
Afterglow of carbon dots: mechanism, strategy and applications
Kai
Jiang
a,
Yuhui
Wang
 a,
Zhongjun
Li
b and
Hengwei
Lin
*ac
a,
Zhongjun
Li
b and
Hengwei
Lin
*ac
aKey Laboratory of Graphene Technologies and Applications of Zhejiang Province & Ningbo Institute of Materials Technology & Engineering (NIMTE), Chinese Academy of Sciences (CAS), Ningbo 315201, China. E-mail: linhengwei@nimte.ac.cn
bCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, 450001, China
cInternational Joint Research Center for Photo-responsive Molecules and Materials, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China
First published on 24th October 2019
Abstract
As a newly emerged carbon-based luminescent nanomaterial, carbon dots (CDs) have attracted considerable attention due to their outstanding features. Their photoluminescence (PL) properties and relevant applications have been extensively studied and reviewed in recent years. However, although their afterglow feature had also been discovered and had seen some important advances, no review article was published to systematically summarize such advances. Considering the scientific importance and promising applications of afterglow materials, herein, we aim to summarize the recent remarkable advances of the afterglow phenomena of CDs, particularly focusing on their room temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF). This review will firstly simply introduce the classification of CDs, and then provide comprehensive discussions regarding the relationships between their structure and afterglow properties. Furthermore, their potential applications in anti-counterfeiting, information protection, sensing, etc., are summarized. Finally, challenges and opportunities in the future of this newly emerged carbon-based afterglow material are proposed.
1. Introduction
As a new class of luminescent nanomaterials, carbon dots (CDs), sometimes being named carbon nanodots (CNDs), carbonized polymer dots (CPDs), carbon quantum dots (CQDs) and graphene quantum dots (GQDs),1–8 have attracted great attention in recent years due to their superior properties.2–5,7–14 Compared to traditional organic fluorescent dyes and semiconductor quantum dots, CDs have numerous advantages, such as facile preparation and purification, tunable photoluminescence (PL) emission and surface functional groups, and excellent photostability and biocompatibility. Consequently, they have demonstrated many potential applications in sensing, anti-counterfeiting, theranostics, catalysis, optoelectronic devices and so on.8,15–29 Although the regulation of the PL properties and potential applications of CDs have been widely explored in the past decade, their long-lived afterglow (i.e., phosphorescence and delayed fluorescence) related characteristics have received relatively less attention. Nevertheless, some efforts still have been devoted to the development of afterglow of CDs and a series of advances has been achieved.30–35It's well known that long-lived afterglow materials play significant roles in certain special fields such as anti-counterfeiting, bioimaging and warning signs, which exhibit more advantages than fluorescent materials.36–41 Nowadays, traditional afterglow materials mainly include rare-earth based phosphors, transition metal based complexes and pure organic compounds.39,42–47 These afterglow materials, however, suffer from the following disadvantages: (i) expensive raw materials, high energy consumption, and/or complicated preparation and purification procedures are usually required in preparation; (ii) stringent conditions are required for effectively producing afterglow, such as the bulk form for rare-earth based afterglow materials and the crystalline state for pure organic compound based afterglow materials; and (iii) generally no afterglow can be observed in solution or the dispersion state.39,42–46,48–51 All these disadvantages greatly hindered their further applications. Therefore, the developments of new types of afterglow materials with facile preparation and superior properties are still highly desirable.
In light of their scientific significance and advanced applications, CDs could be regarded as promising candidates for afterglow materials thanks to their merits of low-cost preparation, excellent biocompatibility and facile activation of afterglow,12,18–20,25,27,52–56 which could evade the shortcomings of traditional afterglow materials. Moreover, the flexibility for design and preparation of CDs endows them with more possibilities in modulation of their afterglow properties (e.g., achieving multiple emission and stimuli responses) and thus could extend the range of their applications.32,33,57–62 To date, numerous review articles have been published to summarize the advances of synthesis methods, PL property regulation and potential applications of CDs.11,12,16,49,52,53,57,63,64 To the best of our knowledge, however, there have been no review articles that discuss the advances of the afterglow properties of these materials. To drive better developments of CD-based afterglow materials, it's significant to summarize the corresponding achievements. In this review article, we firstly provide a brief introduction about the definition, classification and chemical structures of this class of carbon-based luminescent nanomaterials (Section 2). Then, the strategies for realizing afterglow emission and potential applications of these carbon-based afterglow nanomaterials in anti-counterfeiting, information encryption and sensing are comprehensively summarized (Sections 3–5). Finally, challenges of and perspectives on CDs in terms of their afterglow properties are proposed, including possible design strategies and research directions (Section 6).
2. Definition and classification of CDs
There are a lot of debates about the definition and classification of CDs. In a mostly acceptable sense, CDs could contain a broad range of carbon-based luminescent nanomaterials with at least one dimension less than 10 nm, consisting of sp2/sp3 carbon and being doped with heteroatoms like oxygen, nitrogen, and/or sulfur. In terms of their morphologies and chemical structures, CDs have recently been divided into the following three types: CPDs, CQDs and GQDs, which could cover almost all kinds of the reported cases.1 In general, CPDs and CQDs are spherical or quasi-spherical nanoparticles,65,66 but CPDs are composed of amorphous carbon structures or contain a partially crystalline sp2 carbon core surrounded by polymer as a shell, and CQDs are regarded to be composed of almost complete sp2 carbon cores with functional groups anchored on their surfaces.12,14,52,54,67 In addition, GQDs usually refer to nano-sized graphene (monolayer or few layers) with a perfect sp2 carbon structure.68–70 Although CPDs, CQDs and GQDs have distinct structures, they could be collectively called CDs.1–8 Note that the above classifications are not always uniform in the literature and researchers sometimes use random names for their products.71–75 Noteworthily, all of these carbon-based nanomaterials usually have various functional groups such as amino, hydroxyl and/or carboxyl anchored onto their surfaces, endowing them with excellent water solubility and potential for further modification.57,76–79 Last but not least, the introduction of heteroatoms in these materials has been proved to be capable of regulating their PL properties, including improving PL quantum yields (QYs) and tuning emission wavelengths.57,65,663. Afterglow properties of CDs
In recent years, PL related studies about CDs, including their emission property regulation, mechanism and potential applications, have received considerable attention and great advances have been achieved.13,17–20,23,26,27,80,81 By choosing appropriate carbon precursors and preparation methods, PL emission of CDs covering the whole visible to near infrared (NIR) light regions with respectable QYs has been realized, and the corresponding mechanism and applications have also been proposed.5,11,12,14,52,55,63,82 Despite the great achievements in the PL properties and applications of CDs, studies regarding their afterglow phenomenon are still in infancy. Nevertheless, some efforts have been devoted towards realizing afterglow and further exploring their potential applications.30–35 From the mechanistic point of view, the long-lived afterglow of these carbon-based nanomaterials is mainly attributed to their excited triplet state related emissions, i.e., phosphorescence (radiative transition from the lowest triplet state (T1) to ground state (S0)) and delayed fluorescence (DF) (radiative transition from the lowest singlet state (S1) to S0via the reverse intersystem crossing (RISC) process) (Fig. 1).44,83,84 From the literature, it is clear that the afterglow emission of CDs is usually considered to have resulted from the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and CN relevant moieties due to their strong spin–orbit coupling, which are beneficial for production of excited triplet states more effectively via intersystem crossing (ISC).30,85–87 Moreover, by doping heteroatoms such as N, P and halogens, the n → π* transitions of CO and CN could be further promoted, which could favour the ISC process, consequently increasing the portion of triplet excitons.32,43,84,88 It should be noted that, however, in order to produce room temperature afterglow from CDs, matrices have usually been utilized to immobilize the materials and thus to stabilize the produced T1 species, so as to make radiative transitions possible from the spin-forbidden process (i.e., from T1 to S0).31,34,35,39,43,61,62,84,86,87,89–101 More interestingly, self-protective CDs (without matrix immobilization) with RTP have been achieved very recently through appropriate design.32,33,93,102–104
O) and CN relevant moieties due to their strong spin–orbit coupling, which are beneficial for production of excited triplet states more effectively via intersystem crossing (ISC).30,85–87 Moreover, by doping heteroatoms such as N, P and halogens, the n → π* transitions of CO and CN could be further promoted, which could favour the ISC process, consequently increasing the portion of triplet excitons.32,43,84,88 It should be noted that, however, in order to produce room temperature afterglow from CDs, matrices have usually been utilized to immobilize the materials and thus to stabilize the produced T1 species, so as to make radiative transitions possible from the spin-forbidden process (i.e., from T1 to S0).31,34,35,39,43,61,62,84,86,87,89–101 More interestingly, self-protective CDs (without matrix immobilization) with RTP have been achieved very recently through appropriate design.32,33,93,102–104
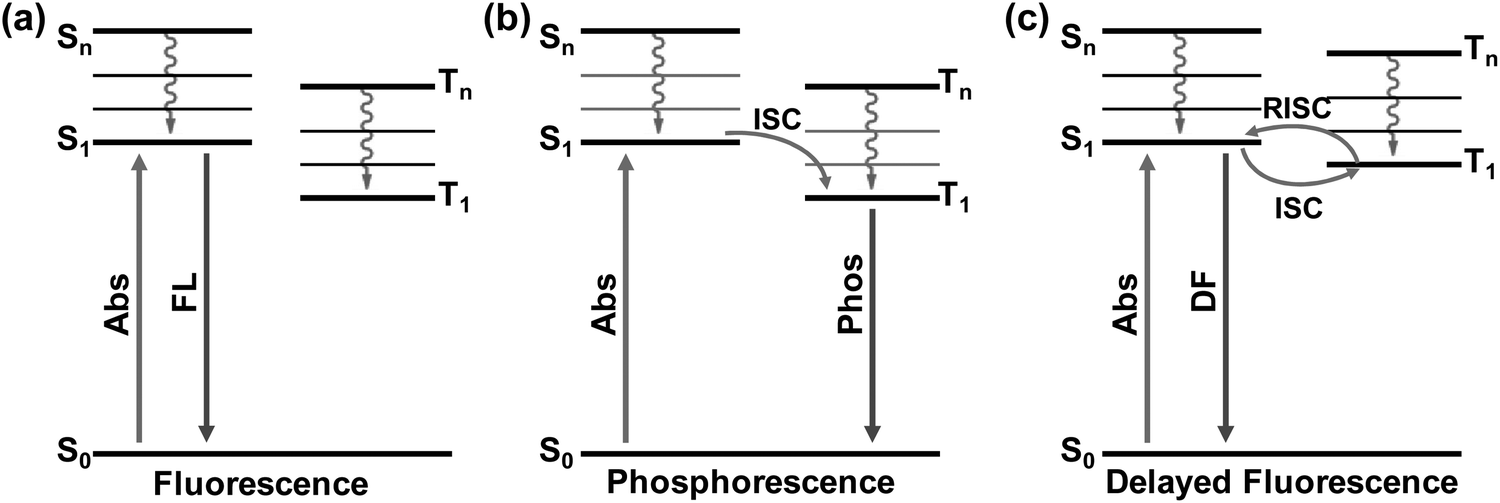 | ||
| Fig. 1 The schematic illustration of the emissive processes of fluorescence (a), phosphorescence (b), and delayed fluorescence (c). | ||
3.1 Room temperature phosphorescence (RTP)
In general, phosphorescence is produced from a spin-forbidden radiative transition (i.e., from T1 to S0) of organic compounds, which shows obvious emission wavelength red-shifts in comparison with fluorescence because of the smaller energy gap between T1 and S0 than that between S1 and S0 (Fig. 1). To generate RTP from organic compounds, effective ISC is critical and meanwhile nonradiative relaxation from T1 has to be evaded. Therefore, RTP of CDs is usually realized in a solid-state matrix that can strongly fix their luminescent subunits and facilitate the radiative relaxation from T1 to S0.44,83,84 As shown in Fig. 2a, Zhao et al. first reported the RTP of CDs by embedding CDs into a poly(vinyl alcohol) (PVA) matrix with a phosphorescence lifetime of 380 ms.31 Moreover, the CO bonds on the surfaces of the CDs were proved to be the origin of the observed RTP emission due to the strong spin–orbit coupling of aromatic carbonyls. Note that no obvious RTP emission was observed when the CDs were dispersed in water and other polymer matrices such as cellulose and polyethylene glycol. So, the PVA was confirmed to play an important role in the generation of RTP of the CDs, attributed to the formation of stabilized networks by hydrogen bonds between the hydroxyl groups on PVA and the CO groups on the CDs, which could obviously prevent the nonradiative relaxation of their T1 states. Likewise, RTP phenomena have also been observed from other CDs by dispersing them into polymer matrices, such as PVA, polyacrylic acid (PAA), polyacrylamide (PAM) and polyurethane (PU) (Fig. 2b).59,61,62,86,94,100,105–107 All these cases were considered to be the same as above due to the formation of hydrogen bonds to fix the luminescent subunits of CDs and consequently trigger RTP. Noteworthily, the CDs that were observed to exhibit RTP in polymer matrices usually contain abundant CO and/or CN groups, which, on one hand, could effectively produce excited triplet states via the ISC due to their strong spin–orbit coupling, and, on the other hand, could form hydrogen bonds between O or N elements and appropriate polymers.42,108–111 As a result, the formation of hydrogen bonds is critical for achieving RTP from certain CDs by suppressing the nonradiative relaxations of the excited triplet species.
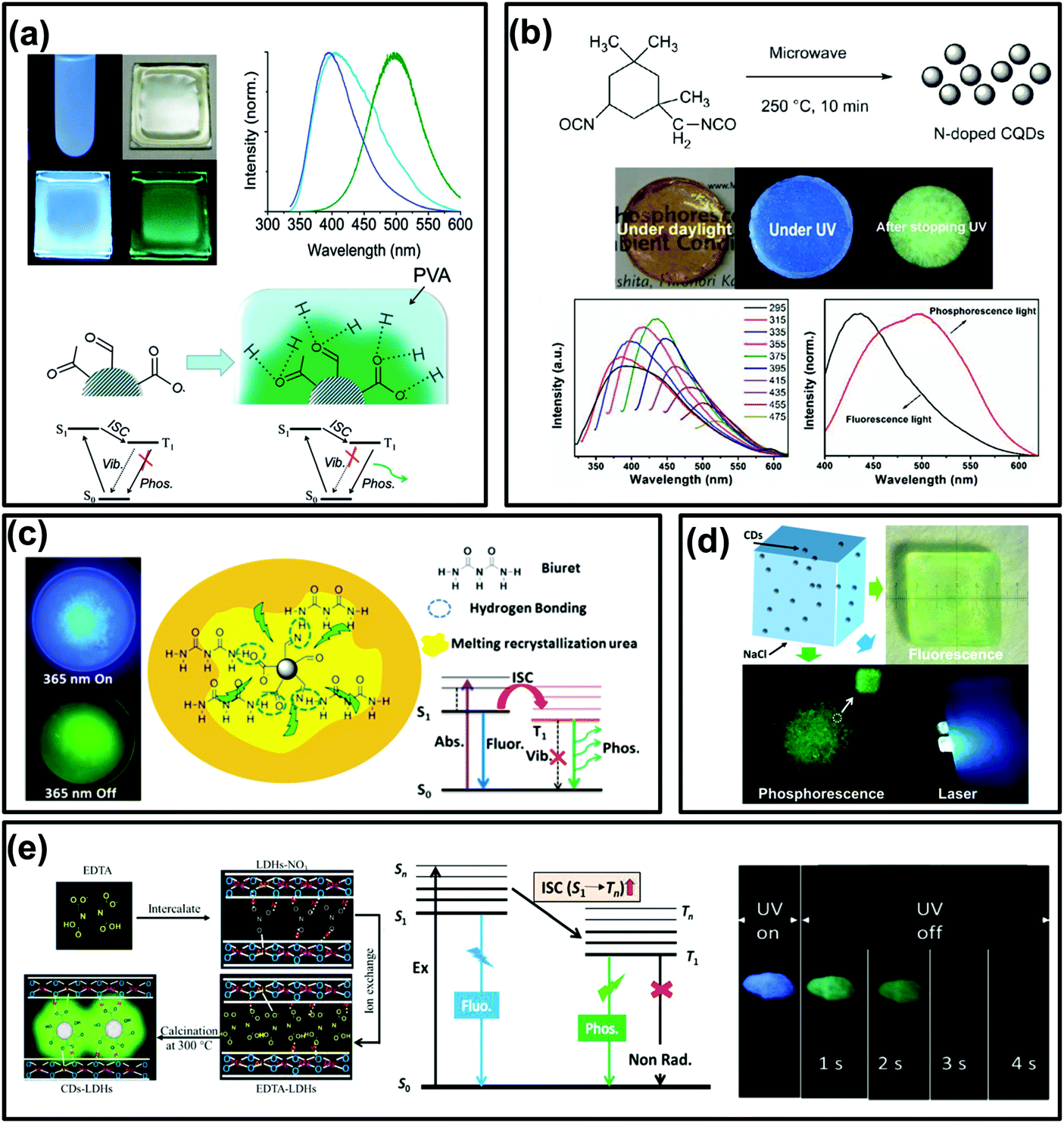 | ||
| Fig. 2 The RTP emissions of CD-based materials. (a) Images and the corresponding spectra of UV excited CDs (in water) and a CD–PVA film under daylight, the formation of hydrogen bonds between PVA and CDs, and the proposed mechanism of RTP emission, respectively. Reprinted from ref. 31 with permission of copyright 2013 Royal Society of Chemistry. (b) Images of the prepared N-doped CQDs re-dispersed in a polymer (PU) matrix under daylight, and with UV light on and off, and their fluorescence and RTP spectra, respectively. Reprinted from ref. 86 with permission of copyright 2016 Royal Society of Chemistry. (c) The images of samples exhibiting FL and RTP emission, and the corresponding transition processes of the formed NCD composite, respectively. Reproduced from ref. 87 with permission of copyright 2016 American Chemical Society. (d) Images of hybrid NaCl crystals and CDs with and without UV radiation and under pumped laser excitation, respectively. Reproduced from ref. 96 with permission of copyright 2017 American Chemical Society. (e) Diagrams of the formation process, the energy level of the photophysical process of CDs–LDHs, and images of CDs–LDHs under UV light on and off, respectively. Reprinted from ref. 99 with permission of copyright 2017 Royal Society of Chemistry. | ||
In many previous studies, the confinement effects of crystalline structures have been demonstrated to be an efficient means for rigidifying T1 species and preventing their nonradiative relaxation, thereby generating RTP.39,42,44,46,112,113 According to this idea, constructing crystalline embedded CDs might be an alternative strategy to obtain CD-based RTP composites. For instance, Zhou et al. fabricated a phosphorescent material through simple heat treatment of urea and N doped CDs (NCDs) (Fig. 2c). The results indicated that CN bonds on the surfaces of the NCDs could be the origin of the observed RTP.87 Moreover, the recrystallized urea and biuret acted as matrices and played an important role in preventing the vibration dissipation of T1 excitons through the formation of crystallized urea and hydrogen bonds between the biuret and NCDs.35,87 Similarly, other crystalline structures, such as NaCl (Fig. 2d),96 Al2(SO4)3,97 KAl(SO4)2·xH2O,101 zeolites,90,114 alkaline earth (Ca, Sr, and Ba) carbonate,92 layered double hydroxides (LDHs) (Fig. 2e),91,95,99,115 boric acid and molten salt,34,89 were also employed to prepare CD-based RTP composites.
3.2 Thermally activated delayed fluorescence (TADF)
As shown in Fig. 1c, if the energy gap (ΔEST) between T1 and S1 is small enough (generally <0.2 eV), the triplet excitons could effectively transfer back from the T1 to S1 excited states (i.e., the RISC process) due to thermal effects, then a radiative transition occurs from S1 to S0. Such an approach is usually called TADF,83,116–118 which displays an identical emission spectrum to that of fluorescence but with a much longer emissive lifetime.119–123 The TADF phenomenon has also been observed from carbon-based luminescent nanomaterials. For example, Tetsuka et al. found that the spin–orbit coupling of amino-functionalized GQDs (af-GQDs) could be enhanced due to the strong resonance between the delocalized π orbital of GQDs and the molecular orbital of –NH2, leading to an effective ISC process. They first observed extraordinarily long delayed fluorescence of GQDs with lifetimes up to a sub-second level (Fig. 3a).124 Later, Ghosh et al. also reported the room temperature delayed fluorescence emission of GQDs in water and dimethyl formamide (DMF) with a relatively short lifetime (10 ± 2 μs).125 In addition, Ding et al. synthesized a class of water-soluble CDs and their small ΔEST value (0.16 eV) was favourable for RISC from T1 to S1 excited states, showing a long-lived TADF at 77 K (Fig. 3b).126 Although the TADF of CDs was claimed to be observed, it is still highly desirable to explore new strategies to achieve long-lifetime TADF under ambient conditions and then to boost their applications. Recently, Yu et al. developed a so-called “dots-in-zeolites” route to facilely prepare CD@zeolite composites.127 They found that the CDs confined in zeolite matrices exhibit an efficient TADF emission with a lifetime of 350 ms, attributed to the effective stabilization of the T1 excited state and RISC process of the CDs (Fig. 3c). The nanometer confined space of zeolites enabled not only the RISC process for TADF, but also the TADF performance in an ambient atmosphere by isolating the excited states from oxygen. Moreover, the abundant dangling hydroxyl groups in the interrupted framework of zeolites could form hydrogen bonds with the CO and CN groups of the CDs, which is another factor in stabilizing the excited triplet species. More recently, Liu et al. demonstrated that room temperature TADF could also be achieved by incorporating CDs into polymers (i.e., PVA) with the assistance of electrospinning technology. According to their investigations, the formation of an ordered mesoporous structure within the CD/PVA nanofibers plays a critical role in further stabilizing the triplet states of CDs, resulting in an effective RISC process for TADF emission.94 In addition, Lin et al. reported the preparation of room temperature long lifetime (0.703 s) afterglow materials via covalent immobilization of CDs onto colloidal nanosilica (nSiO2) (Fig. 3d).85 Different from hydrogen-bond fixation induced RTP from the same CDs (i.e., by PVA), the formation of covalent bonds between CDs and nSiO2 evidently decreased the ΔEST from ∼0.5 eV to ∼0.3 eV, and consequently they confirmed that the afterglow emission consisted of predominantly delayed fluorescence and a portion of phosphorescence. Such a covalent bond fixing strategy not only provides an alternative method to stabilize the excited triplet states of the CDs (i.e., instead of commonly used hydrogen bonds), but could also tune the afterglow properties by altering their ΔEST value. Importantly, they found that the as-prepared CD@nSiO2 composite shows strong afterglow emission even in aqueous media. This very unique phenomenon was attributed to the strong covalent bond fixing emissive species. Actually, this is a significant advantage of the covalent bond fixing in comparison to other weak interaction fixing strategies (e.g., hydrogen bonding), and the latter would be easily destroyed in the presence of water and thus the afterglow properties cannot be employed in aqueous media.
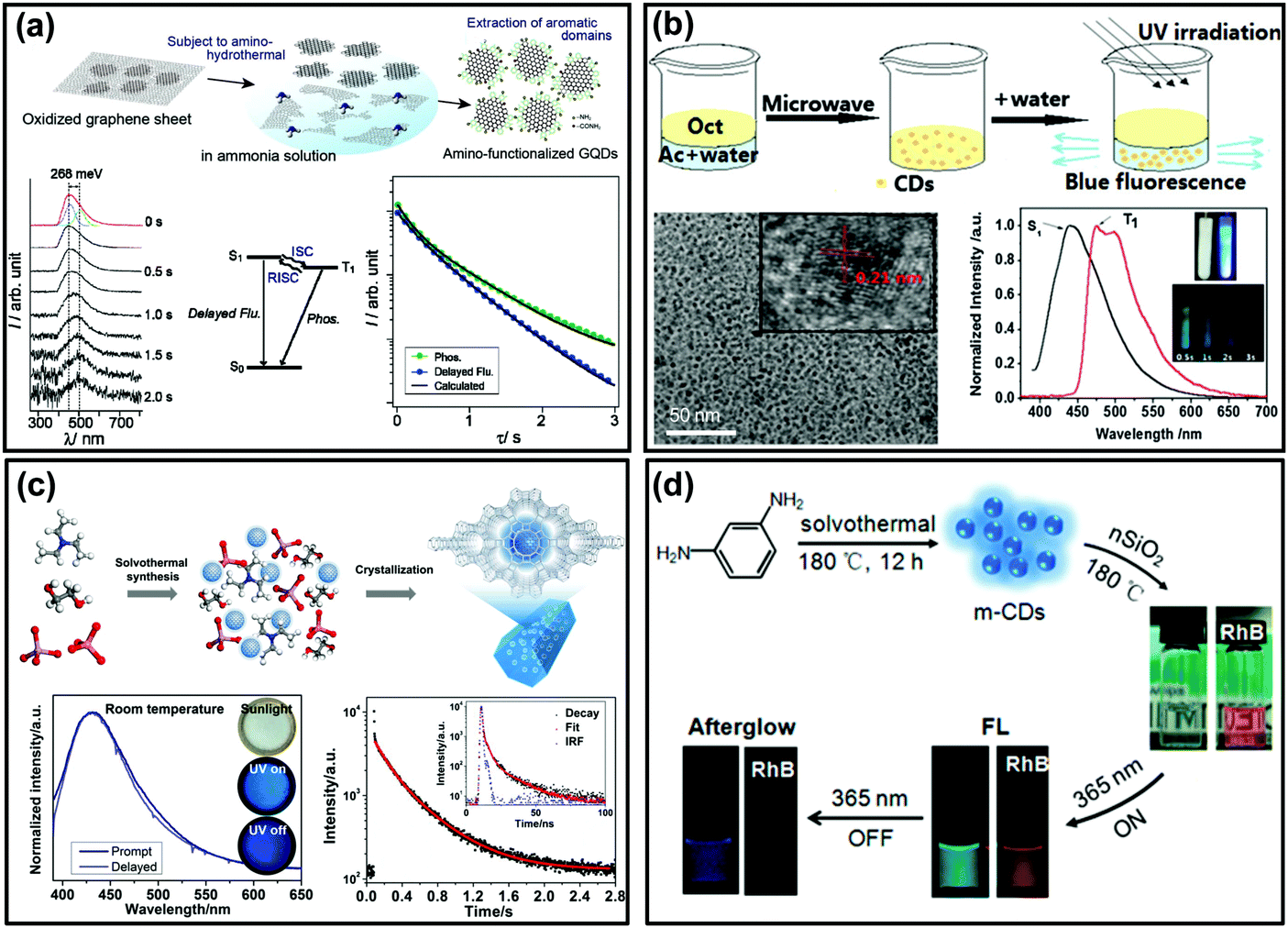 | ||
| Fig. 3 The TADF emission of CD-based materials. (a) The preparation process of af-GQDs and their time-resolved luminescence spectra, Jablonski diagram and decay curves. Reprinted from ref. 124 with permission of copyright 2012 Wiley-VCH. (b) Microwave assisted hydrothermal method for the preparation of crystallized CDs and their TADF emission spectra and images of the sample at 77 K. Reprinted from ref. 126 with permission of copyright 2015 Royal Society of Chemistry. (c) The formation process of the CD@AIPO-5 composite and its images under daylight, and UV light on and off, the corresponding luminescence spectra, and time-resolved decay spectra, respectively. Reprinted from ref. 127 with permission of copyright 2017 Science AAAS. (d) Schematic illustration of the synthesis of CDs with nSiO2 (i.e., m-CDs@nSiO2) for afterglow emission in water. Reproduced from ref. 85 with permission of copyright 2017 American Chemical Society. | ||
4. Structure effects on the afterglow of CDs
According to the above discussion, the afterglow of CDs is greatly affected by their chemical structures, and such control would provide possibilities for achieving spin–orbit coupling and harvesting triplet excitons more effectively by facilitating the ISC process. Therefore, relationships between the chemical structures of CDs and their long lifetime afterglow emissions should be investigated, and this might be helpful for designing and exploiting more efficient CD-based long lifetime afterglow materials.4.1 Halogen doping for improving the afterglow performances of CDs
Heavy atom effects (e.g., halogens) have long been established to be an efficient strategy for improving phosphorescence properties in numerous studies.36,39,42,43,84,108 To obtain halogen doped CDs, Geddes et al. developed a method to incorporate bromine and iodine into CDs which exhibited unique long-lived (92 ± 7 μs) phosphorescence emission.128 However, no phosphorescence was observed if halogens were not incorporated, indicating the vital role of doping halogens into CDs in activating phosphorescence. Therefore, the long-lived phosphorescence of their CDs was ascribed to the heavy atom effects. Halogenated CDs could also be prepared by using some specific precursors containing halogens.103,129 As reported in a previous study, Wang et al. provided an example of preparing bromine doped CDs using 3-bromophenol by a hydrothermal method.129 The RTP emission with a lifetime of 3.7 ms was considered to have resulted from the aromatic ring and doped bromine atom. In addition, Feng et al. prepared fluorine and nitrogen co-doped CDs (FNCDs) via one-step solvothermal treatment of glucose and (C2H5)3N·3HF.103 Although the abundant C–N/CN bonds effectively facilitated the n → π* transitions for the generation of RTP, the emerged C–F bonds with steric protection also greatly contributed to the observed RTP by reducing the nonradiative transition of triplet excitons. These observations present a facile and novel strategy for achieving phosphorescence emission from CDs by doping halogens.
