
Afterglow of carbon dots: mechanism, strategy and applications
 a
Hengwei
Lin
*ac
a
Hengwei
Lin
*ac
Abstract
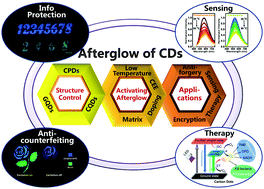
As a newly emerged carbon-based luminescent nanomaterial, carbon dots (CDs) have attracted considerable attention due to their outstanding features. Their photoluminescence (PL) properties and relevant applications have been extensively studied and reviewed in recent years. However, although their afterglow feature had also been discovered and had seen some important advances, no review article was published to systematically summarize such advances. Considering the scientific importance and promising applications of afterglow materials, herein, we aim to summarize the recent remarkable advances of the afterglow phenomena of CDs, particularly focusing on their room temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF). This review will firstly simply introduce the classification of CDs, and then provide comprehensive discussions regarding the relationships between their structure and afterglow properties. Furthermore, their potential applications in anti-counterfeiting, information protection, sensing, etc., are summarized. Finally, challenges and opportunities in the future of this newly emerged carbon-based afterglow material are proposed.
- This article is part of the themed collection: Carbon Dots
 Please wait while we load your content...
Please wait while we load your content...

