CrossRef CAS.
- H. Cong, P. Wang and S. Yu, Chem. Mater., 2013, 25, 3357–3362 CrossRef CAS.
- G. Ge, Y. Zhang, J. Shao, W. Wang, W. Si, W. Huang and X. Dong, Adv. Funct. Mater., 2018, 28, 1802576 CrossRef.
- S. Xia, S. Song and G. Gao, Chem. Eng. J., 2018, 354, 817–824 CrossRef CAS.
- B. J. Kim, D. X. Oh, S. Kim, J. H. Seo, D. S. Hwang, A. Masic, D. K. Han and H. J. Cha, Biomacromolecules, 2014, 15, 1579–1585 CrossRef CAS.
- L. Li, W. Smitthipong and H. Zeng, Polym. Chem., 2015, 6, 353–358 RSC.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338–341 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS.
- L. Han, Y. Zhang, X. Lu, K. Wang, Z. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 29088–29100 CrossRef CAS.
- L. Han, X. Lu, K. Liu, K. Wang, L. Fang, L. T. Weng, H. Zhang, Y. Tang, F. Ren, C. Zhao, G. Sun, R. Liang and Z. Li, ACS Nano, 2017, 11, 2561–2574 CrossRef CAS PubMed.
- J. Shin, J. S. Lee, C. Lee, H. Park, K. Yang, Y. Jin, J. H. Ryu, K. S. Hong, S. Moon, H. Chung, H. S. Yang, S. H. Um, J. Oh, D. Kim, H. Lee and S. Cho, Adv. Funct. Mater., 2015, 25, 3814–3824 CrossRef CAS.
- J. H. Ryu, Y. Lee, W. H. Kong, T. G. Kim, T. G. Park and H. Lee, Biomacromolecules, 2011, 12, 2653–2659 CrossRef CAS.
- C. Gong, C. Lu, B. Li, M. Shan and G. Wu, J. Biomed. Mater. Res., Part A, 2017, 105, 1000–1008 CrossRef CAS.
- B. P. Lee, J. L. Dalsin and P. B. Messersmith, Biomacromolecules, 2002, 3, 1038–1047 CrossRef CAS.
- S. Liu, R. Zheng, S. Chen, Y. Wu, H. Liu, P. Wang, Z. Deng and L. Liu, J. Mater. Chem. C, 2018, 6, 4183–4190 RSC.
- H. S. Kang, S. R. Yang, J.-D. Kim, S.-H. Han and I.-S. Chang, Langmuir, 2001, 17, 7501–7506 CrossRef CAS.
- Z. Deng, Y. Guo, X. Zhao, P. X. Ma and B. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- X. Tong, L. Du and Q. Xu, J. Mater. Chem. A, 2018, 6, 3091–3099 RSC.
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS.
- S. R. Shin, H. Bae, J. M. Cha, J. Y. Mun, Y.-C. Chen, H. Tekin, H. Shin, S. Farshchi, M. R. Dokmeci, S. Tang and A. Khademhosseini, ACS Nano, 2012, 6, 362–372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00582j |
| This journal is © the Partner Organisations 2020 |