4.2 Polymer structure for self-protective RTP CDs
As mentioned in Wang's work, the polymer-like structure of CDs could act as a matrix to suppress the nonradiative relaxation of whose contained emissive moieties, and such specific chemical structures were confirmed to play a critical role in activating their RTP.129 This idea provides an innovative strategy to prepare self-protective RTP CDs. Impressively, self-protective RTP of polymer-like CDs has emerged in recent years. In Liu's report, the as-synthesized self-quenching-resistant nitrogen doped CDs prepared from PVA and ethylenediamine show surprising aggregation-induced RTP features in the solid state.130 It was proposed that the polymeric structure of CDs originated from the polymer precursor; meanwhile, the large amount of PVA-chains on their surfaces played a role of moisture resistance and behaved as a rigidification matrix and an oxygen barrier, thereby giving rise to RTP (Fig. 4a). More interestingly, Song et al. developed a seeded growth method to prepare CDs by controlling the number of seeds and reaction time, which was demonstrated to be an effective way to tune their optical properties. The as-prepared CDs showed not only color-tunable fluorescence by increasing the size of the seed CDs, but also color-tunable RTP over a broad spectral range of 500–600 nm. The resultant RTP was attributed to the presence of the nitrogen-containing groups and the polyvinylpyrrolidone polymer chains at the surfaces of the CDs.102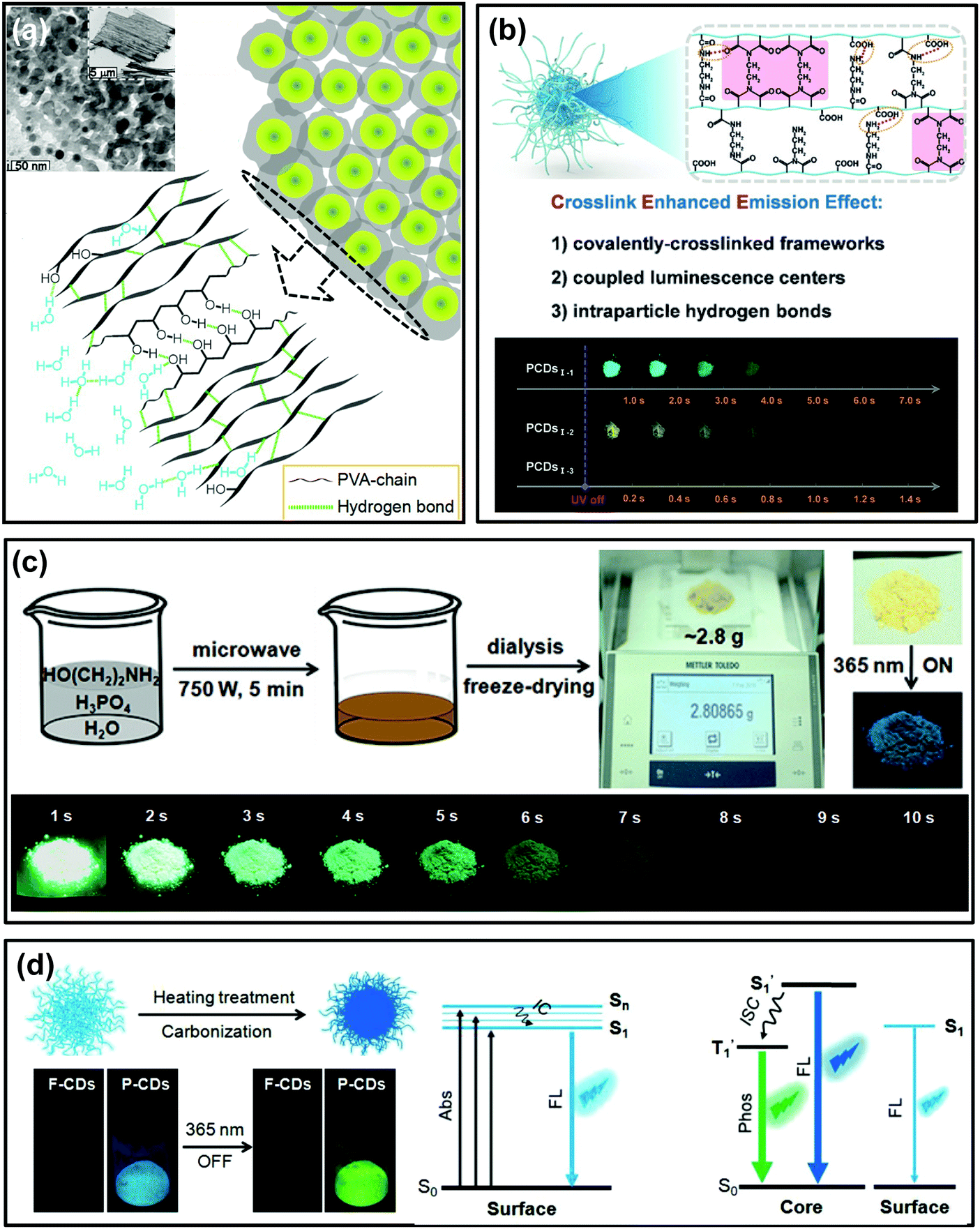 | ||
| Fig. 4 Polymer-structured CDs for self-protective RTP. (a) TEM image and schematic diagram of PVA-chain induced self-protective RTP emission. Reprinted from ref. 130 with permission of copyright 2017 Royal Society of Chemistry. (b) Schematic illustration of the CEE effect and the corresponding photographs of the obtained CDs (PCDs1–1, PCDs1–2, and PCDs1–3) at different delay times after UV irradiation. Reprinted from ref. 33 with permission of copyright 2018 Wiley-VCH. (c) The gram-scale preparation process for ultralong RTP (URTP) CDs and the images of the purified URTP CD powder under daylight, and under a 365 nm UV lamp (on and off), respectively. Reprinted from ref. 32 with permission of copyright 2018 Wiley-VCH. (d) Schematic illustration of the conversion from F-CDs to P-CDs, and their FL and RTP images and the corresponding energy level of the photophysical process. Reprinted from ref. 60 with permission of copyright 2018 Wiley-VCH. | ||
Recently, Yang et al. verified that both the polymer-like structure and crosslink enhanced emission (CEE) effects contributed to the generation of RTP in CDs.33 After carefully selecting the precursors, polymer-like structured CDs with abundant amides or imides were directly constructed through appropriate synthesis routes. The formed covalently-crosslinked framework and intraparticle hydrogen bonds greatly suppressed the nonradiative relaxation of luminescent centers in CDs, and thus long lifetime RTP was achieved (Fig. 4b). This result is believed to provide potential routes for preparation of polymer-like-structured CDs with different RTP properties.
Subsequently, considering the general prerequisites for achieving efficient RTP, Lin et al. reported a facile and gram-scale preparation method for ultralong lifetime RTP CDs via microwave-assisted heat treatment of ethanolamine and phosphoric acid in a few minutes (Fig. 4c).32 The as-prepared CDs showed mainly an amorphous polymer-like structure and exhibited a long RTP lifetime of up to 1.46 s (more than 10 s to the naked eye). Importantly, no RTP can be observed in the prepared CDs under the same reaction conditions but using ethylene glycol to replace ethanolamine or using sulphuric acid to replace phosphoric acid, respectively. Thus, the doping N and P elements are critical for production of RTP, because these elements are known favoured n → π* transition and consequently accelerated ISC process. Consequently, triplet excitons were effectively generated and activated RTP. Furthermore, the intraparticle hydrogen bonds were proposed to play a significant role in producing RTP as well, which could be confirmed by heating treatment of the fluorescent CDs (F-CDs) that were prepared from the same raw materials at a relatively low temperature (i.e. 180 °C). During the heating treatment process (i.e. 280 °C), a large number of intertwined polymers of F-CDs were further crosslinked, dehydrated and carbonized, and finally phosphorescent CDs (P-CDs) formed with compact cores and the excited triplet species were self-stabilized due to the intraparticle hydrogen bonds (Fig. 4d).60
More recently, Kang et al. reported a new strategy to prepare RTP CDs by adding acrylamide to the reaction system. They found that not only does the doping nitrogen element in the CDs effectively increase their fluorescence intensity, but also the produced CN bonds promote the formation of triplet excitons. More importantly, the acrylic amide and the as-generated polyacrylamide on the surfaces of the CDs could easily connect with the as-prepared pyridinic N via the reaction between citric acid and urea, and the formed hydrogen bonds could stabilize the triplet excitons. Hence, the as-obtained CDs exhibited intense RTP emission.131
5. Applications of CD-based afterglow
5.1 Anti-counterfeiting
Counterfeiting is a growing problem that floods around the world. Thus, many efforts have been made towards developing advanced strategies to protect valuable items from being replicated.132–135 Anti-counterfeiting labels are considered to be one of the most efficient ways against counterfeiting by taking advantage of high throughput, easy handling, and facile design, and thus have been widely adopted. CDs with room temperature afterglow characteristics offer a smart choice for use in anti-counterfeiting.31–35,59,60,87,89,93,95,127As illustrated in Fig. 5a, the CD–PVA composite was firstly demonstrated as a time-resolved anti-counterfeiting ink by handwriting numeric codes “60” on a plastic package with an afterglow emission for seconds. Besides, this composite was also applied as tags for information hidden, the written letters “CIOMP” were hardly observable under daylight and UV light due to the background fluorescence of the commercial printing paper, while they were distinctly recognized after switching off the UV light.31 More impressively, inspired by their different colours and long lifetimes of afterglow, CD-based RTP or TADF materials could provide advanced methods for anti-counterfeiting. As shown in Fig. 5b, a dual mode security application based on the TADF of CDs has been realized. Specifically, the flower and stem parts of the pattern were printed with TADF materials (CDs@2D-AIPO) and benzil dye molecules, respectively. Under UV light excitation, the whole pattern exhibited blue and green FL from each part. When the UV excitation was turned off, only the flower part can be recognized by the naked eye due to its long lasting TADF.127 In another example, a colourful butterfly was drawn with both commercial fluorescent materials and long lifetime afterglow CDs on paper. Under UV excitation, the butterfly pattern emitted multi-colour FL; after ceasing the excitation, the encrypted part of the butterfly emerged with green RTP that lasted for several seconds (Fig. 5c).33 Similarly, when a quick response (QR) code was printed onto commercial A4 paper using ultralong RTP CD based ink, the invisible correct code can be observed only with the UV lamp being shut down (Fig. 5d).32 More recently, newly emerged CD-based composites with multicolour afterglow have also been applied for security protection, making CDs promising functional materials for anti-counterfeiting.34,35
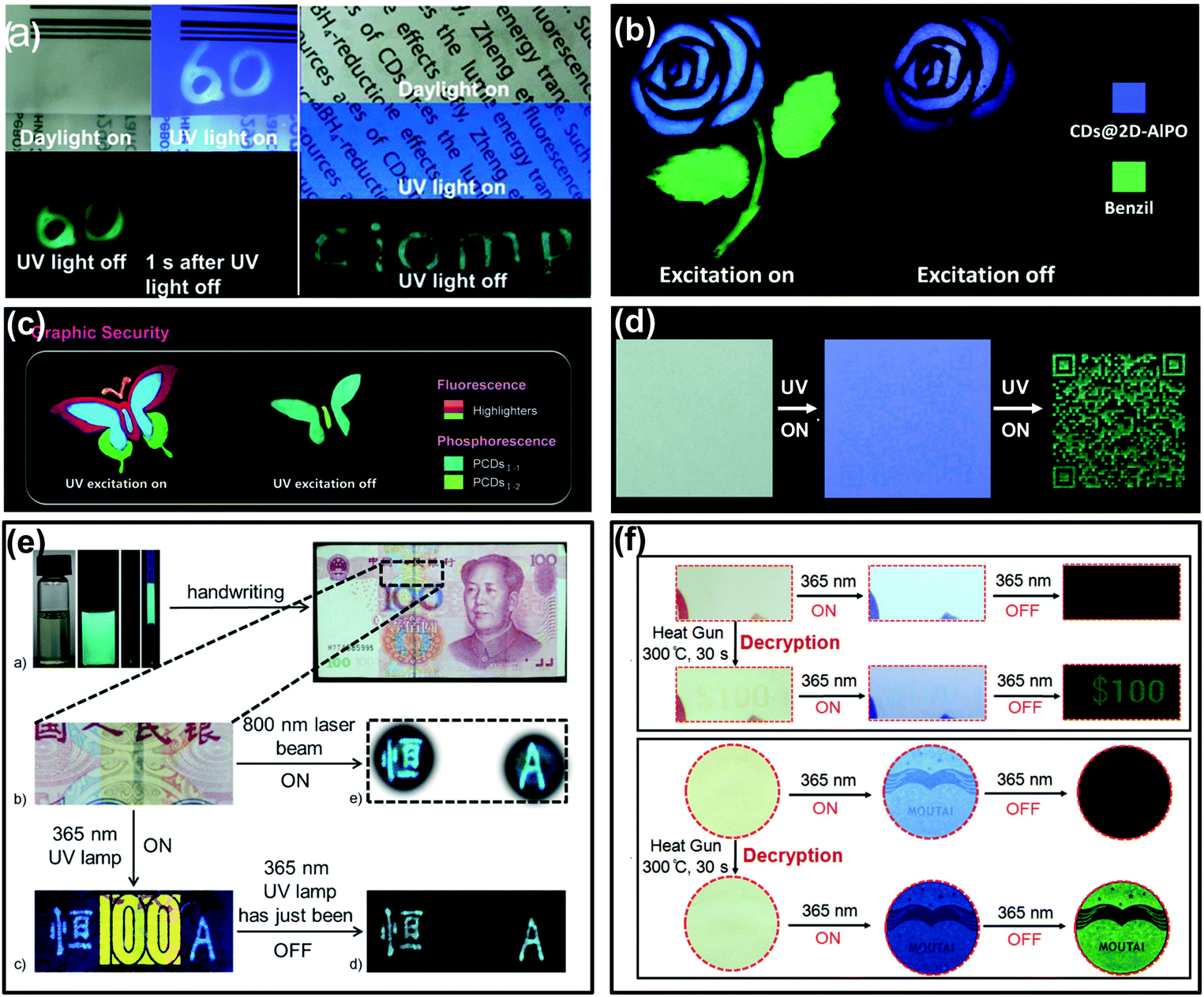 | ||
| Fig. 5 Anti-counterfeiting applications of the afterglow of CDs. (a) The demonstrated applications of the CD–PVA composite in security and information hiding. Reprinted from ref. 31 with permission of copyright 2013 Royal Society of Chemistry. (b) The application in dual-responsive security protection using TADF of CDs@2D-AlPO and fluorescence of commercial benzil dye molecules. Reprinted from ref. 127 with permission of copyright 2017 Science AAAS. (c) Applications in Graphic Security using PCDsI-1, PCDsI-2, and commercial highlighters. Reprinted from ref. 33 with permission of copyright 2018 Wiley-VCH. (d) Ultralong RTP CD based security inks for hiding and identifying the printed QR code. Reprinted from ref. 32 with permission of copyright 2018 Wiley-VCH. (e) The triplet mode emission of m-CD–PVA composites in anti-counterfeiting applications in paper money. Reprinted from ref. 59 with permission of copyright 2016 Wiley-VCH. (f) The heating responsive afterglow of F-CDs for identifying the authenticity of the coupon and the Moutai Liquor as a new kind of anti-counterfeiting method. Reprinted from ref. 60 with permission of copyright 2018 Wiley-VCH. | ||
The triple-mode emission properties of CDs were also used for advanced anti-counterfeiting.59 As shown in Fig. 5e, the characters “heng” in Chinese and “A” in English were handwritten on a banknote (RMB) using m-CDs (prepared from m-phenylenediamine) dispersed in PVA aqueous solution. The blue FL and cyan upconversion photoluminescence of the two characters were observed upon irradiation with a 365 nm UV lamp and an 800 nm femtosecond pulse laser, respectively. In addition, once the UV light was switched off, the characters showed long-lasting blue-green afterglow (RTP), while the yellow fluorescence anti-counterfeiting mark “100” from the banknote itself disappeared immediately. Based on this unique triple-mode emission feature, the m-CD–PVA composite holds promising prospects in security applications, especially rare triple-mode optical authentication.
More recently, stimuli-responsive afterglow of CDs was also employed in the anti-counterfeiting field. Lin et al. demonstrated that polymer-like-structured CD (F-CD) aqueous dispersion could be used as a heat-stimuli-responsive security ink toward anti-counterfeiting.60 As shown in Fig. 5f, the digit pattern “$100” printed on a coupon and the logo of Moutai Liquor printed on wood paper using the F-CDs as ink were almost invisible under daylight. Under UV excitation, the logo of Moutai Liquor was recognizable due to the FL emission of the F-CDs, but the printed digit pattern “$100” was hardly identified because of the strong background FL of the commercial paper. In addition, both the digit pattern and the logo cannot be seen upon ceasing the UV excitation. However, if the digit pattern and the logo were treated using a heat gun for 30 s, the “$100” on the coupon and the logo of Moutai Liquor can be identified, attributed to the heating treatment that converted the fluorescent CDs (i.e., F-CDs) into phosphorescent CDs (i.e., P-CDs). Such a heat-stimuli-responsive production of long-lived afterglow emission offers a facile and high-level anti-counterfeiting strategy.
5.2 Information encryption
Information encryption is a process of recognizing hidden information through certain stimuli-responses. Due to their potential multiple emissive and environmentally friendly features, CDs could be ideal candidates for the design of smart information encryption and decryption systems. The most distinct property of CDs is their interaction-dependent luminescence: production of afterglow through embedding, crosslink or immobilization.31,34,59,124,127 Consequently, CDs could show stimuli-responsive afterglow emission by moisture, pH or heating,60–62,85,91,136 which are especially suitable for their application in information encryption and decryption systems.Zhao et al. demonstrated the application of an ultralong and efficient RTP emissive nitrogen doped CD (NCD) composite in information encryption.87 As shown in Fig. 6a, to encrypt the security information “2468” in “12345678”, the “1357” was printed using a NCD–starch composite (with only fluorescence properties), while “2468” was patterned with a mixture of the RTP NCD composite and the NCD–starch composite. As a result, the hidden information (i.e., “2468”) cannot be recognized under either daylight or UV excitation, but which was readily visualized with the UV light being switched off. Similar modes were also demonstrated in other studies, as shown in Fig. 6b; letters “Just Love U” were written with short-lifetime fluorescent materials, among which “J L U” was filled with long-lived RTP CDs. Under 365 nm UV light excitation, the blue fluorescent “Just Love U” was observed. However, the hidden information “J L U” emerged as green phosphorescence which lasted for several seconds when the excitation light was switched off.33 Likewise, as shown in Fig. 6c, the codes that were printed on wood paper using blue fluorescent b-CDs and ultralong RTP CDs were invisible under daylight, while upon excitation with a 365 nm UV lamp, false information (a blue emissive pattern of “8888”) on the non-FL background paper was observed. When the UV lamp was turned off, the hidden information (“0710”) could be clearly recognized due to the ultralong lifetime RTP nature of CDs.32
 | ||
| Fig. 6 Information encryption applications of afterglow of CDs. (a) Information encryption application of the phosphorescent N-CD composite. Reproduced from ref. 87 with permission of copyright 2016 American Chemical Society. (b) Photographs of information encryption made from PCDs1–1, and commercial FL CDs. Reprinted from ref. 33 with permission of copyright 2018 Wiley-VCH. (c) Data encryption based on the ultralong RTP of CDs and interfering fluorescent b-CDs. Reprinted from ref. 32 with permission of copyright 2018 Wiley-VCH. (d) Moisture-responsive reversible RTP-to-TADF switching model for information protection application (encryption and decryption). Reproduced from ref. 85 with permission of copyright 2017 American Chemical Society. (e) Heating conversion from FL to RTP of CDs for information encryption applications. Reprinted from ref. 60 with permission of copyright 2018 Wiley-VCH. (f) Photographs of the overlapped patterns realized with three kinds of composites after annealing at 80, 150, and 200 °C, under UV light excitation (left) and after switching the UV light off (right). Reprinted from ref. 62 with permission of copyright 2018 Wiley-VCH. (g) Fluorescence (“UV on”) and phosphorescence (“UV off”) images of 200 °C annealed CD@PVA composite films before and after being sprayed with water, and those of films further annealed at 100 °C, and the application of the temperature dependent RTP for information protection. Reprinted from ref. 61 with permission of copyright 2019 Royal Society of Chemistry. | ||
Advanced applications of CD-based afterglow have also been realised by taking advantage of their moisture sensitive or insensitive RTP properties. As described in Lin et al.'s report, the formed covalent bonds in the m-CD@nSiO2 composite offer stronger interaction in comparison to hydrogen bonds for stabilizing triplet states, which exhibits unique long lifetime afterglow in aqueous solution.85 As they described in their report, the green RTP from the m-CD@PVA composite disappeared after being wetted with moisture, but the blue afterglow from m-CDs@nSiO2–PVA was still observable. As a result, the hidden data or information that needs protection could be decrypted by observing the afterglow emission from wetting the filter paper (Fig. 6d). This work provides new insight into the application of the long afterglow of CDs in advanced information protection.
In another study of Lin's group, they have demonstrated the conversion of fluorescence CDs (F-CDs) to ultralong RTP CDs (P-CDs) by a further heating treatment.60 Consequently, they designed a heating-stimuli responsive strategy for information protection. As shown in Fig. 6e, the hidden information was first printed on a wood-free paper using F-CD ink, and then the interfering information was printed using a common ink with only blue fluorescence properties. Due to its ultralong lifetime, RTP emission could be observed from the F-CDs after been subjected to a heating treatment (e.g., heat gun for 30 s); the hidden information was observed only under the RTP mode.
Recently, Qu et al. investigated the effects of temperature on the RTP properties of a CD–PVA composite.61,62 They found that the RTP intensity of the CD–PVA composite could be improved to a recognizable level upon thermal treatment over 150 °C, and thus a concept of heating-stimuli responsive data encryption was proposed. As shown in Fig. 6f, three overlapped patterns were written with different composites and were gradually heated at 80 °C, 150 °C and 200 °C; the encrypted information (i.e., “CDots”) could only be observed under thermal treatment at 200 °C.62 Furthermore, RTP emissive films were prepared using the CD–PVA composite via post-synthetic thermal annealing at 200 °C. After quenching the RTP with water, the films could be used as “paper” for IR laser writing. Fig. 6g shows the characters “CD” written using an IR laser, which can be distinctly recognized by reading out the RTP signal.61
5.3 Sensing
The triplet state induced afterglow has been established for use in the optical sensing field due to the capability for effective avoidance of auto-fluorescence and scattering light from matrices, so it's possible to achieve higher sensing sensitivity than traditional fluorescence methods. In addition, afterglow emission is usually sensitive to oxygen, moisture and temperature.39,46,83,84 Therefore, CD based afterglow materials have been proposed in sensing fields.For example, Lu et al. applied LDHs as a matrix to immobilize CDs, and the as-prepared composite (i.e., CDs@LDHs) exhibited RTP with a lifetime of 386.6 ms.99 Based on the property of RTP being sensitive to oxygen, they observed that the phosphorescence intensity of CDs@LDHs gradually decreased upon increasing the concentration of oxygen from 0% (pure N2) to 100%, indicating a potential application for the detection of oxygen (Fig. 7a). In another study, CDs were fabricated as a fluorescent temperature sensor.95 However, Liu et al. observed that the phosphorescence exhibited different temperature-responsive behaviour from fluorescence. As shown in Fig. 7b, the intensity of phosphorescence and fluorescence showed double-exponential and linear decays, respectively, as the temperature increased from 30 to 90 °C, demonstrating that the phosphorescence is more sensitive to temperature than that of fluorescence.130
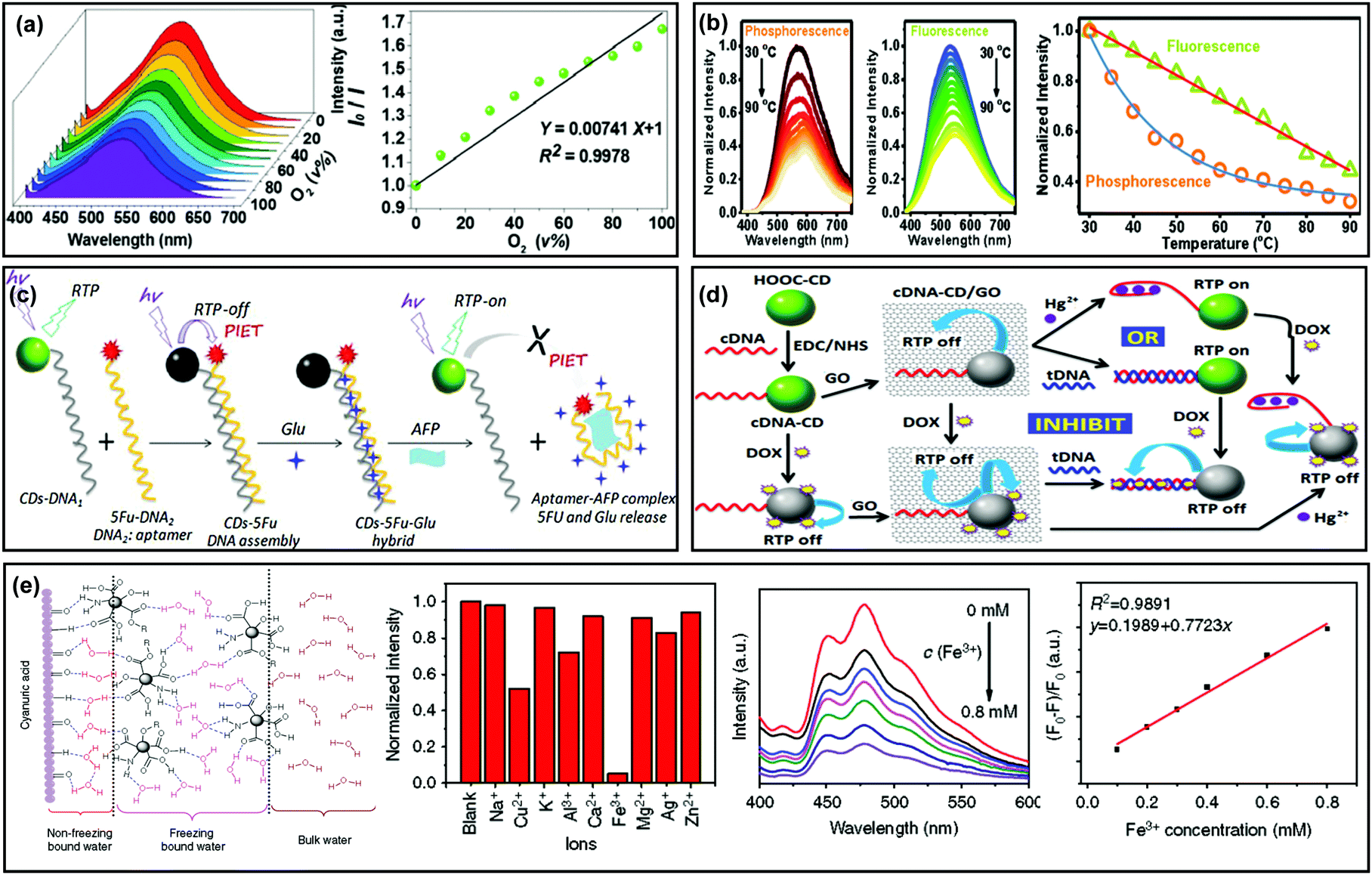 | ||
| Fig. 7 Sensing applications of the afterglow properties of CDs. (a) Phosphorescence emission spectra of CD–LDH composites at different oxygen concentrations. Reprinted from ref. 99 with permission of 2017 Royal Society of Chemistry. (b) Phosphorescence and fluorescence spectra of the NCD powder in the temperature range from 30 to 90 °C. Reprinted from ref. 130 with permission of 2017 Royal Society of Chemistry. (c) Preparation of CD–5Fu–Glu composites, and their applications in AFP sensing and glutamic release. Reprinted from ref. 139 with permission of copyright 2018 Elsevier. (d) RTP CD-based logic gates: preparation, Hg2+ sensing and doxorubicin release. Reprinted from ref. 140 with permission of copyright 2015 from the Royal Society of Chemistry. (e) Schematic illustration of molecular interactions between the CDs, CA particles and water molecules, and the analytical performance for Fe3+ detection based on the RTP CDs. Reprinted from ref. 141 with permission of copyright 2018 Nature. | ||
Benefitting from the abundant functional groups on the surfaces of CDs, they showed specific binding affinity to various metal ions. By using such a feature, CDs have been widely applied for the detection of metal ions (or certain analytes that can strongly bind with metal ions).15–17,63,78,80,137 Recently, sensing applications have been expanded by use of the RTP properties of CDs, which exhibited advantages for improving detection signal-to-noise ratios. For example, Chen et al. reported the RTP of a CD-based composite in aqueous media and the RTP intensity was found to be significantly quenched by the presence of metal ions due to the formation of non-luminescent chelates.138 Moreover, the quenched RTP of Fe3+–CDs was recovered upon addition of phosphate-containing molecules such as adenosine triphosphate (ATP), attributed to the higher affinity of Fe3+ to phosphate ions than CDs. Therefore, an “off–on” approach for ATP detection with a linear range from 20 to 200 mM and a detection limit of 14 mM was obtained. In addition, CD–DNA nanohybrids were designed and developed for the detection of proteins (e.g., alpha fetal protein, AFP) and metal ions (e.g., Hg2+) by the use of the RTP properties of CDs (Fig. 7c and d).139,140
Subsequently, Zhou et al. found that cyanuric acid (CA) could be applied to immobilize CDs through forming robust bridge-like hydrogen bonded networks, which strongly stabilized the CO bonds on the surfaces of the CDs (Fig. 7e). This finding offered a method to realize RTP of CDs in aqueous media. Furthermore, they demonstrated an application of their design for Fe3+ sensing by taking advantage of the RTP properties of CDs.141 Very recently, Yang et al. synthesized nitrogen and phosphorus co-doped CDs (P-CDs) by a facile microwave method, in which triethanolamine served as the carbon source and phosphoric acid as the dopant. The P-CDs showed both bright-blue fluorescence emission and green-RTP emission. Then, they applied the P-CDs to, respectively, assay pH ranging from 9.15 to 13.55 and 2.29 to 13.55 by virtue of their pH-responsive fluorescence and RTP.142 In another Yang et al.'s study, they prepared RTP CDs using a similar strategy (with n-aminoethyltrimethoxysilane as a carbon source and phosphoric acid as a dopant). The as-obtained CDs could interact with tetracyclines (TCs) due to the presence of –COOH and –OH groups on their surfaces, resulting in quenching of both fluorescence and RTP due to an inner filter effect. Therefore, a dual-channel strategy was established for assaying TCs.143
5.4 Others
Due to the highly efficient photosensitization effects of excited triplet species to molecular oxygen, the afterglow properties of CDs have been proposed for use in the fields of biotechnology, such as photodynamic therapy (PDT) and enzyme engineering.144,145 For example, Ge et al. presented a GQD-based PDT agent in 2014, which exhibited not only a broad absorption spanning from the UV to visible light regions, but also the capability to produce singlet oxygen (1O2) via a photosensitization process. Since the energy gap between T1 and S0 of the GQDs was larger than the formation energy of 1O2, their T1 states could behave as an extra pathway to participate in the generation of 1O2, resulting in an overall 1O2 production quantum yield even higher than 1.0. Therefore, GQDs could be potentially applied as a PDT agent, allowing simultaneous imaging and highly efficient cancer therapy.144In addition, Wu et al. developed a highly efficient photosensitizer by the use of phosphorescent CDs. Compared to traditional photosensitizers, such as other types of carbon nanomaterials and molecular photosensitizers, phosphorescent CDs exhibited higher photo-oxidation activity and the activities were found to be correlated well with their phosphorescence quantum yields. Thus, the reported CDs could act as a promising type of light-driven oxidase-mimicking nanozyme, and the generation of singlet oxygen was further explored for photodynamic antimicrobial chemotherapy.145
6. Summary and outlook
In recent years, some exciting advances in the long-lived room temperature afterglow of CDs have been achieved. Such novel optical properties made them suitable in diverse and significant fields of application. Benefiting from the numerous merits of CDs, especially facile preparation, low toxicity and abundant surface functional groups, they could represent one promising class of afterglow materials in the future. To realize afterglow of CDs, the ISC process should be improved and which is critical for production of triplet excitons more effectively. Meanwhile, post-treatments of CDs with hosts or dispersion into rigid matrices is usually required to stabilize and isolate the triplet excitons. Of course, self-protective RTP CDs could also be realized by appropriate designs. Note that for generating TADF, the ΔEST between the T1 and S1 excited states should be reduced to a relatively small value (i.e., <0.2 eV) for an efficient RISC process, which could be realized by strong interactions (e.g., covalent bonds) or embedding in specific matrices. These strategies about structure design and control for generation of long lifetime afterglow of CDs would open new directions for research and understanding their photophysical properties, which may have profound influences on developing pure organic afterglow materials.Although research studies have put many efforts towards the synthesis and application of CD based afterglow materials, they are actually still in their infancy stage. To drive further applications of these materials, it is proposed that particular attention be paid to the following aspects: (1) the emission efficiency (i.e., QYs) and lifetime of CD based afterglow materials should be further improved; (2) effective methods for achieving afterglow of CD based materials in aqueous media should be developed, and this is important for expanding their range of applications (e.g., bioimaging and sensing); (3) achievements of multicolor and oil dispersible self-protective room temperature afterglow of CDs are significant, which would be critical for their applications in optoelectronic devices; (4) expanding the excitation wavelength range for the afterglow of CD based materials from the UV region to the visible light region is necessary for their application in biologically relevant fields; and (5) preparation of CD based afterglow materials with unique properties, such as stimuli-response and excitation-wavelength dependence, is also interesting for expanding their applications.
To realize the abovementioned aims, appropriate carbon precursor selection, synthetic methods, structure design, post-treatment and adjustment of rigid hosts are necessary. Moreover, in-depth understanding of the afterglow emission mechanism and the relationships between structures and long-lived excitons in CDs are highly important, which would be favorable for their rational design, controllable synthesis and practical applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (51872300 and U1832110), the S&T Innovation 2025 Major Special Programme of Ningbo (2018B10054), the China Postdoctoral Science Foundation (BX20190338), and the W. C. Wong Education Foundation (rczx0800).Notes and references
- S. Tao, T. Feng, C. Zheng, S. Zhu and B. Yang, J. Phys. Chem. Lett., 2019, 10, 5182–5188 CrossRef CAS.
- K. Nekoueian, M. Amiri, M. Sillanpää, F. Marken, R. Boukherroub and S. Szunerits, Chem. Soc. Rev., 2019, 48, 4281–4316 RSC.
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS.
- O. Kozák, M. Sudolská, G. Pramanik, P. Cígler, M. Otyepka and R. Zbořil, Chem. Mater., 2016, 28, 4085–4128 CrossRef.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626–14637 CrossRef CAS.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620–1636 CrossRef CAS.
- L. Xiaoming, R. Muchen, S. Jizhong, S. Zihan and Z. Haibo, Adv. Funct. Mater., 2015, 25, 4929–4947 CrossRef.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- Y. Wang and A. Hu, J. Mater. Chem. C, 2014, 2, 6921–6939 RSC.
- R. Wang, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- Z. L. Wu, Z. X. Liu and Y. H. Yuan, J. Mater. Chem. B, 2017, 5, 3794–3809 RSC.
- L. Xiao and H. Sun, Nanoscale Horiz., 2018, 3, 565–597 RSC.
- K. J. Mintz, Y. Zhou and R. M. Leblanc, Nanoscale, 2019, 11, 4634–4652 RSC.
- X. Sun and Y. Lei, TrAC, Trends Anal. Chem., 2017, 89, 163–180 CrossRef CAS.
- M. Li, T. Chen, J. J. Gooding and J. Liu, ACS Sens., 2019, 4, 1732–1748 CrossRef CAS.
- S. Huang, W. Li, P. Han, X. Zhou, J. Cheng, H. Wen and W. Xue, Anal. Methods, 2019, 11, 2240–2258 RSC.
- K. O. Boakye-Yiadom, S. Kesse, Y. Opoku-Damoah, M. S. Filli, M. Aquib, M. M. B. Joelle, M. A. Farooq, R. Mavlyanova, F. Raza, R. Bavi and B. Wang, Int. J. Pharm., 2019, 564, 308–317 CrossRef CAS.
- M. Hassan, V. G. Gomes, A. Dehghani and S. M. Ardekani, Nano Res., 2018, 11, 1–41 CrossRef CAS.
- J. Du, N. Xu, J. Fan, W. Sun and X. Peng, Small, 2019, 15, 1805087 CrossRef.
- K. D. Patel, R. K. Singh and H.-W. Kim, Mater. Horiz., 2019, 6, 434–469 RSC.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- K.-W. Chu, S. L. Lee, C.-J. Chang and L. Liu, Polymers, 2019, 11, 689 CrossRef CAS.
- S. Sharma, V. Dutta, P. Singh, P. Raizada, A. Rahmani-Sani, A. Hosseini-Bandegharaei and V. K. Thakur, J. Cleaner Prod., 2019, 228, 755–769 CrossRef CAS.
- F. Yuan, S. Li, Z. Fan, X. Meng, L. Fan and S. Yang, Nano Today, 2016, 11, 565–586 CrossRef CAS.
- C. Hu, M. Li, J. Qiu and Y.-P. Sun, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- J. Li, B. Wang, H. Zhang and J. Yu, Small, 2019, 15, 1805504 CrossRef.
- M. Semeniuk, Z. Yi, V. Poursorkhabi, J. Tjong, S. Jaffer, Z.-H. Lu and M. Sain, ACS Nano, 2019, 13, 6224–6255 CrossRef CAS.
- T. Yuan, T. Meng, P. He, Y. Shi, Y. Li, X. Li, L. Fan and S. Yang, J. Mater. Chem. C, 2019, 7, 6820–6835 RSC.
- M. L. Mueller, X. Yan, J. A. McGuire and L.-s. Li, Nano Lett., 2010, 10, 2679–2682 CrossRef CAS.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751 RSC.
- K. Jiang, Y. Wang, X. Gao, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2018, 57, 6216–6220 CrossRef CAS.
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 CrossRef CAS.
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, Angew. Chem., Int. Ed., 2019, 58, 7278–7283 CrossRef CAS.
- C. Lin, Y. Zhuang, W. Li, T.-L. Zhou and R.-J. Xie, Nanoscale, 2019, 11, 6584–6590 RSC.
- H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601 CrossRef.
- Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, S. Liu, Q. Yu, Q. Zhao and W. Huang, Adv. Funct. Mater., 2016, 26, 4386–4396 CrossRef CAS.
- X. Zhou, H. Liang, P. Jiang, K. Y. Zhang, S. Liu, T. Yang, Q. Zhao, L. Yang, W. Lv, Q. Yu and W. Huang, Adv. Sci., 2016, 3, 1500155 CrossRef.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CrossRef CAS.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685 CrossRef CAS.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef.
- S. Mukherjee and P. Thilagar, Chem. Commun., 2015, 51, 10988–11003 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef.
- A. Lv, W. Ye, X. Jiang, N. Gan, H. Shi, W. Yao, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2019, 10, 1037–1042 CrossRef CAS.
- M. L. Liu, B. B. Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC.
- T. V. de Medeiros, J. Manioudakis, F. Noun, J.-R. Macairan, F. Victoria and R. Naccache, J. Mater. Chem. C, 2019, 7, 7175–7195 RSC.
- T. Feng, S. Zhu, Q. Zeng, S. Lu, S. Tao, J. Liu and B. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 12262–12277 CrossRef CAS PubMed.
- M. Shamsipur, A. Barati and S. Karami, Carbon, 2017, 124, 429–472 CrossRef CAS.
- X.-T. Tian and X.-B. Yin, Small, e1901803 Search PubMed.
- L. Li and T. Dong, J. Mater. Chem. C, 2018, 6, 7944–7970 RSC.
- F. Arcudi, L. Đorđević and M. Prato, Acc. Chem. Res., 2019, 52, 2070–2079 CrossRef CAS.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Adv. Mater., 2018, 30, 1800783 CrossRef.
- X. Bao, E. V. Ushakova, E. Liu, Z. Zhou, D. Li, D. Zhou, S. Qu and A. L. Rogach, Nanoscale, 2019, 11, 14250–14255 RSC.
- Z. Tian, D. Li, E. V. Ushakova, V. G. Maslov, D. Zhou, P. Jing, D. Shen, S. Qu and A. L. Rogach, Adv. Sci., 2018, 5, 1800795 CrossRef.
- X. Hai, J. Feng, X. Chen and J. Wang, J. Mater. Chem. B, 2018, 6, 3219–3234 RSC.
- Q. Xu, W. Li, L. Ding, W. Yang, H. Xiao and W.-J. Ong, Nanoscale, 2019, 11, 1475–1504 RSC.
- Q. Xu, T. Kuang, Y. Liu, L. Cai, X. Peng, T. Sreenivasan Sreeprasad, P. Zhao, Z. Yu and N. Li, J. Mater. Chem. B, 2016, 4, 7204–7219 RSC.
- F. Li, D. Yang and H. Xu, Chem. – Eur. J., 2019, 25, 1165–1176 CrossRef CAS.
- S. Tao, S. Zhu, T. Feng, C. Xia, Y. Song and B. Yang, Mater. Today Chem., 2017, 6, 13–25 CrossRef.
- H. Lu, W. Li, H. Dong and M. Wei, Small, 2019, 15, 1902136 CrossRef.
- M. J. Sweetman, S. M. Hickey, D. A. Brooks, J. D. Hayball and S. E. Plush, Adv. Funct. Mater., 2019, 29, 1808740 CrossRef.
- Y. Yan, J. Gong, J. Chen, Z. Zeng, W. Huang, K. Pu, J. Liu and P. Chen, Adv. Mater., 2019, 31, 1808283 CrossRef.
- Y. Dong, H. Pang, H. B. Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS.
- X. Chen, Q. Jin, L. Wu, C. Tung and X. Tang, Angew. Chem., Int. Ed., 2014, 53, 12542–12547 CAS.
- Z. Song, T. Lin, L. Lin, S. Lin, F. Fu, X. Wang and L. Guo, Angew. Chem., Int. Ed., 2016, 55, 2773–2777 CrossRef CAS.
- S. Zhu, X. Zhao, Y. Song, S. Lu and B. Yang, Nano Today, 2016, 11, 128–132 CrossRef CAS.
- S. Gu, C.-T. Hsieh, Y. Ashraf Gandomi, J.-K. Chang, J. Li, J. Li, H. Zhang, Q. Guo, K. C. Lau and R. Pandey, J. Mater. Chem. C, 2019, 7, 5468–5476 RSC.
- S. Chandra, S. H. Pathan, S. Mitra, B. H. Modha, A. Goswami and P. Pramanik, RSC Adv., 2012, 2, 3602–3606 RSC.
- A. M. Schwenke, S. Hoeppener and U. S. Schubert, Adv. Mater., 2015, 27, 4113–4141 CrossRef CAS.
- W. Liu, C. Li, Y. Ren, X. Sun, W. Pan, Y. Li, J. Wang and W. Wang, J. Mater. Chem. B, 2016, 4, 5772–5788 RSC.
- B. B. Chen, M. L. Liu, C. M. Li and C. Z. Huang, Adv. Colloid Interface Sci., 2019, 270, 165–190 CrossRef CAS.
- P. Devi, P. Rajput, A. Thakur, K.-H. Kim and P. Kumar, TrAC, Trends Anal. Chem., 2019, 114, 171–195 CrossRef CAS.
- M. Pirsaheb, S. Mohammadi and A. Salimi, TrAC, Trends Anal. Chem., 2019, 115, 83–99 CrossRef CAS.
- X. Shi, H. Meng, Y. Sun, L. Qu, Y. Lin, Z. Li and D. Du, Small, 2019, 15, 1901507 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- K. Jiang, Y. Wang, C. Cai and H. Lin, Chem. Mater., 2017, 29, 4866–4873 CrossRef CAS.
- J. Tan, R. Zou, J. Zhang, W. Li, L. Zhang and D. Yue, Nanoscale, 2016, 8, 4742–4747 RSC.
- Q. Li, M. Zhou, Q. Yang, Q. Wu, J. Shi, A. Gong and M. Yang, Chem. Mater., 2016, 28, 8221–8227 CrossRef CAS.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334–337 CrossRef CAS.
- C. Wang, Y. Chen, T. Hu, Y. Chang, G. Ran, M. Wang and Q. Song, Nanoscale, 2019, 24, 11967–11974 RSC.
- B. Wang, Y. Mu, H. Zhang, H. Shi, G. Chen, Y. Yu, Z. Yang, J. Li and J. Yu, ACS Cent. Sci., 2019, 5, 349–356 CrossRef CAS.
- X. Kong, X. Wang, H. Cheng, Y. Zhao and W. Shi, J. Mater. Chem. C, 2019, 7, 230–236 RSC.
- D. C. Green, M. A. Holden, M. A. Levenstein, S. Zhang, B. R. G. Johnson, J. Gala de Pablo, A. Ward, S. W. Botchway and F. C. Meldrum, Nat. Commun., 2019, 10, 206 CrossRef.
- C. Xia, S. Tao, S. Zhu, Y. Song, T. Feng, Q. Zeng, J. Liu and B. Yang, Chem. – Eur. J., 2018, 24, 11303–11308 CrossRef CAS.
- J. He, Y. He, Y. Chen, X. Zhang, C. Hu, J. Zhuang, B. Lei and Y. Liu, Chem. Eng. J., 2018, 347, 505–513 CrossRef CAS.
- L. Bai, N. Xue, Y. Zhao, X. Wang, C. Lu and W. Shi, Nano Res., 2018, 11, 2034–2045 CrossRef CAS.
- H. Liu, F. Wang, Y. Wang, J. Mei and D. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 18248–18253 CrossRef CAS.
- J. Julin and A. A. Anappara, ChemistrySelect, 2017, 2, 4058–4062 CrossRef.
- J. Joseph and A. A. Anappara, Phys. Chem. Chem. Phys., 2017, 19, 15137–15144 RSC.
- L. Q. Bai, N. Xue, X. R. Wang, W. Y. Shi and C. Lu, Nanoscale, 2017, 9, 6658–6664 RSC.
- J. Tan, J. Zhang, W. Li, L. Zhang and D. Yue, J. Mater. Chem. C, 2016, 4, 10146–10153 RSC.
- X. Dong, L. Wei, Y. Su, Z. Li, H. Geng, C. Yang and Y. Zhang, J. Mater. Chem. C, 2015, 3, 2798–2801 RSC.
- J. Zhu, X. Bai, X. Chen, H. Shao, Y. Zhai, G. Pan, H. Zhang, E. V. Ushakova, Y. Zhang, H. Song and A. L. Rogach, Adv. Opt. Mater., 2019, 7, 1801599 CrossRef.
- P. Long, Y. Feng, C. Cao, Y. Li, J. Han, S. Li, C. Peng, Z. Li and W. Feng, Adv. Funct. Mater., 2018, 28, 1800791 CrossRef.
- Y. Gao, H. Han, W. Lu, Y. Jiao, Y. Liu, X. Gong, M. Xian, S. Shuang and C. Dong, Langmuir, 2018, 34, 12845–12852 CrossRef CAS.
- K. Patir and S. K. Gogoi, ACS Sustainable Chem. Eng., 2018, 6, 1732–1743 CrossRef CAS.
- H. Feng, M. Zheng, H. Dong, B. Lei, H. Zhang, Y. Xiao and Y. Liu, Opt. Mater., 2014, 36, 1787–1791 CrossRef CAS.
- H. Gou, Y. Liu, G. Zhang, Q. Liao, X. Huang, F. Ning, C. Ke, Z. Meng and K. Xi, Nanoscale, 2019, 11, 18311–18319 RSC.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS.
- H. A. Al-Attar and A. P. Monkman, Adv. Funct. Mater., 2012, 22, 3824–3832 CrossRef CAS.
- G.-P. Yong, Y.-M. Zhang, W.-L. She and Y.-Z. Li, J. Mater. Chem., 2011, 21, 18520–18522 RSC.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS.
- B. Wang, Y. Yu, H. Zhang, Y. Xuan, G. Chen, W. Ma, J. Li and J. Yu, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.2019110035.
- W. Shi, J. Yao, L. Bai and C. Lu, Adv. Funct. Mater., 2018, 28, 1804961 CrossRef.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- Y. Liu, S. Zhou, D. Tu, Z. Chen, M. Huang, H. Zhu, E. Ma and X. Chen, J. Am. Chem. Soc., 2012, 134, 15083–15090 CrossRef CAS PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- H. Tetsuka, R. Asahi, A. Nagoya, K. Okamoto, I. Tajima, R. Ohta and A. Okamoto, Adv. Mater., 2012, 24, 5333–5338 CrossRef CAS PubMed.
- S. Sarkar, D. Gandla, Y. Venkatesh, P. R. Bangal, S. Ghosh, Y. Yang and S. Misra, Phys. Chem. Chem. Phys., 2016, 18, 21278–21287 RSC.
- J. Hou, L. Wang, P. Zhang, Y. Xu and L. Ding, Chem. Commun., 2015, 51, 17768–17771 RSC.
- J. Liu, N. Wang, Y. Yu, Y. Yan, H. Zhang, J. Li and J. Yu, Sci. Adv., 2017, 3, e1603171 CrossRef.
- R. Knoblauch, B. Bui, A. Raza and C. D. Geddes, Phys. Chem. Chem. Phys., 2018, 22, 15518–15527 RSC.
- W.-S. Zou, Y.-J. Ji, X.-F. Wang, Q.-C. Zhao, J. Zhang, Q. Shao, J. Liu, F. Wang and Y.-Q. Wang, Chem. Eng. J., 2016, 294, 323–332 CrossRef CAS.
- Y. Chen, J. He, C. Hu, H. Zhang, B. Lei and Y. Liu, J. Mater. Chem. C, 2017, 5, 6243–6250 RSC.
- H. Li, S. Ye, J.-q. Guo, J.-t. Kong, J. Song, Z.-h. Kang and J.-l. Qu, J. Mater. Chem. C, 2019, 7, 10605–10612 RSC.
- J. M. Soon and L. Manning, Food Res. Int., 2019, 123, 135–143 CrossRef.
- R. Arppe and T. J. Sørensen, Nat. Rev. Chem., 2017, 1, 0031 CrossRef CAS.
- P. Kumar, S. Singh and B. K. Gupta, Nanoscale, 2016, 8, 14297–14340 RSC.
- B. Yoon, J. Lee, I. S. Park, S. Jeon, J. Lee and J.-M. Kim, J. Mater. Chem. C, 2013, 1, 2388–2403 RSC.
- J. Tan, Y. Ye, X. Ren, W. Zhao and D. Yue, J. Mater. Chem. C, 2018, 6, 7890–7895 RSC.
- V. Sharma, P. Tiwari and S. M. Mobin, J. Mater. Chem. B, 2017, 5, 8904–8924 RSC.
- X. Yan, J.-L. Chen, M.-X. Su, F. Yan, B. Li and B. Di, RSC Adv., 2014, 4, 22318–22323 RSC.
- R. Gui, W. He, H. Jin, J. Sun and Y. Wang, Sens. Actuators, B, 2018, 255, 1623–1630 CrossRef CAS.
- R. Gui, H. Jin, Z. Wang, F. Zhang, J. Xia, M. Yang, S. Bi and Y. Xia, Nanoscale, 2015, 7, 8289–8293 RSC.
- Q. Li, M. Zhou, M. Yang, Q. Yang, Z. Zhang and J. Shi, Nat. Commun., 2018, 9, 734 CrossRef.
- Q. Su, C. Lu and X. Yang, Carbon, 2019, 152, 609–615 CrossRef CAS.
- C. Lu, Q. Su and X. Yang, Nanoscale, 2019, 11, 16036–16042 RSC.
- J. Ge, M. Lan, B. Zhou, W. Liu, L. Guo, H. Wang, Q. Jia, G. Niu, X. Huang, H. Zhou, X. Meng, P. Wang, C.-S. Lee, W. Zhang and X. Han, Nat. Commun., 2014, 5, 4596 CrossRef CAS.
- J. Zhang, X. Lu, D. Tang, S. Wu, X. Hou, J. Liu and P. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 40808–40814 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |