
Polysaccharide-containing block copolymers: synthesis and applications
Anastasia S.
Volokhova
ab,
Kevin J.
Edgar
 bc and
John B.
Matson
*ab
bc and
John B.
Matson
*ab
aDepartment of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: jbmatson@vt.edu
bMacromolecules Innovation Institute, Virginia Tech, Blacksburg, Virginia 24061, USA
cDepartment of Sustainable Biomaterials, Virginia Tech, Blacksburg, Virginia 24061, USA
First published on 18th October 2019
Abstract
Polysaccharide-based block copolymers have been studied since the early 1960s and command growing interest due to the increasing importance of sustainable materials. Polysaccharides are renewable, often biodegradable, generally nontoxic, and offer wide structural variety, yielding remarkably diverse materials that provide a wide range of properties and applications. Block copolymers containing one or more polysaccharide blocks and one or more synthetic blocks can be synthesized using a variety of methods, either through direct coupling or chain extension approaches. Here we review current synthetic methods for the preparation of polysaccharide-containing block copolymers. We also discuss established and emerging applications of this class of block copolymers, such as their use as drug delivery vehicles, hydrogels for localized drug delivery, and surfactants/interfacial agents for stabilizing interfaces, including those in polysaccharide/synthetic polymer blends.
 From left to right: Kevin J. Edgar, John B. Matson and Anastasia S. Volokhova | Anastasia S. Volokhova received her undergraduate degree in Chemistry at Virginia Tech in 2017, and she continues her studies there as 3rd year graduate student in Professor Matson's research group. She works on synthesizing block copolymers that contain polysaccharide derivatives, utilizing them in a variety of polymer blends and composites to study properties of sustainable materials. Kevin J. Edgar is a professor in the Virginia Tech Department of Sustainable Biomaterials, and adjunct in the Department of Chemistry. He is an active member of the Macromolecules Innovation Institute, with its internationally renowned Macromolecular Science and Engineering interdisciplinary graduate program, and a leader of the VT Center for Engineered Health. He is associate Dean in the VT Graduate School. His interests include synthesis of novel polysaccharide derivatives, unraveling their structure property relationships, and exploiting their useful properties in demanding, high-value applications. Edgar received his BS in Chemistry from Bucknell University, and his PhD in Organic Chemistry from Duke University. John B. Matson is currently an associate professor in the Virginia Tech Department of Chemistry. He is also a member of the Macromolecules Innovation Institute and the Virginia Tech Center for Drug Discovery. He received his undergraduate degree majoring in Chemistry and German at Washington University in St. Louis before moving to Caltech to pursue a PhD in the area of organic and polymer chemistry with Professor Bob Grubbs. He then moved to Northwestern University, where he worked on peptide-based biomaterials with Professor Sam Stupp. In 2012 he began his independent career at Virginia Tech in the Department of Chemistry, where he was promoted to Associate Professor in 2018. His group focuses on macromolecular and supramolecular chemistry with applications in biology, medicine, and sustainability. |
1. Introduction
Polysaccharides, in particular cellulosic polymers, were among the first substances studied in polymer science, and presently they are under increased scrutiny as the effort to increase sustainability of plastics intensifies.1 In fact, nitrocellulose was first synthesized in 1844 (it was called “guncotton” at the time) and commercialized in the 1860s,2,3 long before Bakelite, which is recognized as the first synthetic polymer material, was commercialized in 1909.4,5 Polysaccharides are highly abundant, biosynthesized organic polymers, and their synthetic derivatives are often biodegradable and are biocompatible with many tissues in many circumstances. Their benign nature and diversity bring environmentally friendly solutions to a variety of applications.6 Polysaccharides display remarkable structural variety in terms of architecture, oxidation state, pendant groups, and linkages between repeating units. This structural variety creates a wide range of properties, for example solubility ranging from hydrocarbon-soluble to water-soluble. Of particular interest to synthetic polymer chemists, the side groups present along the polymer backbone (mostly hydroxyl groups) can be readily derivatized, permitting fine-tuning of the thermal and mechanical properties of the resulting polymer.1 Polysaccharides have commercial applications including in coatings, food additives, emulsifiers, textiles, membranes, tissue scaffolds, drug delivery vehicles, bioadhesives, and several other material and biomedical applications.1,7–15While polysaccharide homopolymers enable a number of applications, utilization of the natural abundance of polysaccharides is quite limited; only cellulose, starch, and a few others are used significantly as materials. This is due in part to inherent properties; their hydrophilic nature, difficult processability, relative brittleness and lack of toughness, and other issues limit applications. It is clear that polysaccharide properties and applications could be extended if they were used as components in block copolymers. Block copolymers contain segments of two or more structurally different types of repeating units, which may form microphase-separated domains, often leading to enhanced material properties compared to those of the two pure homopolymers. For example, block copolymers containing both low Tg and high Tg blocks may form thermoplastic elastomers, which combine the strength of the high Tg thermoplastic with the toughness of the low Tg elastomer.16 An example of this principle is the commercial block copolymer styrene–butadiene–styrene (SBS) with the trade name Kraton,17 which behaves like a physically crosslinked elastomer while maintaining processability. Linking the two blocks therefore provides superior properties, supporting applications inaccessible to either homopolymer by itself.
Synthetic block copolymers are generally prepared by sequential addition of monomers. Common methods for block copolymer synthesis include anionic polymerization,18 ring-opening polymerization,19 and reversible-deactivation radical polymerization (RDRP) techniques such as atom-transfer radical polymerization (ATRP),20 reversible addition–fragmentation chain transfer polymerization (RAFT),21,22 and nitroxide-mediated radical polymerization (NMRP).23 These polymerization techniques, which are living or have living characteristics, provide good control over molecular weight and dispersity, enabling finely tuned properties.16 However, these bottom-up techniques are not generally applicable to polysaccharides, which are usually isolated from natural sources as high molecular weight polymers, then modified to make useful polymeric materials in a top-down approach.
Utilization of polysaccharide units within block copolymers can provide beneficial properties, not only potentially allowing for incorporation of biodegradability into polymers with other functionalities, but also providing high Tg segments (polysaccharide derivatives typically have Tg > 100 °C), rigidity, and hydrophilicity. Therefore, synthetic approaches to make polysaccharide-containing block copolymers are needed to more effectively exploit these valuable, renewable polymeric feedstocks. Due to the top-down approach to polysaccharide homopolymer synthesis, synthetic approaches to formulating linear polysaccharide-containing block copolymers often differ from conventional block copolymer sequential methods. Most polysaccharide-containing block copolymers are prepared either by chain extension of a synthetic polymer block newly grown from existing polysaccharide end groups, or by coupling preformed blocks onto a polysaccharide unit. In addition to their potential applications as degradable materials, polysaccharide-containing block copolymers have also recently gained traction in specialty polymer areas, with potential applications including drug delivery,24,25 surfactants,26 emulsifiers,27 and polymer blend compatibilizers.28 The field of polysaccharide copolymers was reviewed in 2010 by Schatz and Lecommandoux,29 and it continues to grow, with new synthetic methods and new polysaccharide-based block copolymers being developed over the past decade. Here we review both well-known and recently developed methods of preparing polysaccharide-containing block copolymers, with a focus on potential applications of these materials.
2. Synthetic methods
When synthesizing block copolymers from monomers, for example by RDRP methods, one chain end contains a masked radical, and chain extension to add the second block begins with revealing this radical. Therefore, there is a well-defined point of attachment for the second block. In polysaccharides this is not as obvious, especially in unfunctionalized polysaccharides, which contain a large number of secondary and/or primary hydroxyl groups. All polysaccharide hydroxyl groups have relatively low reactivity (due to limited approach angles) and relatively small reactivity differences from one another. Fortunately, the saccharide residue on the ω-chain end (the so-called reducing end) has reactivity that differs from all of the others, creating the potential for selective derivatization. While all modern synthetic strategies rely on reducing-end functionalization in some form to add on one or more additional blocks, early efforts employed more crude chain-breaking methods to generate initiating sites.The earliest reported method for producing linear polysaccharide-containing block copolymers was described in 1961 by Ceresa.30 This synthetic strategy involved mechanical scission of polysaccharide backbones using a variety of techniques to generate radicals capable of initiating polymerization of vinyl monomers. For example, one method involved mechanically degrading cellulose acetate (CA) in the presence of vinyl acetate (VAc), thereby generating radicals on the CA backbone that initiated free radical polymerization, generating CA-b-PVAc. Other methods developed by Ceresa included vapor-phase swelling and aqueous freezing forces, which provided alternative ways to mechanically rupture the polysaccharide backbone. Both block copolymers and homopolymers were obtained in this process, as confirmed by IR spectroscopy; more complex copolymer structures (e.g., ABA triblock copolymers and potentially branched structures) also likely formed. While these techniques lacked molecular weight control, numerous solvent extractions enabled isolation of the desired polysaccharide-containing block copolymers in poor to fair yields.
Aside from specialized enzymatic polymerization methods, in which enzymes directly polymerize monosaccharide or oligosaccharide units (sometimes resulting in sequence-controlled polysaccharides),31,32 most recent efforts have employed the reducing chain end of the polysaccharide, affording enhanced selectivity versus Ceresa's approach. The equilibrium between ring-closed and ring-opened forms in polysaccharides means that aldehydes are transiently available on the reducing end (Scheme 1). Selective reactions of this single aldehyde can produce new functional groups for chain extension, coupling, and other types of reactions. Common methods include azide–alkyne cycloaddition reactions, reductive amination, oxime ligation, and other chain-end modification reactions to install pre-made polymer blocks, initiators or chain transfer agents, or other reactive functional groups. These synthetic methods are described in the sections below, organized according to method of chain-end functionalization.
 | ||
| Scheme 1 Depiction of the ring-closed and ring-opened form of a cellulose reducing end monosaccharide, displaying the aldehyde functional group available for further reactions. The figure employs the numbering convention for C1–C6 of a monosaccharide unit (glucopyranose here). | ||
Maltoheptaose chain-end functionalization
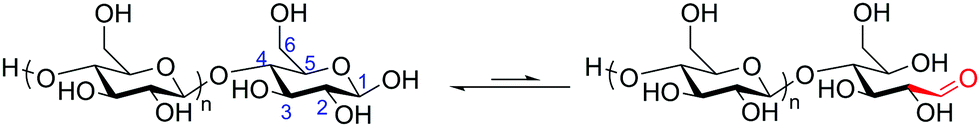
Prior to production of polysaccharide block copolymers, several studies were conducted with oligosaccharides, demonstrating useful techniques that were in some cases later applied to polysaccharides. These precursors to polysaccharide-based copolymers were first explored by Haddleton et al., who utilized β-cyclodextrin to prepare a block copolymer via ATRP with several common vinyl monomers.33 They first fully acetylated cyclodextrin, and then opened the ring to form a linear heptasaccharide (Scheme 2A). They next site-specifically hydrolyzed the peracetylated maltoheptaose to produce a hydroxyl group on the reducing end. An ATRP initiator was attached to the hydroxyl group, and the oligosaccharide was finally chain-extended using various monomers, including methyl methacrylate (MMA) (Scheme 2B). The resulting maltoheptaose-b-PMMA polymers and related products were also hydrolyzed to remove the cyclodextrin acetyl groups, exhibiting control over hydrophilicity of the heptasaccharide component using this method.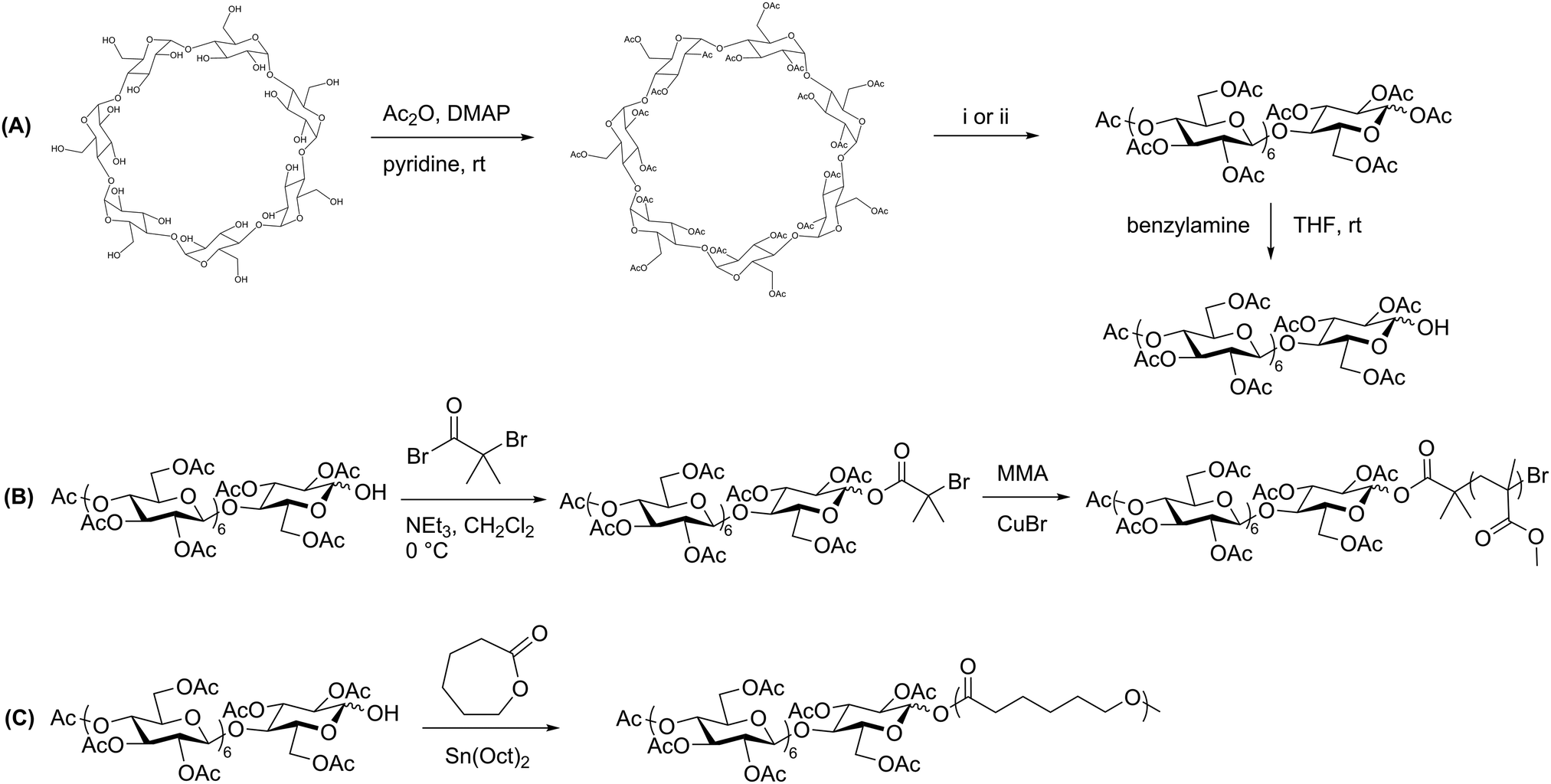 | ||
| Scheme 2 (A) Derivatization of β-cyclodextrin to produce peracetylated maltoheptaose with a reactive reducing-end hydroxyl group. (B) Chain extension by ATRP with MMA. (C) Chain extension by ROP with ε-caprolactone. (i) H2SO4 in Ac2O, 55 °C; (ii) FeCl3·6H2O in Ac2O, 70 °C. Adapted from ref. 33 and 34. Copyrights ACS 2000, Elsevier 2008, respectively. | ||
A similar approach was explored by Li and Zhang, who produced maltoheptaose-b-PCL (PCL = poly(ε-caprolactone)) via β-cyclodextrin derivatization followed by ring-opening polymerization (ROP).34 In this synthetic pathway, acetylated maltoheptaose with a hydroxyl terminus was synthesized using a method similar to that described above. The reducing-end hydroxyl substituent then initiated ROP of ε-caprolactone (Scheme 2C). Again, deacetylation of the maltoheptaose component afforded the amphiphilic maltoheptaose-b-PCL product, which self-assembled into spherical micelles in water. Although the maltoheptaose components in these polymers were oligosaccharides, the effectiveness of these synthetic approaches paved the way for incorporation of polysaccharides into block copolymers.
Reductive amination
Reductive amination is used widely in organic synthesis because it is selective, generally fast, and simple to carry out. This two-step reaction involves the condensation of a primary amine with an aldehyde to form an imine intermediate. A reducing agent such as sodium borohydride is typically included, which quickly and selectively reduces the imine as it forms, generating a secondary amine. Control over pH is key in this process, and a pH of 6–8 is ideal for most reductive amination reactions.35 Recognizing that the reducing-end aldehyde on polysaccharides is available for reductive amination, researchers have used this method extensively for chain-end functionalization of polysaccharides.36The first applications of reductive amination in polysaccharide chemistry involved attachment of small molecules and proteins to cellulose, alginate, and dextran to prepare solid supports for affinity chromatography.37 In 2002, this method was applied by Bosker et al. to prepare dextran-b-PS (PS = polystyrene) block copolymers.38 In this synthesis, the researchers employed 12 kg mol−1 PS containing a terminal primary amine, which was coupled via reductive amination with dextran through the C1 aldehyde at the reducing end (Scheme 3A). These reactions were performed in polar solvents and exhibited good yields on the basis of equivalents of amine used, because dextran was used in large excess. Several washing and filtration steps were then carried out to remove the unreacted dextran and isolate the dextran-b-PS block copolymer. The scope was limited with regard to dextran molecular weight, as dextrans above 6000 g mol−1 could not be coupled using this method.
 | ||
| Scheme 3 Synthesis of dextran-containing block copolymers using (A) reductive amination alone and (B) a combination of reductive amination and CuAAC. Adapted from ref. 38 and 48. Copyrights ACS 2003, Wiley 2009. The dashed bonds on the 3-OH units indicate branches present in dextran. | ||
Reductive amination and click chemistry
While polysaccharide-containing block copolymers may be constructed using reductive amination alone, a more powerful synthetic strategy is to couple reductive amination with click chemistry. Click chemistry describes reactions characterized by high efficiency, simple methodology, and functional group tolerance. Defined by Sharpless in 2001, a click reaction must exhibit high yields, a wide substrate scope, simple product isolation, and modularity.39 Polymer click reactions have additional requirements, including equimolarity between reacting groups and fast reaction kinetics.40 Due to their functional diversity, click reactions have been used to develop facile methods of obtaining complex polymer architectures, including block, graft, and star polymers, as well as introducing new functionalities to commodity polymers.41 The most commonly used click reaction remains the Huisgen 3+2 copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction,42,43 although thiol–ene and other click reactions are also used in polymer synthesis and polymer–polymer couplings.44–47There exist several noteworthy examples of sequential usage of reductive amination and click chemistry.49 One particular case from Schatz et al. involves the synthesis of a polysaccharide-b-polypeptide block copolymer, which was prepared from dextran and poly(γ-benzyl L-glutamate).48 First, the polypeptide was prepared by ROP of γ-benzyl L-glutamate initiated by 1-azido-3-aminopropane, which provided end-functionalization with an azide. A 6600 g mol−1 dextran component was end-functionalized using reductive amination to place a propargyl group on the reducing end, and the polypeptide was coupled to it using CuAAC (Scheme 3B). Dextran was used in two-fold excess to ensure full conversion of the azide-terminated polypeptide, and the unreacted dextran was removed via dialysis. The yield was quantitative with respect to polypeptide, as indicated by key signals in 1H NMR and IR spectra of the product. These amphiphilic block copolymers assembled into small (radius = 45 nm), glycoprotein-like vesicles similar to those in living cells, making these compounds ideal for mimicking viral morphologies.
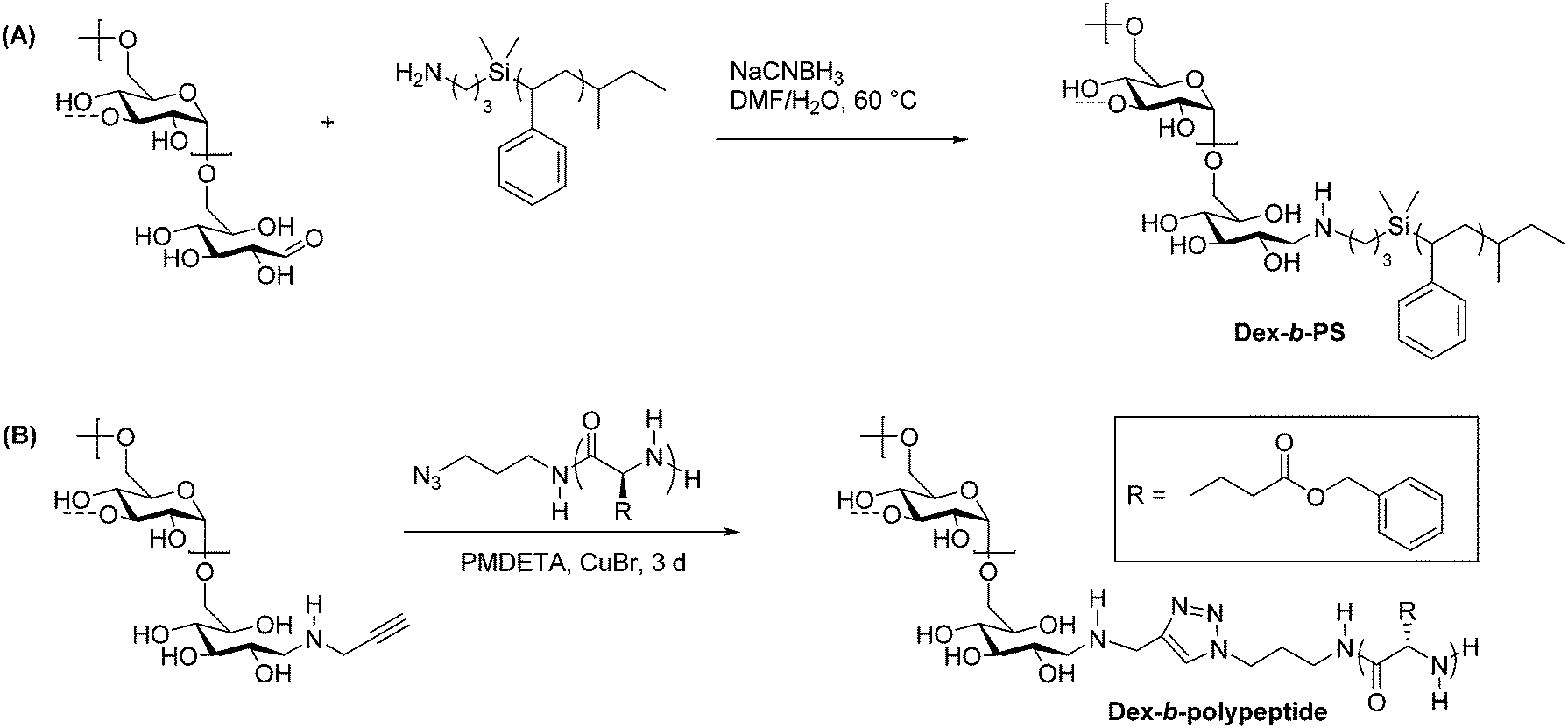
Thiol–ene chemistry in tandem with reductive amination is another click approach that enables the preparation of polysaccharide-containing block copolymers. Recently, thiol–ene click reactions performed by Wang et al. facilitated the coupling of hydroxypropyl methyl cellulose (HPMC) and poly-L-lactic acid (PLLA).50 The synthesis of this block copolymer began with ring-opening polymerization of L-lactide initiated by allyl alcohol, yielding an olefin-terminated PLLA block (Scheme 4A). Separately, HPMC was end-functionalized with a thiol via reductive amination with 2-aminoethanethiol (Scheme 4B). The two end-functionalized polymers were then coupled using UV irradiation to promote a thiol–ene click reaction, and the resulting block copolymers were purified by several precipitation and centrifugation cycles. The HPMC-b-PLLA block copolymers self-assembled into 50–100 nm micelles that displayed relatively low CMCs ranging from 0.12 to 0.15 g L−1, which makes this block copolymer potentially useful as an injectable carrier of hydrophobic drugs. Thiol–ene chemistry applied to polysaccharide-containing block copolymers may offer advantages over other click methods such as CuAAC due to the lack of a metal catalyst and easy access to functional groups required for coupling.
 | ||
| Scheme 4 Synthesis of block copolymers of HPMC and PLLA using reductive amination followed by thiol–ene click. Conditions: (i) toluene, 15 h, 80 °C; (ii) DTT, 60 °C, 6 d; (iii) DMPA, 10 h. Adapted from ref. 50. Copyright Wiley 2017. | ||
As previously mentioned, ATRP, RAFT, and NMRP are common ways of preparing conventional synthetic block copolymers from monomers with good control over molecular weight and dispersity. Some of these techniques have also been applied in the preparation of polysaccharide-containing block copolymers, typically by installing an initiator or chain transfer agent on the polysaccharide reducing end. This chain-extension approach permits the synthesis of much higher molecular weight block copolymers than coupling methods because coupling high molecular weight polymers together that diffuse relatively slowly is often slow and inefficient. Often a combination of reductive amination and click chemistry is employed to functionalize the polysaccharide with an initiator. For example, in 2008 Charleux et al. synthesized dextran-b-PVAc (PVAc = poly(vinyl acetate)) block copolymers through a combination of reductive amination, CuAAC, and RAFT (Scheme 5).27 First, dextran was functionalized on the reducing end by reductive amination with propargylamine. Next, an azide-terminated xanthate was added by CuAAC to place a chain transfer agent for RAFT on the dextran reducing end. Polymerization of VAc from this end proceeded via RAFT to give dextran-b-PVAc. Emulsion polymerization of PVAc in the presence of this block copolymer was improved compared with that of native dextran as indicated by the smaller particle diameter and narrower particle size distribution.
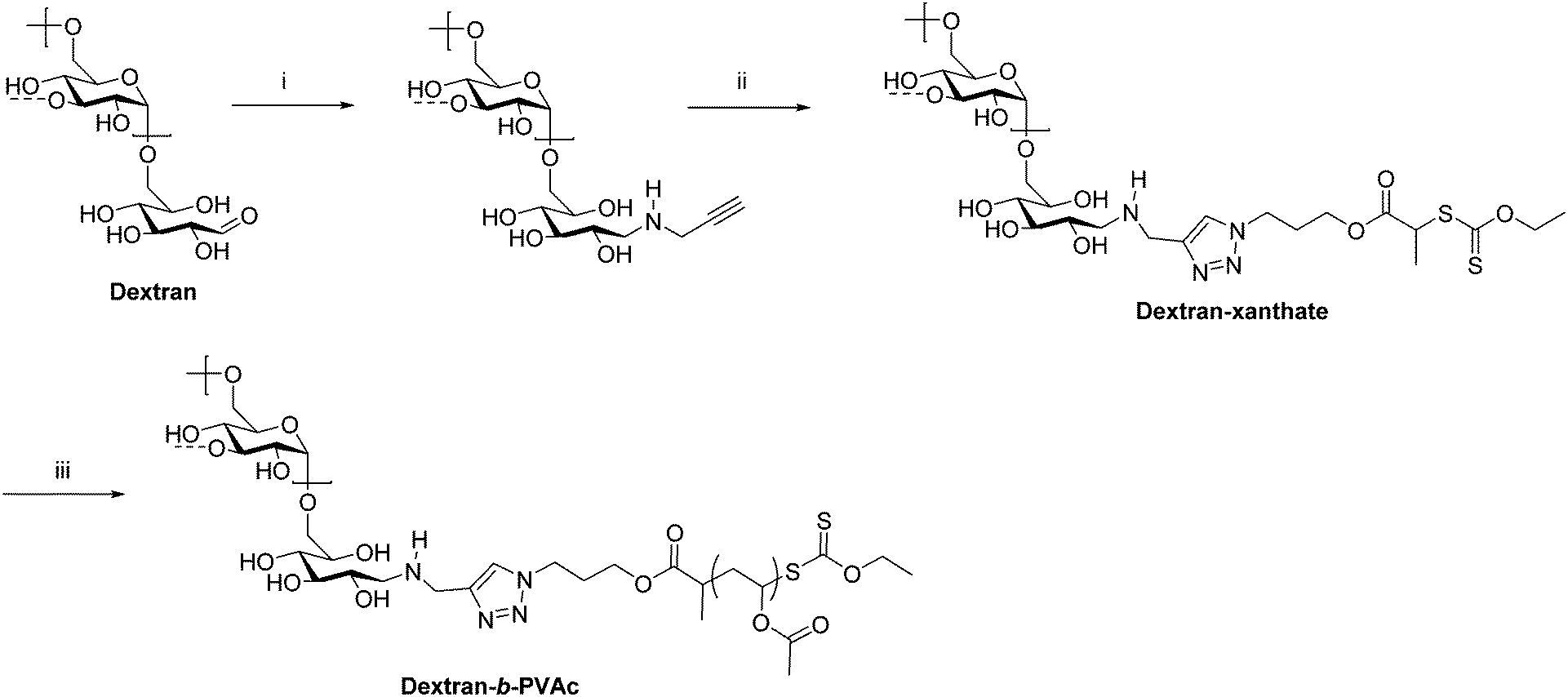 | ||
| Scheme 5 Chain extension of dextran via RAFT producing the diblock copolymer dextran-b-PVAc. (i) Propargylamine, NaBH3CN, phosphate buffer pH = 7, 37 °C, 7 d; (ii) azido-xanthate chain transfer agent, CuSO4, ascorbic acid, PMDETA, DMSO/H2O, 25 °C, 48 h; (iii) AIBN, VAc, trioxane, 60 °C. The dashed bonds on the 3-OH units indicate occasional branches present in dextran. Adapted from ref. 27. Copyright Wiley 2008. | ||
Reductive amination and enzymatic polymerization
While the majority of pathways to polysaccharide-containing block copolymers discussed here involve modification of a biosynthesized polysaccharide chain, there exist a few examples of generating the polysaccharide component by enzyme-catalyzed polymerization of monosaccharides. An enzymatic approach for production of amylose derivatives was first introduced by Phannenmuler et al. in 1975,51 and it has also been applied in more recent work.52–54 Enzymatic polymerization is typically carried out using α-D-glucose-1-phosphate and maltoheptaose in the presence of potato phosphorylase, which effectively produces a homopolymer of amylose. Reductive amination can then be used to either chain-extend or couple the amylose block with another polymer block. One example of this approach utilized reducing end-functionalization of maltopentaosylamine with MPEO-p-nitrophenylcarbonate (MPEO = ω-methoxypoly(ethylene oxide)) via reductive amination, followed by enzymatic polymerization, which inserted additional α-D-glucose units into the amylose block (Scheme 6).55 Similar enzymatic approaches were also used to prepare amylose-b-polystyrene linear block copolymers,53,56 in addition to several graft architectures produced with amylose macromonomers.57–59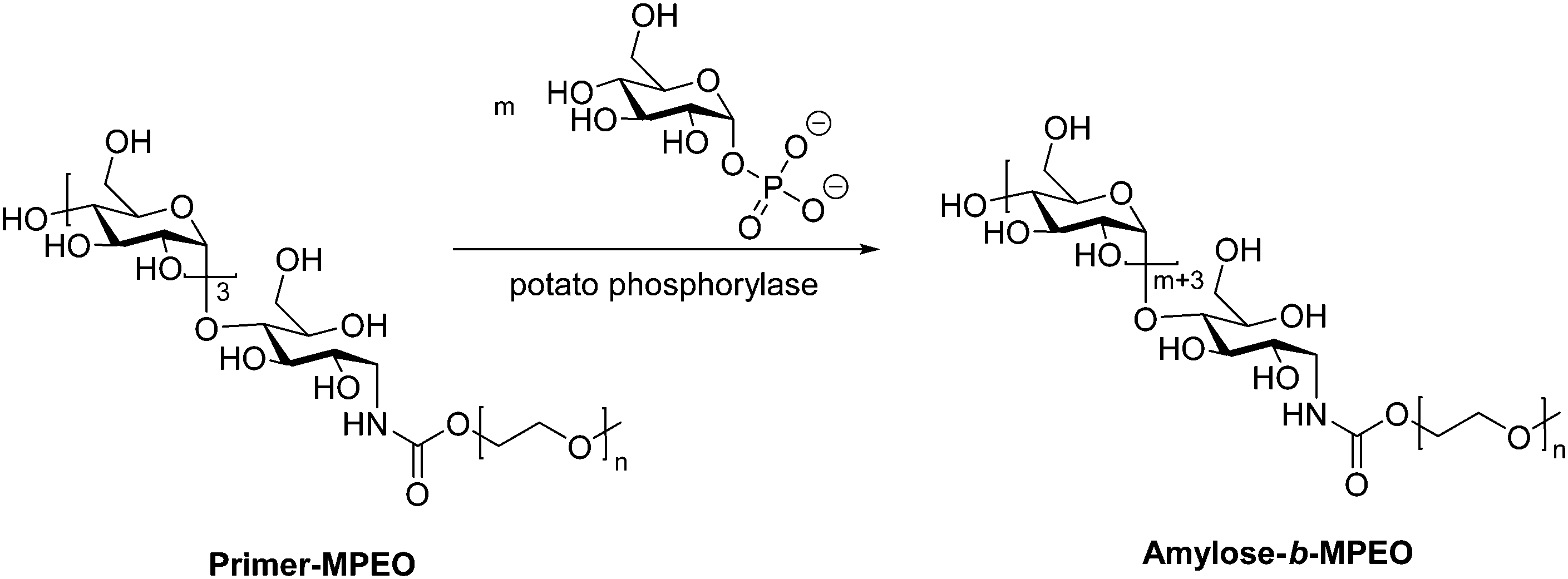 | ||
| Scheme 6 Enzymatic polymerization of amylose using potato phosphorylase. Adapted from ref. 55. Copyright Wiley 1999. | ||
Reductive amination remains one of the simplest and most effective methods for producing polysaccharide-containing block copolymers, but it has several limitations. For example, multi-day reaction times are required due to low amounts of free aldehyde available at equilibrium compared to the ring-closed hemiacetal, as shown in Scheme 1. Additionally, equimolar reactions between polymer blocks of even moderate molecular weights often afford incomplete conversions due to the low concentrations of the complementary reactive chain ends in this 2nd order reaction. Incomplete conversion in polymer coupling reactions necessitates purification steps to separate unreacted homopolymer(s) from the target diblock copolymer; such separations may be tedious at best, or in some cases may be impossible. Dialysis or preparative size exclusion chromatography are often required, which are expensive, scale-limiting, and time-consuming. Generally, functionalizing polysaccharide reducing-ends first with propargyl amine or another functional amine, followed by reaction with an end-functionalized (e.g., with an alkyne) synthetic polymer, is more effective than one-step reductive amination with an amine-terminated polymer. The two-step method is more effective because a large excess of reactive, small molecule amine can be added to achieve full reducing-end conversion. The reactive end groups can be chosen, as in the CuAAC examples, to quickly and selectively react with one another. These features reduce the need for polymer–polymer separations after addition of the second block. To avoid two-step procedures, polymer chemists have sought out synthetic methods that achieve near quantitative conversion to block copolymer products in a single step.
Oxime ligation
A direct method of preparing polysaccharide-containing block copolymers was developed by Novoa-Carballal and Müller in 2012 using oxime ligation, sometimes referred to as oxime click.60 Their initial approach involved coupling a pre-formed polymer to an aldehyde-terminated polysaccharide. A major advantage of this approach is that it utilizes the reducing end aldehyde without requiring prior modification. Oxime ligation with amine-functionalized PEG was demonstrated with dextran, chitosan, and hyaluronic acid, yielding a one-pot synthetic route to diblock copolymers without the need for a catalyst, harsh reaction conditions, or intermediate products. Remarkably, polysaccharides with MWs exceeding 40 kg mol−1 were employed here, although low MW PEG (5 kg mol−1) was used and purification steps were still needed to isolate the desired block copolymer products. Since these types of block polymers may contain charged polysaccharide blocks, they could prove useful as interpolyelectrolyte complexes for protein and gene carriers.A similar coupling technique was also used to prepare diblock copolymers by attachment of PEG or poly(sarcosine) (PSar) to dextran or pullulan (Scheme 7A).61 The PEG and PSar components were synthesized with a terminal primary alkoxyamine and coupled to the reducing end aldehyde on either dextran or pullulan via oxime ligation. Each homopolymer exhibited a molecular weight of approximately 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, producing block copolymers with a total molecular weight of 40000 g mol−1. A small amount (<15%) of unreacted polysaccharide was noted in each diblock copolymer, again likely due to low concentrations of end groups in this 2nd order reaction. Due to the immiscibility between the polysaccharide and synthetic polymer blocks in these diblock copolymers, they formed vesicle-like aggregates in aqueous solutions with hydrodynamic radii up to 20 μm. The polymers comprising two hydrophilic blocks were dubbed “aquanelles”; they offer promising possibilities for drug delivery and nanoreactors due to their aggregation properties.
000 g mol−1, producing block copolymers with a total molecular weight of 40000 g mol−1. A small amount (<15%) of unreacted polysaccharide was noted in each diblock copolymer, again likely due to low concentrations of end groups in this 2nd order reaction. Due to the immiscibility between the polysaccharide and synthetic polymer blocks in these diblock copolymers, they formed vesicle-like aggregates in aqueous solutions with hydrodynamic radii up to 20 μm. The polymers comprising two hydrophilic blocks were dubbed “aquanelles”; they offer promising possibilities for drug delivery and nanoreactors due to their aggregation properties.
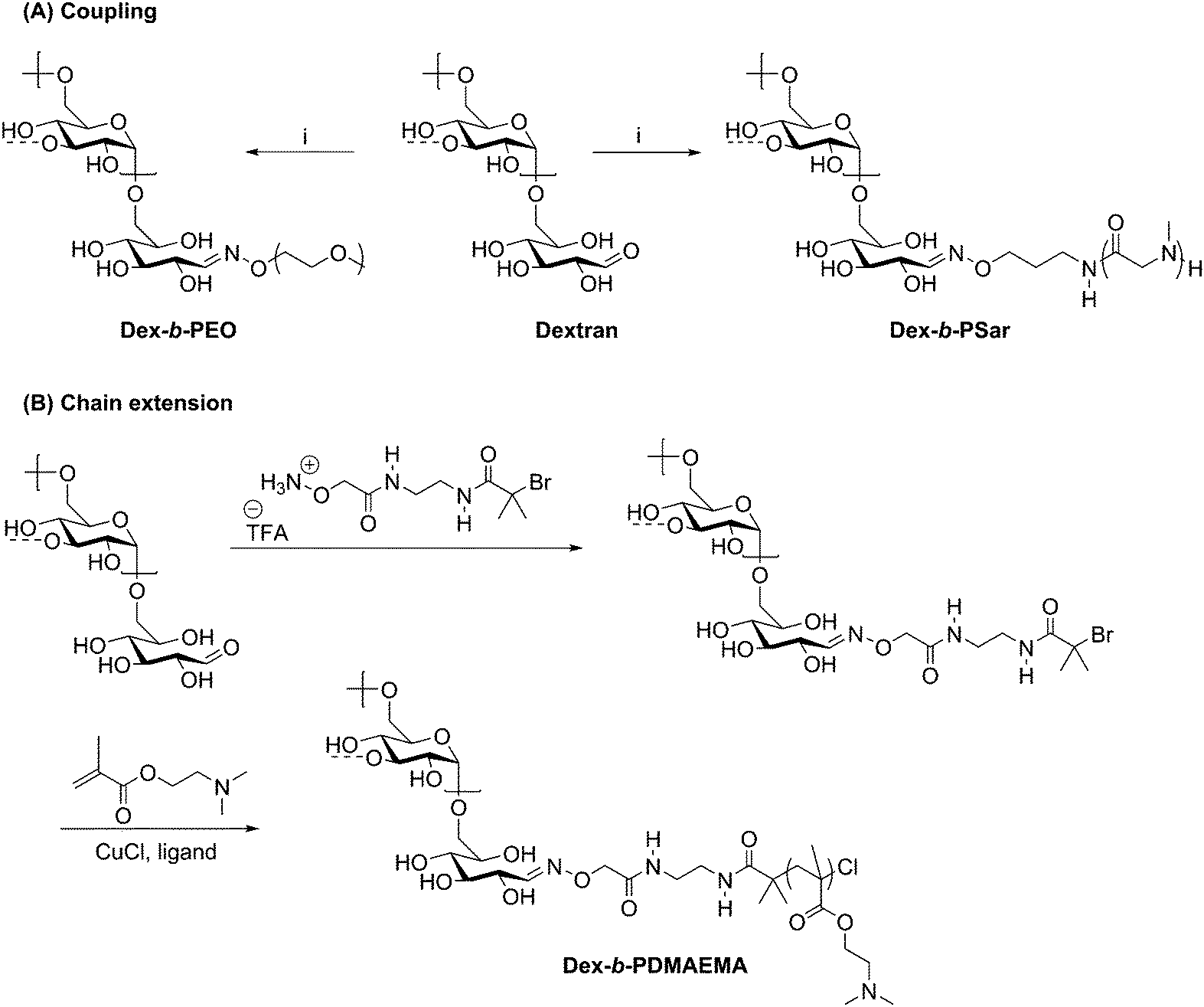 | ||
| Scheme 7 Synthesis of polysaccharide-containing block copolymers by oxime ligation where (A) displays a coupling method with a preformed polymer block and (B) shows the attachment of an ATRP initiator for chain extension. (i) H2NO-PEG or H2NO-poly(sarcosine), citric acid (pH = 3); H2O/DMSO (1:1); 45 °C, 5 d. The dashed bonds on the 3-OH units indicate occasional branches present in dextran. Adapted from ref. 61 and 62. Copyrights Wiley 2015, Royal Society of Chemistry 2013. | ||
The versatility of oxime ligation in polysaccharide-containing block copolymer synthesis is illustrated by its ability to permit chain extension of polysaccharide blocks, in addition to the coupling methods described above. In 2013, shortly after Novoa-Carballal and Müller described coupling polymers via oxime ligation, they reported use of oxime ligation to synthesize copolymers of dextran via attachment of an ATRP initiator to grow poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) from the dextran reducing end (Scheme 7B).62 ATRP reactions were carried out for multiple days with various ligands and either CuBr and CuCl as a copper source, with conversions varying from 25% to 65%. Macroinitiator efficiency reached a maximum at only 76%, requiring separation of the dextran macroinitiator from the desired diblock copolymer product. To carry out this separation, the lower critical solution temperature behavior of DMAEMA was exploited by heating the mixture to 40 °C (above the cloud point) and dialyzing at pH 11, effectively isolating the diblock copolymer within the dialysis tube, such that little desired product was lost during purification. The isolated diblock copolymer was complexed with poly(styrenesulfonate) to investigate the potential for stabilization of polyelectrolyte complexes, which may be promising for specialized drug delivery systems.
Compared with reductive amination, oxime ligation is more effective in direct coupling reactions, allowing for the construction of larger block copolymers with shorter reaction times. This is likely due to the enhanced nucleophilicity of hydroxylamines compared with amines as a result of the alpha effect. However, synthesis of the oxime is usually necessary because these derivatives are not typically commercially available, and low concentrations of the polysaccharide aldehydes at the reducing ends because of hemiacetal/aldehyde equilibrium is still an issue that leads to long reaction times for the oxime ligation reactions as well.
Chain degradation-nucleophilic displacement
The Matson lab has recently developed a method for preparing polysaccharide-containing ABA triblock copolymers using polysaccharide end-group modification and ring-opening metathesis polymerization (ROMP).28 The synthesis employed cellulose triacetate (CTA), but it could be broadly applied to other polysaccharide derivatives provided that they exhibit organic solubility. The anomeric linkages of commercial CTA were first reacted with HBr, affording cellulose acetates with 1-bromo groups at reducing end termini (Scheme 8). The bromide was then displaced using a terminally unsaturated alcohol, generating a mono-olefin-functionalized CTA. The resulting terminal olefins (RHC = CH2, where R is the substituted polysaccharide) were used as chain transfer agents in ROMP of unhindered, strained cyclic olefins, placing functional groups (R) on both chain ends.63 ROMP of cyclooctadiene in the presence of olefin-functionalized CTA generated CTA-b-PB-b-CTA ABA triblock copolymer, where PB is poly(1,4-butadiene), structurally identical to poly(cyclooctadiene). This synthetic sequence is significantly faster than the reductive amination and oxime ligation strategies, but it is currently limited to production of ABA triblock copolymers. The CTA-b-PB-b-CTA triblock copolymer was applied as an interfacial agent (i.e., compatibilizer) in blends of CTA and PB, offering exciting opportunities for creating new applications of polysaccharides through blending. We are currently employing this approach in the synthesis of other polysaccharide-containing block copolymers, and applications of these materials are under further exploration in our lab.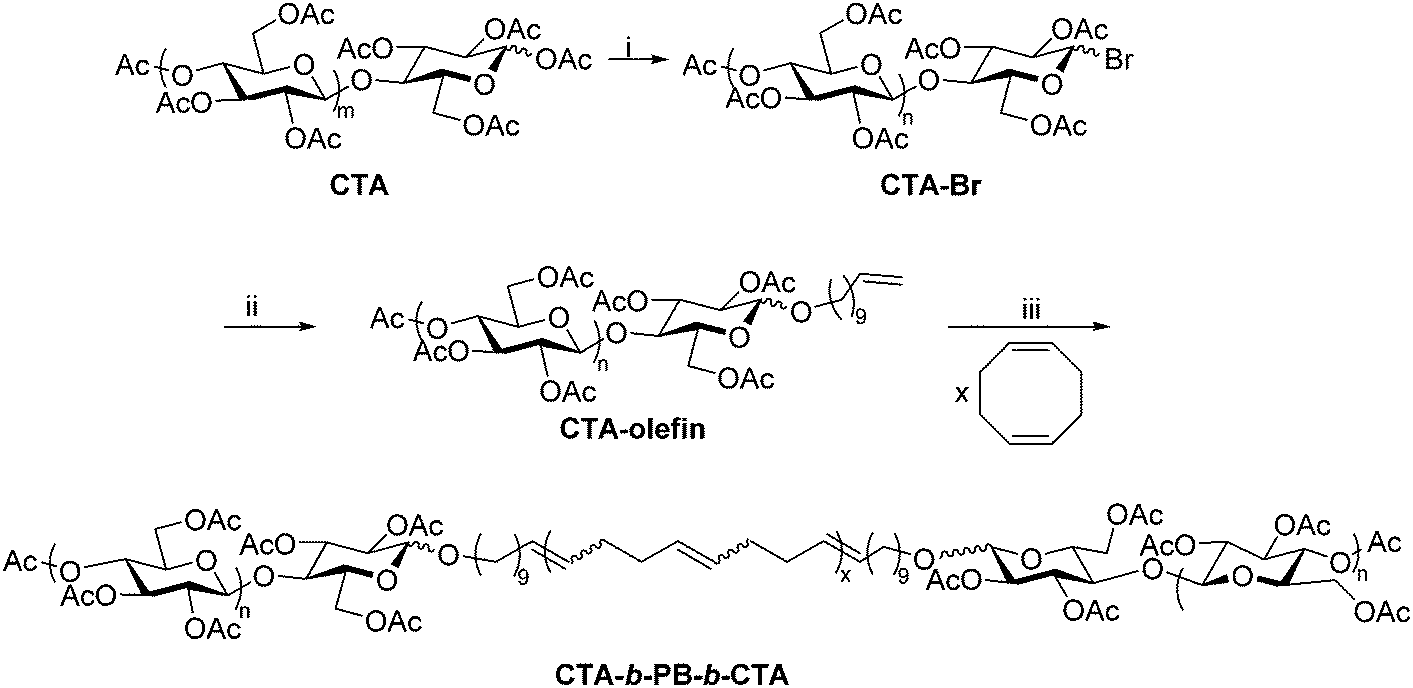 | ||
| Scheme 8 Synthetic route to cellulose triacetate-containing ABA triblock using ROMP. (i) 33 wt% HBr in acetic acid, CHCl3, rt, 30 min; (ii) undec-10-en-1-ol, DBU, CHCl3, rt, 4 h; (iii) HG2, CHCl3, rt, 24 h. Copied from ref. 28. Copyright ACS 2019. | ||
3. Applications of polysaccharide-containing block copolymers
Researchers have explored many potential applications for polysaccharide-containing block copolymers due to their potential for degradability, useful polysaccharide properties (e.g., high Tg, gas barrier properties), and general lack of toxicity of the polysaccharide components. Biomedical applications of these materials have thus far dominated this field, primarily including drug delivery vehicles and hydrogels for localized drug release. In other fields, polysaccharide-containing block copolymers have been applied as surfactants as well as compatibilizers in polymer blends. Here we discuss some specific examples of polysaccharide-containing block copolymer applications.Amphiphilic block copolymers and drug delivery vehicles
The assembly of amphiphilic block copolymers into micelles has been thoroughly studied with synthetic polymers; however, incorporation of polysaccharides into these assemblies brings new advantages for drug delivery. The polysaccharide blocks contribute to low immunogenicity, biocompatibility with many tissues in many situations,64 and often biodegradability, which are essential for a micellar drug delivery vehicle.65 While drug delivery applications of polysaccharide-containing polymers are dominated by grafted, brush-like architectures,65–69 block copolymers have also been investigated. A large contributor to their usefulness is the possibility to generate amphiphilic block copolymers capable of self-assembly by incorporating hydrophilic and hydrophobic blocks. Additionally, controlling functionalization of the polysaccharide backbone and the nature of the synthetic block can aid in achieving desired polymer solubility, as well as providing responsiveness to stimuli such as pH or temperature in some cases. Many polysaccharides contain free hydroxyl groups which may be functionalized with carboxylic acid or hydroxyl-containing drugs, including cholesterol and cholic acid.70–73 In this section, some recent manipulations of these components are discussed, in addition to the uses of the resulting amphiphilic block copolymers.A recent example from Wich et al. demonstrates how polysaccharide-based micelles provide advantages over synthetic block copolymers. Amphiphilicity requires one component to be hydrophobic, while the other is hydrophilic, and this can be achieved in polysaccharides through differential derivatization of backbone hydroxyl substituents in two separate polysaccharide blocks. In this example, the authors prepared a diblock copolymer from dextran and a dextran acetal through CuAAC coupling (Scheme 9).74 These dextran-based block copolymers self-assembled into micelles in aqueous solution and disassembled at pH 5–6 due to cleavage of the acid-labile acetals, making this polymer potentially suitable for drug delivery in mildly acidic environments such as inflammation sites and tumor tissue. The authors explored these micelles in the delivery of the hydrophobic model drug curcumin. The micelle was loaded with curcumin, and 95% of encapsulated drug was released after 24 h of dialysis at pH 5.5. This study highlights the potential in medicine for polysaccharide-based block copolymers, specifically those based on dextran, and may inspire continued development of dextran-based drug delivery vehicles.
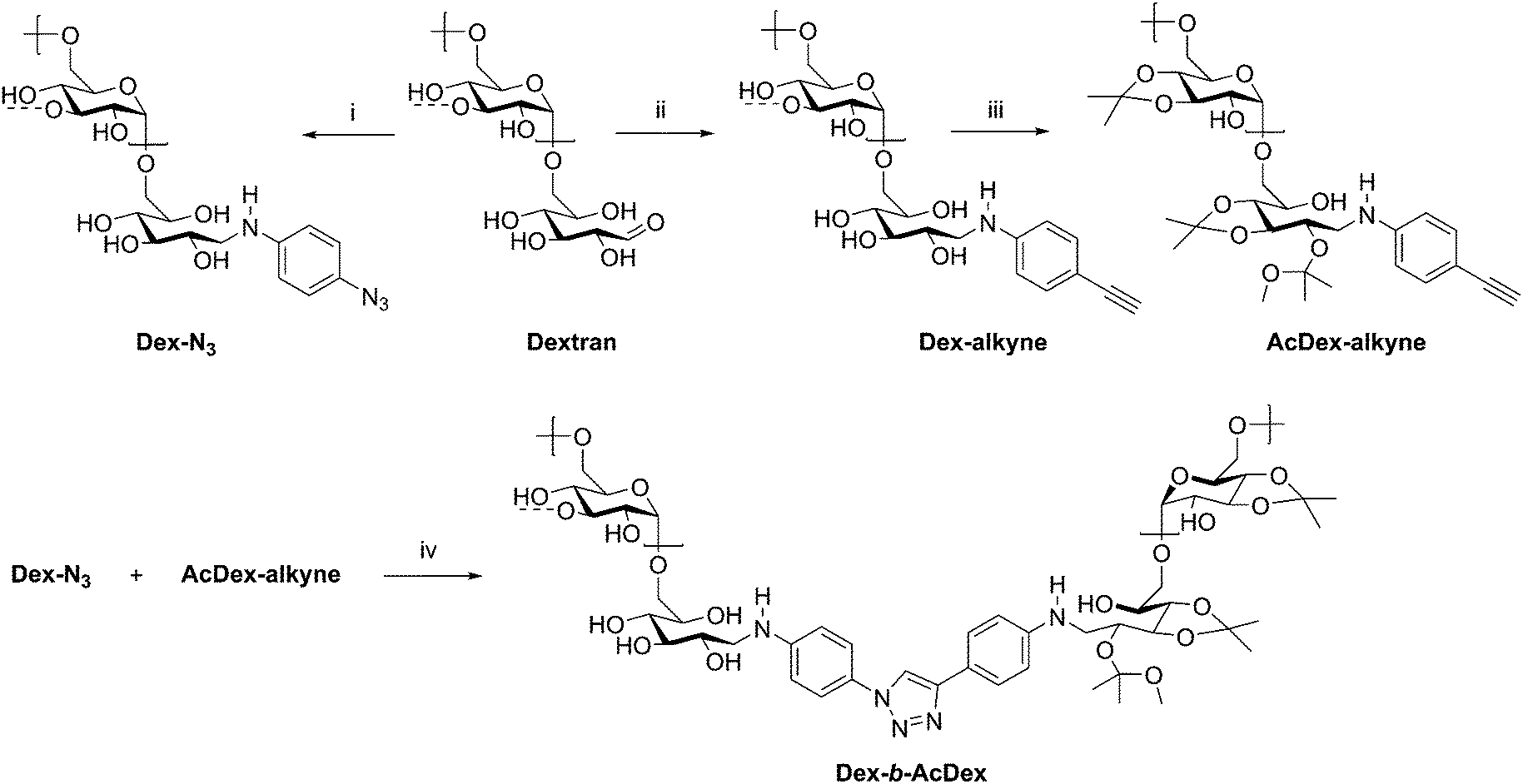 | ||
| Scheme 9 Synthetic route to diblock copolymer of dextran and acetalated dextran. (i) 4-Azidoaniline·HCl, NEt3, NaCNBH3, B(OH)3-buffer/MeOH, microwave, 4 h, 30 °C; (ii) 4-ethynylaniline, NaCNBH3, B(OH)3-buffer/MeOH, microwave, 4 h, 50 °C; (iii) PPTS, 2-methoxypropene, DMSO, 10 min, rt; (iv) CuBr, PMDETA, 72 h, 50 °C, Ar, DMSO. The dashed bonds on the 3-OH units indicate occasional branches present in dextran. Adapted from ref. 74. Copyright ACS 2017. | ||
Cancer therapy remains among the most investigated applications of amphiphilic block copolymers due to the ability of nanosized objects to deliver chemotherapeutics to tumor sites with greater specificity than that of pure drugs. Natural polysaccharides can be advantageous as degradable components of the delivery vehicles. One example is a dextran-b-PCL diblock copolymer, which was synthesized via disulfide exchange (Scheme 10).68 Dextran was first end-functionalized with cysteamine, followed by pyridyl disulfide to make Dex-SS-Py. Next, thiol-terminated PCL was reacted with Dex-SS-Py through an exchange reaction, producing the desired diblock copolymer with a molecular weight of 9100 g mol−1. Micelles prepared from this block copolymer were loaded with the chemotherapy drug doxorubicin (Dox). Upon exposure to a reductive environment, such as in the cytosol, the disulfide bond linking the two blocks was cleaved, readily removing the outer dextran shell of the micelle. The micelles demonstrated 70% drug loading efficiency and displayed quantitative Dox release over 10 h, resulting in the drug successfully reaching the cell nucleus. These diblock copolymer micelles may have advantages over PEG-based drug delivery vehicles given the drawbacks of PEG, including interaction with the immune system, degradation under stress, and bioaccumulation.75
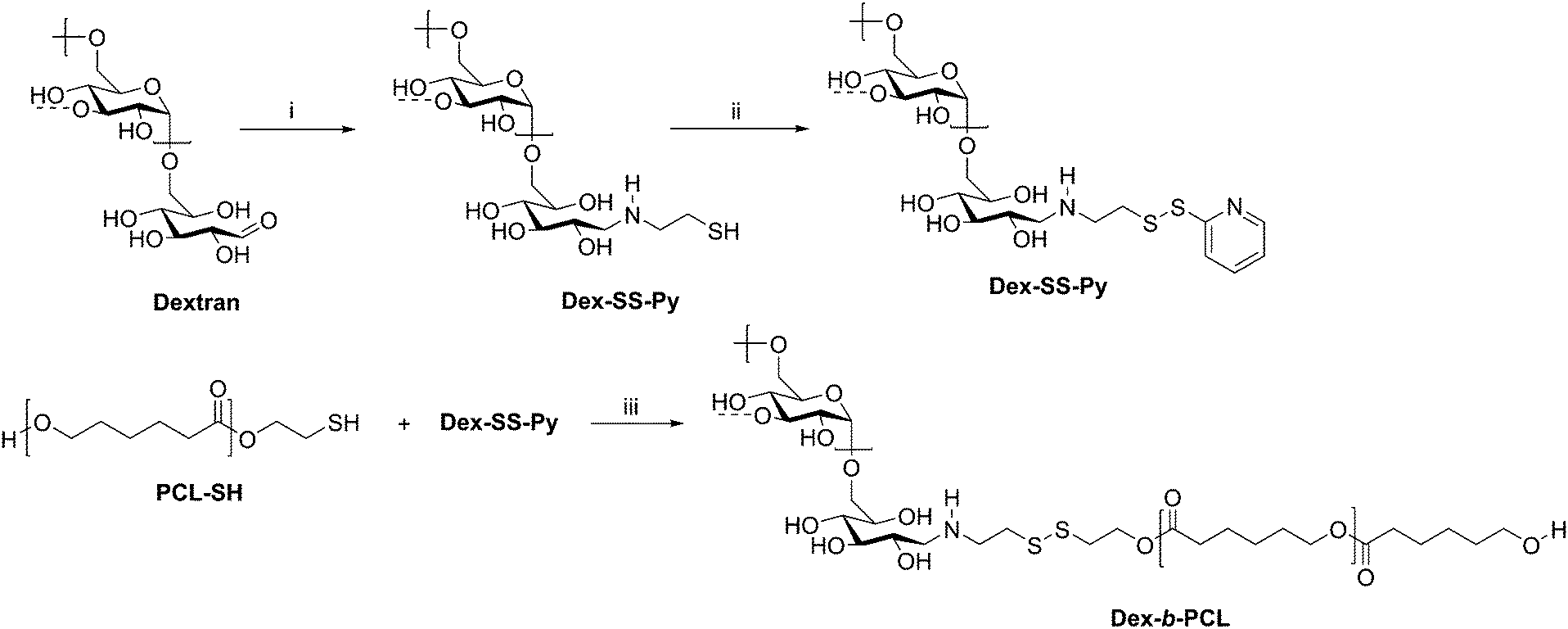 | ||
| Scheme 10 Synthesis of diblock copolymer dextran-b-PCL via disulfide exchange. (i) Cysteamine, NaCNBH3, DMSO/H2O, 60 °C for 2 d and 40 °C for 2 d; (ii) Py-SS-Py, pH 2, H2O, rt, 24 h; (iii) DMF/LiCl, acetic acid, 40 °C, 24 h. The dashed bonds on the 3-OH units indicate occasional branches present in dextran. Adapted from ref. 24. Copyright ACS 2010. | ||
In related work, control over Dox release was demonstrated with micelles constructed from dextran-b-PDLA and dextran-b-PLLA block copolymers (PDLA = poly(D-lactide) and PLLA = poly(L-lactide)), which were synthesized using click chemistry.25 PDLA and PLLA were synthesized using ROP and monofunctionalized with an azide on the ω-chain end. The dextran was then functionalized with propargylamine on the reducing end. The dextran-alkyne and PLA-azide polymers were coupled via CuAAC, yielding the diblock copolymers. Utilization of both D and L stereoisomers of PLA in solution led to stereocomplex formation, which aided in the stability of micelles and contributed to the slow release of Dox over 3 h (Fig. 1). These fully degradable block copolymers leave behind no polymeric or other harmful residues, and these types of micelle structures hold potential for delivering hydrophobic drugs for cancer therapy.
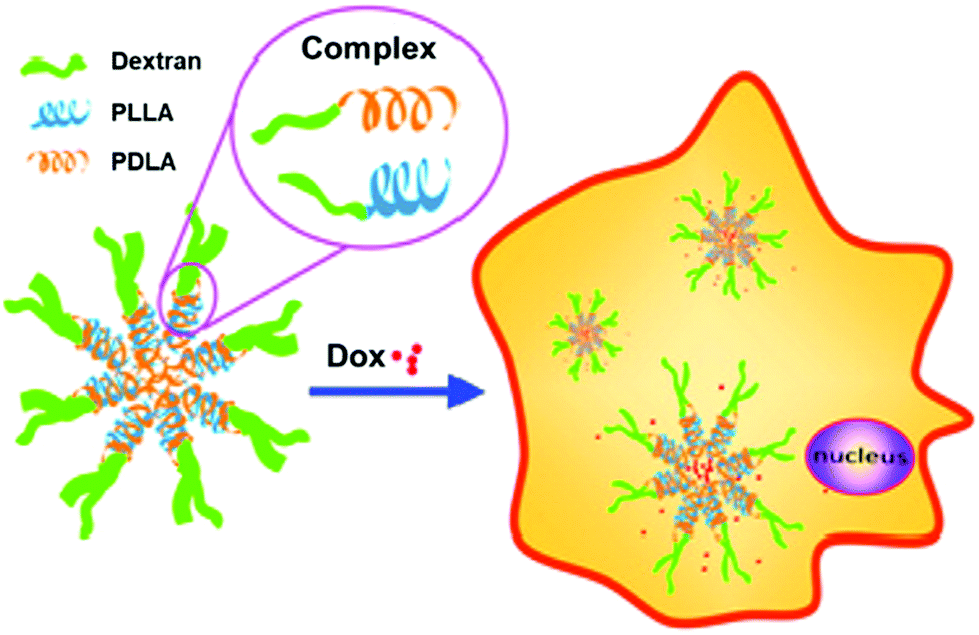 | ||
| Fig. 1 Illustration of stereocomplex formation between dextran-b-PLLA and dextran-b-PDLA, and subsequent loading and release of Dox. Adapted from ref. 25. Copyright ACS 2013. | ||
Hydrogels
Hydrogels are swollen networks of water-soluble polymers, and they have a variety of commercial applications, ranging from diapers to contact lenses. Beyond these uses, they play a special role in biomedical applications including tissue engineering and drug delivery due to their abilities to swell, encapsulate cells, and release drugs. Drug-loaded hydrogels can be injected or implanted at a desired site and release their payloads in a slow and controlled fashion. Additional control may come from inclusion of a stimuli-responsive unit triggered by pH, temperature, or light to alter the release profile.76–79 Polysaccharide hydrogels could be biodegradable competitors to commercial superabsorbent materials, such as polyacrylate polymers in diapers, and researchers are now exploring polysaccharide-containing diblock copolymer hydrogels for localized drug delivery.80–82Ganji and Abdekhodaie reported a chitosan-b-PEG diblock copolymer, which formed a temperature-responsive hydrogel.83 This block copolymer was synthesized by radical-induced fragmentation of chitosan in the presence of PEG-acrylate. In aqueous solution at room temperature, this block copolymer exhibited low viscosity. At 35 °C, the LCST for chitosan, a thermoreversible sol–gel transition was observed, making this material suitable for injection. Solutions of 2–3 wt% easily passed through a 22 gauge needle and required 6–11 minutes to fully reach the gel state, depending on the PEG/chitosan ratio of the block copolymer. These types of hydrogels hold potential as advanced materials for tissue engineering, cell encapsulation, adsorbents, and for localized drug delivery.
Functional materials: surfactants and interfacial agents
Development of non-biomedical applications for polysaccharide-containing block copolymers is at an early stage. For example, a block copolymer composed of xyloglucan and poly(dimethylsiloxane) (PDMS) was tested as a surfactant in aqueous solution, and it minimized surface tension at the air/water interface.26 Due to its neutral charge, this type of block copolymer may be attractive for industrial surfactant applications because it can be used in conjunction with other common uncharged surfactants. Additionally, Bosker et al. found that block copolymers of dextran and polystyrene, synthesized as described earlier, formed monolayers at the air/water interface.38 Hydrogen bonding in the dextran block led to slow absorption of the chains and the formation of aggregates at the interface in compression/decompression cycles.Beyond stabilizing air/water interfaces, block copolymers can also stabilize interfaces in blends of immiscible polymers. With the ability to anchor into both bulk phases, block(y) copolymers are used widely as compatibilizers in polymer blends. However, examples of blends of polysaccharides with synthetic polymers are rare. This concept was first explored in 2010, when blends of cellulose and polystyrene were compatibilized using a cellulose-b-polystyrene block copolymer.84 Cellulose/polystyrene blends were investigated using optical microscopy, and blends containing the block copolymer compatibilizer had reduced domain sizes compared with uncompatibilized blends, indicating that the compatibilizer stabilized the interfaces. The Matson lab also very recently investigated the role of the ABA triblock copolymer CTA-b-PB-b-CTA as a compatibilizer.28 Adding it to blends of CTA and PB consistently increased toughness, with a 60-fold increase in toughness compared with uncompatibilized blends and a 4-fold increase in toughness compared with CTA alone. Additionally, the PB domain sizes significantly decreased upon addition of the ABA triblock copolymer, suggesting that it was successful in decreasing interfacial tension between CTA and PB (Fig. 2). Blends of polysaccharides and synthetic polymers, compatibilized using polysaccharide-containing block copolymers, have seen little attention thus far, but their applications may be extensive. We plan to explore more blends and optimize compatibilizer structure in the future.
 | ||
| Fig. 2 SEM images portraying 90:10 wt% blends of CTA and PB containing (A) 0% CTA-b-PB-b-CTA compatibilizer and (B) 5% CTA-b-PB-b-CTA compatibilizer. (C) Display of the optical clarity of blends containing the compatibilizer. Adapted from ref. 28. Copyright ACS 2019. | ||
4. Conclusions and perspectives
Several synthetic methods have been developed to synthesize polysaccharide-containing block copolymers, enabling a variety of applications (Fig. 3). Maltoheptaose chain-end functionalization was an early method to form functionalized oligosaccharides that could be chain extended using various polymerization techniques. Currently the most popular methods for polysaccharide chain-end functionalization are reductive amination and oxime ligation, often aided by click chemistry. Both approaches enable coupling and chain extension pathways to diblock copolymers originating from a polysaccharide chain end. Reducing end functionalization with a leaving group (accompanied by degradation of degree of polymerization) followed by nucleophilic displacement provides an alternative route to polysaccharide chain end functionalization that avoids the long reaction times characteristic of other methods.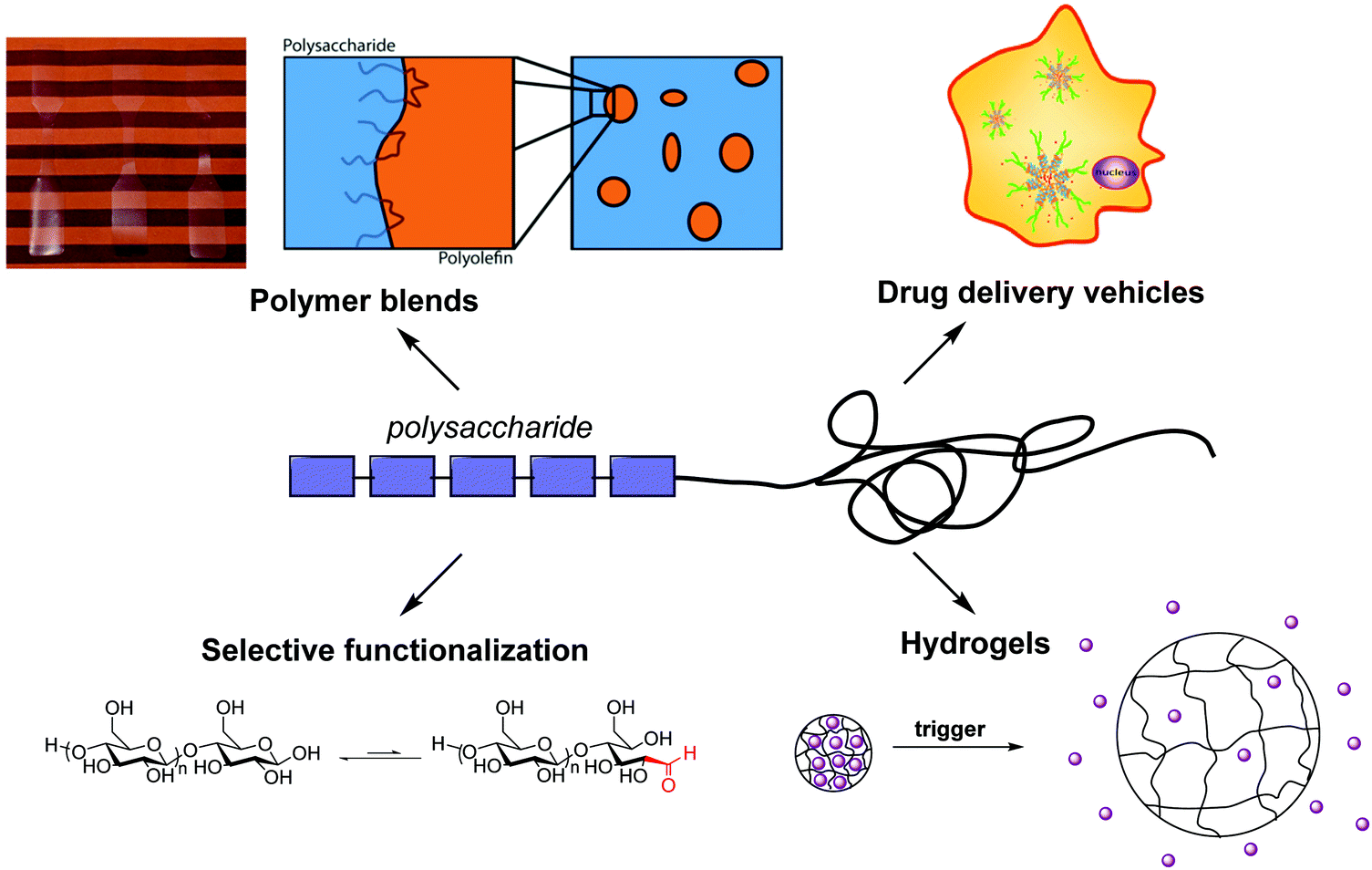 | ||
| Fig. 3 Representation of current research in polysaccharide-containing polymers and their applications. Adapted from ref. 25, 28 and 85. | ||
Challenges remain for those interested in synthesis of polysaccharide-based block copolymers. Some difficulties are shared with methods for preparation of fully synthetic block copolymers, most notably challenging separations of the desired block copolymer from other polymeric segments. In the case of polymer–polymer coupling methods, isolation of the desired copolymer from homopolymeric impurities is needed, except in cases of quantitative, stoichiometric couplings, which are rare and generally limited to low MW homopolymers. In the case of chain-extension methods, in which the modified polysaccharide is used as a macroinitiator (for example in the RDRP chemistries described earlier), separations may also be needed if quantitative initiation from the polysaccharide block is not achieved. In practice, one is frequently limited to synthesis of diblock copolymers where polymeric impurities can be effectively removed to afford pure diblock copolymer products. Use of highly efficient coupling methods not yet applied to polysaccharide chemistry may simplify these tedious purification steps.
Biodegradability is often cited as a useful property of polysaccharides; however, polysaccharide biodegradability varies with substitution. All native polysaccharides readily biodegrade. Depolymerization of polysaccharides is usually performed by a variety of depolymerases, which are comprised of glycoside hydrolases, glycosyl transferases, phosphorylases and lyases.86 Several factors may influence the tendency of derivatives to biodegrade, a major one being side chain substitution and DS. For example, native cellulose readily biodegrades because it has no substituents and the enzymes that break it down are highly abundant in nature.87 However, degradation of cellulose ethers and esters is more complex. Cellulose ether degradation typically is initiated by cellulases on monosaccharides that are unsubstituted or lightly substituted; more highly substituted cellulose ethers biodegrade slowly if at all, presumably due to lack of similarity to the natural cellulase substrate.88,89 Cellulose ester degradation begins with the action of lipases; since many lipases have long chain glycerol esters as their natural substrates, the reasons for the observed structure–property relationships (slower degradation of cellulose esters with longer chain acids, or with higher DS (ester)) for cellulose ester biodegradation are not clear.90,91 Cellulose “diacetate” (DS 2.5) biodegrades in a composting environment over the course of weeks, while cellulose triacetate and cellulose acetate propionate (DS(propionyl) 2.6), undergo little degradation under comparable conditions.92 Another issue with biodegradability in the context of polysaccharide-containing block copolymers is that any synthetic polymer blocks may be very slow to biodegrade. Finally, one must consider the breakdown products of these synthetic blocks. For example, while PEG is generally regarded as safe, polystyrene can break down to produce toxic monomers and oligomers. The formation of toxic byproducts in polysaccharide-containing block copolymers requires significant consideration, especially when discussing applications in the body and food packaging.
Nearly all of the current methods for making polysaccharide-containing block copolymers take advantage of the unique properties of the anomeric carbon (C1) of the reducing end monosaccharide, created because it is in equilibrium between hemiacetal and aldehyde (the only aldehyde in the molecule). In order to expand the structural possibilities of polysaccharide block copolymers, it would be greatly desirable to develop methods that would allow functionalization of the non-reducing chain terminus. There are no aldehydes on that terminal monosaccharide, and it is difficult to imagine how one might distinguish its primary alcohol (present in common monosaccharides like glucose, mannose, galactose, and 2-amino-2-deoxyglucose) from those on other monosaccharides, or likewise the secondary alcohols from those on other monosaccharides. One possibility may be highly selective enzyme-catalyzed modification, for example using glycosynthases for polysaccharides like vertebrate hyaluronan, whose chain is grown from the non-reducing end.93 Beyond enzymatic selectivity, chemo- and regioselective methods could take advantage of the hydroxyl that is involved in linkages in all other monosaccharides except the terminal one on the non-reducing end (for example, the 4-OH of 1,4-linked polysaccharides like amylose and cellulose). Chemistries like acetal formation might be able to take advantage of this structural feature.94,95 This is an important frontier in polysaccharide-based copolymer chemistry that may enable the construction of even more complex structures than the AB and ABA block copolymers described here.
Additional challenges involve analytical chemistry. Specifically, it can be difficult to quantify modification of the single reducing end of a high MW polysaccharide. For example, accurate determination of ratios of functionalized end group protons to those of the polysaccharide backbone by 1H NMR spectroscopy, a workhorse technique in polysaccharide chemistry, may be limited by the relatively very low intensity of signals from a single end-group substituent. Challenges unique to polysaccharides in determination of end-group functionalization and coupling efficiency by 1H NMR spectroscopy can include significant overlap of some substituent or copolymer segment signals (e.g., the methylene protons of PEG segments) with those of the polysaccharide backbone. It can also be challenging to quantify purity of a product block copolymer; in many cases size exclusion chromatography (SEC) is the only effective technique, and it requires both adequate signal and good chromatographic separation between diblock copolymer and homopolymer impurities.
New synthetic methods and better analytical methods for polysaccharide-containing block copolymer synthesis and characterization will enable new applications. Thus far, applications have focused primarily on drug delivery. Amphiphilic block copolymers self-assemble in water into spherical or other morphologies, which have been explored in several drug delivery applications. Surfactants and interfacial agents are more recent applications of polysaccharide-containing block copolymers with exciting potential in stabilizing air/water interfaces and phase domain boundaries in polymer blends. More complex block copolymers and potentially other polymer architectures may expand applications of polysaccharide-containing copolymers in these areas. Beyond drug delivery vehicles and interfacial agents, the biodegradability of polysaccharides may enable environmentally friendly detergents and cosmetics. We also envision that future work in this area may promote discoveries in cell–cell communication and cell–virus interactions. Overall, the continued development of synthetic methods for making complex polysaccharide-containing block copolymers will enable more applications, from household items to bio-inert drug delivery vehicles to complex biological mimics.
Conflicts of interest
There are no conflicts to declare.References
- R. D. Gilbert, Cellulosic polymers, blends and composites, Hanser, 1994 Search PubMed.
- R. E. Oesper, J. Chem. Educ., 1929, 6, 677 CrossRef CAS.
- Bericht über die Verhandlungen der Naturforschenden Gesellscahft in Basel, ed. W. Haas, Naturforschende Gesellschaft in Basel, Basel, 1844 Search PubMed.
- L. Redman and A. Mory, Ind. Eng. Chem., 1931, 23, 595–597 CrossRef CAS.
- L. H. Baekeland, US Pat., 942699, 1909 Search PubMed.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- J. A. Manson, Polymer blends and composites, Springer Science & Business Media, 2012 Search PubMed.
- C. García-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425–1438 CrossRef.
- I. Brückle, The Book and Paper Group Annual, 1996, vol. 15, pp. 25–26 Search PubMed.
- Z. Shi, Y. Zhang, G. O. Phillips and G. Yang, Food Hydrocolloids, 2014, 35, 539–545 CrossRef CAS.
- E. M. Renkin, J. Gen. Physiol., 1954, 38, 225–243 CAS.
- F. A. Müller, L. Müller, I. Hofmann, P. Greil, M. M. Wenzel and R. Staudenmaier, Biomaterials, 2006, 27, 3955–3963 CrossRef PubMed.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688 CrossRef CAS.
- K. J. Edgar, Cellulose, 2007, 14, 49–64 CrossRef CAS.
- H. C. Arca, L. I. Mosquera-Giraldo, V. Bi, D. Xu, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2018, 19, 2351–2376 CrossRef CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer chemistry, CRC Press, 2007 Search PubMed.
- M. Furuta and T. Maruyama, US Pat., 5071912, 1991 Search PubMed.
- N. Hadjichristidis, M. Pitsikalis, S. Pispas and H. Iatrou, Chem. Rev., 2001, 101, 3747–3792 CrossRef CAS.
- O. Nuyken and S. D. Pask, Polymers, 2013, 5, 361–403 CrossRef.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- A. Kermagoret and D. Gigmes, Tetrahedron, 2016, 72, 7672–7685 CrossRef CAS.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS.
- Z. Zhao, Z. Zhang, L. Chen, Y. Cao, C. He and X. Chen, Langmuir, 2013, 29, 13072–13080 CrossRef CAS.
- S. Halila, M. Manguian, S. Fort, S. Cottaz, T. Hamaide, E. Fleury and H. Driguez, Macromol. Chem. Phys., 2008, 209, 1282–1290 CrossRef CAS.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- K. J. Arrington, J. V. Haag Iv, E. V. French, M. Murayama, K. J. Edgar and J. B. Matson, ACS Macro Lett., 2019, 8, 447–453 CrossRef CAS.
- C. Schatz and S. Lecommandoux, Macromol. Rapid Commun., 2010, 31, 1664–1684 CrossRef CAS.
- R. J. Ceresa, Polymer, 1961, 2, 213–219 CrossRef CAS.
- S. Kobayashi, K. Kashiwa, J. Shimada, T. Kawasaki and S.-i. Shoda, Makromol. Chem., Macromol. Symp., 1992, 54–55, 509–518 CrossRef.
- V. Codera, K. J. Edgar, M. Faijes and A. Planas, Biomacromolecules, 2016, 17, 1272–1279 CrossRef CAS.
- D. M. Haddleton and K. Ohno, Biomacromolecules, 2000, 1, 152–156 CrossRef CAS.
- B.-G. Li and L.-M. Zhang, Carbohydr. Polym., 2008, 74, 390–395 CrossRef CAS.
- R. F. Borch, M. D. Bernstein and H. D. Durst, J. Am. Chem. Soc., 1971, 93, 2897–2904 CrossRef CAS.
- D. S. Dalpathado, H. Jiang, M. A. Kater and H. Desaire, Anal. Bioanal. Chem., 2005, 381, 1130–1137 CrossRef CAS.
- O. Larm and E. Scholander, Carbohydr. Res., 1977, 58, 249–251 CrossRef CAS.
- W. T. E. Bosker, K. Ágoston, M. A. Cohen Stuart, W. Norde, J. W. Timmermans and T. M. Slaghek, Macromolecules, 2003, 36, 1982–1987 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- B. Worrell, J. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- M. Arslan, G. Acik and M. A. Tasdelen, Polym. Chem., 2019, 10, 3806–3821 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- J. Collins, Z. Xiao, M. Müllner and L. A. Connal, Polym. Chem., 2016, 7, 3812–3826 RSC.
- C. Schatz, S. Louguet, J. F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS.
- X. Meng and K. J. Edgar, Prog. Polym. Sci., 2016, 53, 52–85 CrossRef CAS.
- J. Wang, M. Caceres, S. Li and A. Deratani, Macromol. Chem. Phys., 2017, 218, 1600558 CrossRef.
- B. Pfannemüller, Naturwissenschaften, 1975, 62, 231–233 CrossRef.
- K. Kobayashi, S. Kamiya and N. Enomoto, Macromolecules, 1996, 29, 8670–8676 CrossRef CAS.
- K. Loos and R. Stadler, Macromolecules, 1997, 30, 7641–7643 CrossRef CAS.
- N. Enomoto, S. Furukawa, Y. Ogasawara, H. Akano, Y. Kawamura, E. Yashima and Y. Okamoto, Anal. Chem., 1996, 68, 2798–2804 CrossRef CAS.
- K. Akiyoshi, M. Kohara, K. Ito, S. Kitamura and J. Sunamoto, Macromol. Rapid Commun., 1999, 20, 112–115 CrossRef CAS.
- K. Loos and A. H. Müller, Biomacromolecules, 2002, 3, 368–373 CrossRef CAS.
- K. Kobayashi, S. Kamiya and N. Enomoto, Macromolecules, 1996, 29, 8670–8676 CrossRef CAS.
- V. v. Braunmuehl, G. Jonas and R. Stadler, Macromolecules, 1995, 28, 17–24 CrossRef.
- S. Kamiya and K. Kobayashi, Macromol. Chem. Phys., 1998, 199, 1589–1596 CrossRef CAS.
- R. Novoa-Carballal and A. H. E. Müller, Chem. Commun., 2012, 48, 3781–3783 RSC.
- S. M. Brosnan, H. Schlaad and M. Antonietti, Angew. Chem., Int. Ed., 2015, 54, 9715–9718 CrossRef CAS.
- R. Novoa-Carballal, A. Pfaff and A. H. Müller, Polym. Chem., 2013, 4, 2278–2285 RSC.
- K. J. Arrington, J. B. Waugh, S. C. Radzinski and J. B. Matson, Macromolecules, 2017, 50, 4180–4187 CrossRef CAS.
- D. F. Williams, Biomaterials, 2014, 35, 10009–10014 CrossRef CAS.
- N. Zhang, P. Wardwell and R. Bader, Pharmaceutics, 2013, 5, 329–352 CrossRef CAS.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS.
- S. Maiti, S. Ranjit and B. Sa, Int. J. PharmTech Res., 2010, 2, 1350–1358 CAS.
- Y. Liu, J. Sun, P. Zhang and Z. He, Curr. Med. Chem., 2011, 18, 2638–2648 CrossRef CAS.
- Y. Dong, L. I. Mosquera-Giraldo, L. S. Taylor and K. J. Edgar, Polym. Chem., 2017, 8, 3129–3139 RSC.
- K. Akiyoshi, S. Yamaguchi and J. Sunamoto, Chem. Lett., 1991, 1263–1266 CrossRef CAS.
- T. Ngawhirunpat, N. Wonglertnirant, P. Opanasopit, U. Ruktanonchai, R. Yoksan, K. Wasanasuk and S. Chirachanchai, Colloids Surf., B, 2009, 74, 253–259 CrossRef CAS PubMed.
- Y. Xiong, J. Qi and P. Yao, Macromol. Biosci., 2012, 12, 515–524 CrossRef CAS.
- Y. Dong, D. C. Novo, L. I. Mosquera-Giraldo, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2019, 221, 37–47 CrossRef CAS.
- B. B. Breitenbach, I. Schmid and P. R. Wich, Biomacromolecules, 2017, 18, 2839–2848 CrossRef CAS.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS.
- H. Meng and J. Hu, J. Intell. Mater. Syst. Struct., 2010, 21, 859–885 CrossRef CAS.
- M. Prabaharan and J. F. Mano, Macromol. Biosci., 2006, 6, 991–1008 CrossRef CAS.
- A. M. Kloxin, C. J. Kloxin, C. N. Bowman and K. S. Anseth, Adv. Mater., 2010, 22, 3484–3494 CrossRef CAS.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS.
- J. Kuang, K. Y. Yuk and K. M. Huh, Carbohydr. Polym., 2011, 83, 284–290 CrossRef CAS.
- H. Li, J. Yang, X. Hu, J. Liang, Y. Fan and X. Zhang, J. Biomed. Mater. Res., Part B, 2011, 98, 31–39 CrossRef.
- A. Pourjavadi, V. Aghajani and H. Ghasemzadeh, J. Appl. Polym. Sci., 2008, 109, 2648–2655 CrossRef CAS.
- F. Ganji and M. J. Abdekhodaie, Carbohydr. Polym., 2008, 74, 435–441 CrossRef CAS.
- S. Yagi, N. Kasuya and K. Fukuda, Polym. J., 2010, 42, 342 CrossRef CAS.
- I. P. Harrison and F. Spada, Pharmaceutics, 2018, 10, 71 CrossRef.
- B. A. Stone, B. Svensson, M. E. Collins and R. A. Rastall, in Glycoscience: Chemistry and Chemical Biology, ed. B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 2325–2375 Search PubMed.
- I. Homma, H. Fukuzumi, T. Saito and A. Isogai, Cellulose, 2013, 20, 2505–2515 CrossRef CAS.
- J. Karlsson, D. Momcilovic, B. Wittgren, M. Schülein, F. Tjerneld and G. Brinkmalm, Biopolymers, 2002, 63, 32–40 CrossRef CAS.
- S. D. Seneker and J. E. Glass, Adv. Chem. Ser., 1996,(248), 125–137 CrossRef CAS.
- C. M. Buchanan, D. Dorschel, R. M. Gardner, R. J. Komarek, A. J. Matosky, A. W. White and M. D. Wood, J. Environ. Polym. Degrad., 1996, 4, 179–195 CrossRef CAS.
- C. M. Buchanan, D. D. Dorschel, R. M. Gardner, R. J. Komarek and A. W. White, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 683–697 CrossRef.
- C. M. Buchanan, C. N. Boggs, D. D. Dorschel, R. M. Gardner, R. J. Komarek, T. L. Watterson and A. W. White, J. Environ. Polym. Degrad., 1995, 3, 1–11 CrossRef CAS.
- S. Bodevin-Authelet, M. Kusche-Gullberg, P. E. Pummill, P. L. DeAngelis and U. Lindahl, J. Biol. Chem., 2005, 280, 8813–8818 CrossRef CAS.
- E. M. Bachelder, T. T. Beaudette, K. E. Broaders, J. Dashe and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 10494–10495 CrossRef CAS PubMed.
- G. Vessella, S. Traboni, D. Cimini, A. Iadonisi, C. Schiraldi and E. Bedini, Biomacromolecules, 2019, 20, 3021–3030 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |