
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inert macrocyclic Eu3+ complex with affirmative paraCEST features†
Tanja
Gambino
 a,
Laura
Valencia
b,
Paulo
Pérez-Lourido
b,
David
Esteban-Gómez
c,
Moritz
Zaiss
de,
Carlos
Platas-Iglesias
*c and
Goran
Angelovski
*a
a,
Laura
Valencia
b,
Paulo
Pérez-Lourido
b,
David
Esteban-Gómez
c,
Moritz
Zaiss
de,
Carlos
Platas-Iglesias
*c and
Goran
Angelovski
*a
aMR Neuroimaging Agents, MPI for Biological Cybernetics, Tuebingen, Germany. E-mail: goran.angelovski@tuebingen.mpg.de
bDepartamento de Química Inorgánica, Facultad de Ciencias, Universidade de Vigo, As Lagoas, Marcosende, 36310 Pontevedra, Spain
cCentro de Investigacións Científicas Avanzadas (CICA) and Departamento de Química, Facultade de Ciencias, Universidade da Coruña, 15071 A Coruña, Spain. E-mail: carlos.platas.iglesias@udc.es
dHigh-Field Magnetic Resonance, MPI for Biological Cybernetics, Tuebingen, Germany
eDepartment of Neuroradiology, University Clinic Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Erlangen, Germany
First published on 16th March 2020
Abstract
We report on a macrocyclic platform based on an 18-membered macrocycle that forms kinetically highly inert paramagnetic complexes and possesses an excellent outlook for the development of bioresponsive paraCEST (paramagnetic chemical exchange saturation transfer) contrast agents. The investigated europium(III) chelate is non-hydrated and contains four amide groups, each possessing two paramagnetically shifted proton resonances distant from bulk water. The X-ray crystal structure and solution studies indicate that the metal ion is ten-coordinated, being directly bound to the six N atoms of the macrocycle and the four amide O atoms of the pendant arms. The complex presents an excellent inertness with respect to dissociation, being stable under a variety of harsh conditions, including highly acidic and basic media or elevated temperatures. The amide protons are in slow-to-intermediate exchange with bulk water, which gives rise to the generation of a strong CEST effect at low probe concentration and saturation powers (∼25% at 5 mM, B1 = 5 μT, 37 °C). We demonstrate the potential of this platform for mapping pH in its microenvironment and foresee potential for the development of diverse paraCEST probes and sensors.
Introduction
Efficient detection and monitoring of pathological processes is a key step in ensuring the timely diagnosis of numerous diseases. Various molecular imaging techniques aim for developing reliable protocols to visualize such biological events, with magnetic resonance imaging (MRI) being at the forefront of this progress. In addition to the ability to cover larger volumes or enable investigations of tissues at any depth, MRI possesses outstanding potential and versatility to study functional processes with an unprecedented specificity and spatial resolution.1,2 This was initially recognized slightly over two decades ago with the first reports on the ‘smart’ contrast agents;3,4 meanwhile different nuclei, MRI protocols or bioresponsive probes have been employed in attempts to assess the functional state of tissues,5,6 especially the environmental pH as one of the best physiological markers of different diseases, including inflammation,7 infection,8 ischemia9 and cancer.10Chemical exchange saturation transfer (CEST) is an emerging MRI methodology appreciated due to a few advantageous features that circumvent the current shortcomings of other existing methods.11 CEST takes advantage of the magnetization transfer between the exchanging species operating at two different frequencies (most frequently a pool of protons from the CEST probe and bulk water), thus reducing the magnetization of the latter once the former species is irradiated with a radiofrequency (RF) pulse of defined energy. The great advantage of this strategy is the ability of providing signal response at will: the CEST effect or image will be generated only with the application of desired saturation pulses. Moreover, the existence of at least two separate exchange processes (e.g. two separate pools of protons in exchange with protons from bulk water) paves the way for highly specific, multifrequency and consequently multicolour CEST measurements. It also favours ratiometric approaches, i.e. exploitation of these separate exchange processes for providing information on the influence of an ion or a molecule in the probe microenvironment, but independent on probe concentration.12 Finally, since the exchange rates are intrinsically tied to the resulting intensity of CEST, any alteration on the exchange rates will ultimately modulate the recorded CEST effect. For instance, pH changes in the microenvironment of the CEST pair(s) affect the saturation transfer process and thus the CEST MR image.
Several useful approaches have been reported in attempts to develop CEST probes and methods enabling pH mapping in tissue,13–21 or for development of bioresponsive agents that can help observing various biological processes.22–25 For instance, a recent report demonstrated the use of ratiometric CEST MRI to map renal pH using a diaCEST agent in vivo.26
Irrespective if they are comprised of diamagnetic molecules or those with paramagnetically shifted protons (dia- or paraCEST probes, respectively), their common feature is the presence of the CEST-active pool sufficiently distant from bulk water frequency. This ensures effective magnetization transfer (MT) and adjustment of the exchange rates in the slow exchange regime, thus allowing low energy saturation pulses to produce detectable CEST effects. In fact, this is a critical aspect for the use of any CEST-based probe in vivo: due to the specific absorption rate (SAR) limitations, i.e. the energy deposition in tissue, it is essential to use low energy saturation pulses (low B1 fields).27,28 Despite an advantageous feature of the currently available paraCEST agents to shift the CEST frequency sufficiently away from the semi-solid MT (tissue background CEST), most of them still lack the optimal exchange regimes to provide the most efficient labelling efficiency (α) and hence to be used at reduced probe concentrations at low B1 fields. These limitations of the currently available probes are certainly the main obstacle for the translation of CEST agents to clinical practice, in spite of the intense research efforts in this field during the last two decades.29–31
Taking these demands into consideration, we developed a paramagnetic platform that encompasses the majority of the parameters necessary for a prospective paraCEST probe. We built upon the previous work on the 18-membered macrocyclic molecule that provides exceptionally inert Pr3+, Eu3+ or Yb3+ complexes and exhibits the paraCEST effect.32 We improved the previous system by incorporating amide instead of hydroxyl groups (Fig. 1), thus obtaining 8 instead of 4 exchanging protons. Furthermore, being geometrically oriented in a different manner, we anticipated their grouping into two pairs of protons. This opens the possibility of getting two CEST effects at two different frequencies. An additional favourable property of this 18-membered macrocycle platform is the formation of Ln3+ complexes that lack coordinated water molecules.32–35 The presence of coordinated water molecules in exchange with bulk water shortens the longitudinal relaxation times of the bulk water signal, which is detrimental to the CEST effect.36 Concurrently, this feature increases the number of coordination bonds between the multidentate chelator and the metal ion, thus significantly improving inertness of the obtained chelate.
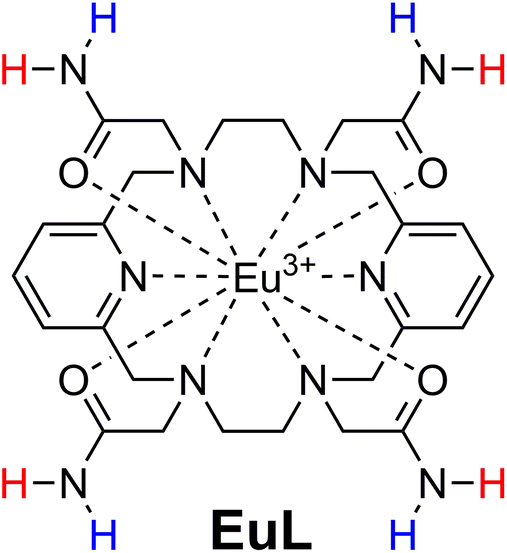 | ||
| Fig. 1 Chemical structure of the complex studied in this work. NMR-equivalent protons on amides were deliberately marked with same colour. | ||
Experimental section
General remarks
1H NMR spectra of the ligand and complexes were recorded on a Bruker ARX400 spectrometer, while the CEST experiments were performed on a Bruker Avance III NMR spectrometer (Bruker, Ettlingen, Germany). Chemical shifts were referenced by using the residual solvent proton signals.37 Elemental analysis was performed in a Carlo-Erba EA 1108 microanalyzer. Attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) spectra were recorded as KBr discs on a Bruker VECTOR 22 spectrometer. ESI experiments were performed on an microTOF(focus) mass spectrometer (Bruker Daltonics, Bremen, Germany) or an ion trap SL 1100 system from Agilent (Germany). For the former, the ions were generated using an ApolloII (ESI) source and ionization was achieved by electrospray.UV-Vis absorption spectra were recorded on a Jasco V-650 spectrometer using 1 cm quartz cells. The excitation and emission spectra in the UV-Vis region were obtained with a Horiba FluoroMax Plus-P spectrofluorometer equipped with a 150 W ozone-free xenon arc lamp and a R928P photon counting emission detector. All spectra were corrected for the instrumental response provided by the manufacturer. An integration time of 0.1 s was used in all steady state measurements. Luminescence decays were measured on the same instrument working in the phosphorescence mode using a xenon flash lamp. Emission lifetimes were obtained by least-squares fits of the decay data using monoexponential decay functions. The emission quantum yield of EuL was obtained with optically diluted solutions using the trisdipicolinate complex Cs3[Eu(pic)3] (Φ = 13.5% in TRIS buffer, pH 7.4) as reference.38,39
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and ν(CN)py], 1676 (s) [ν(CO)], 3165 (m) [ν(NH)]. MS (ESI-MS, m/z, found (calculated)): 555.3 (555.3) [L + H]+. 1H NMR (CD3OD, δ, ppm): H1 7.70 (t, 2H), H2 7.25 (d, 4H, 3J = 7.7 Hz), H4 3.37 (b, 8H), H5 2.75 (b, 8H), H6 3.14 (b, 8H).
C) and ν(CN)py], 1676 (s) [ν(CO)], 3165 (m) [ν(NH)]. MS (ESI-MS, m/z, found (calculated)): 555.3 (555.3) [L + H]+. 1H NMR (CD3OD, δ, ppm): H1 7.70 (t, 2H), H2 7.25 (d, 4H, 3J = 7.7 Hz), H4 3.37 (b, 8H), H5 2.75 (b, 8H), H6 3.14 (b, 8H).
General procedure for the preparation of the complexes
A solution of Ln(NO3)3·xH2O (0.04 mmol) in methanol (5 mL) was added to a stirred solution of L·H2O (0.023 g, 0.04 mmol) in the same solvent (10 mL) and the mixture was slightly heated. Slow concentration of the methanolic solutions gave crystalline products that were isolated by filtration and dried.C) and ν(CN)py], 1651 (s) [ν(CO)], 3262 (m), 3162 (m) ν(NH2), 1314 (s), 829 (m) [ν(NO3−)] cm−1. Crystals suitable for X-ray diffraction were obtained by slow evaporation of an aqueous solution of the complex.
C) and ν(CN)py], 1661 (s) [ν(CO)], 3169 (m) ν(NH2), 1321 (s), 888 (m), 747 (m) [ν(NO3−)] cm−1. Crystals with formula [YL](NO3)3·3H2O suitable for X-ray diffraction were obtained by slow evaporation of an aqueous solution of the complex.
Crystal structure determinations
Crystallographic data of [EuL](NO3)3·3H2O were collected at 293(2) K on a BRUKER Smart-CCD-1000 diffractometer using Graphite monochromated Mo-Kα radiation. All data were corrected by Lorentz and polarization effects. For [YL](NO3)3·3H2O, crystallographic data were measured at room temperature using a Bruker Smart 6000 CCD detector and Cu-Kα radiation (λ = 1.54178 Å) generated by an Incoatec microfocus source equipped with Incoatec Quazar MX optics. The software APEX2 was used for collecting frames of data, indexing reflections, and the determination of lattice parameters, while SAINT was used for integration of the intensity of reflections.41 The software SADABS was used in all cases for scaling and empirical absorption correction.42 All structures were solved by using the SHELXT program and refined by a full-matrix least-squares based on F2.43 The Squeeze program was used to correct the reflection data for the diffuse scattering due to the disordered nitrate ions and water molecules present in the unit cell of [EuL](NO3)3·3H2O.44 Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in idealized positions and refined with isotropic displacement parameters. CCDC 1886847 and 1886848† contain the supplementary crystallographic data for this paper. Molecular graphics were generated using ORTEP-3.45CEST experiments and the optimal kex
For each temperature, data were collected by varying the saturation power whilst the saturation time remained constant (5 s for experiments reported in Fig. 4, or 15 s for the experiments reported in Fig. 5, ESI Fig. S8–11† and Table 1). The saturation field strengths used were: 2.5, 3, 5, 6, 7.5, 10 and 15 μT for experiments reported in the Fig. 4, or 1, 2.5, 5, 7.5, 10, 12.5 and 15 μT for the experiments reported in Fig. 5, S8–11† and Table 1. Longitudinal relaxation times were obtained in an independent experiment using the standard inversion-recovery method with 1% gradient to eliminate the radiation damping effect. The CEST experiments at variable pH (experiments reported in Fig. 5, ESI Fig. S8–11† and Table 1) were performed using a 5 mm NMR tube with EuL dissolved in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture H2O:D2O (v/v). The CEST experiments with shorter saturation (5 s) were performed using a 5 mm NMR tube filled with an aqueous solution of EuL or iopamidol and a smaller 2 mm NMR tube (1.6 mm inner diameter) filled with D2O. For data evaluation, Z-spectra were normalized by an unsaturated fully relaxed water signal M0. The CEST effect was calculated employing the inverse asymmetry analysis of the normalized Z-magnetization. The inverse difference of the magnetization transfer, MTRind, was calculated according to eqn (1), where M0 is the unsaturated water magnetization, while Mz+ and Mz− are magnetizations of the on-resonance at the frequency +Δω and of the off-resonance at the frequency −Δω relative to bulk water, respectively.46
:1 mixture H2O:D2O (v/v). The CEST experiments with shorter saturation (5 s) were performed using a 5 mm NMR tube filled with an aqueous solution of EuL or iopamidol and a smaller 2 mm NMR tube (1.6 mm inner diameter) filled with D2O. For data evaluation, Z-spectra were normalized by an unsaturated fully relaxed water signal M0. The CEST effect was calculated employing the inverse asymmetry analysis of the normalized Z-magnetization. The inverse difference of the magnetization transfer, MTRind, was calculated according to eqn (1), where M0 is the unsaturated water magnetization, while Mz+ and Mz− are magnetizations of the on-resonance at the frequency +Δω and of the off-resonance at the frequency −Δω relative to bulk water, respectively.46
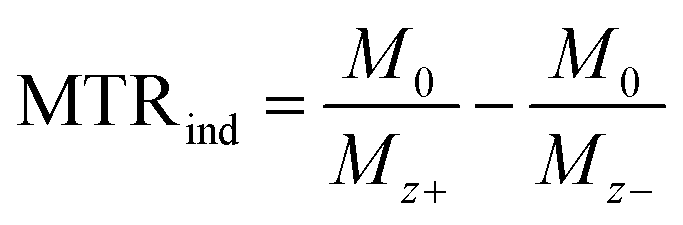 | (1) |
| pH | Peak at 8 ppm | Peak at 14 ppm |
|---|---|---|
| 6.0 | 138 ± 19 | 66 ± 23 |
| 6.4 | 321 ± 30 | 162 ± 31 |
| 6.8 | 941 ± 75 | 487 ± 69 |
| 7.2 | 2631 ± 130 | 1364 ± 101 |
| 7.6 | 5631 ± 225 | 2977 ± 150 |
| 8.0 | 15481 ± 1792 |
7368 ± 1002 |
Simultaneous multi-B1 Z-spectra fitting was performed as described previously,47 using the tool published online.48
Theoretically, the CEST effect that can be acquired with a B1 of 10 μT (2π·400 Hz ≅ 2700 Hz) directly depends on the exchange rate by the labeling efficiency relation (eqn (2)):
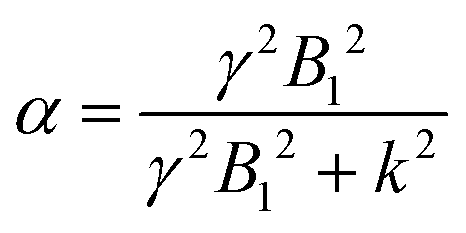 | (2) |
It is also well known and easy to calculate that this function has a maximum of 0.5 for k = γ × B1. Thus for the limit of B1 = 10 μT, the optimal CEST effect can be achieved with a CEST exchange rate of k = γ × 10 μT = 2700 Hz.
For mimicking a grey brain matter environment of the paraCEST agents, the water and semisolid MT pool were added to the CEST simulation using a water longitudinal relaxation time T1w = 1.67 s, and a transverse relaxation time T2w = 0.043 s. The semisolid MT pool (Super-Lorentzian lineshape) was defined by its fractional concentration χMT = 0.05, transverse relaxation time T2MT = 9.1 μs, exchange rate kex(MT) = 40 Hz; the chemical shift was set to δMT = 0 ppm for simplicity.49 The reported CEST effects were obtained by subtracting the simulated Z-spectrum that assumed the water, MT and one or two paraCEST pools with the simulated Z-spectrum that assumed the water and MT pools only.
MRI experiments
MRI measurements on tube phantoms were performed on a Bruker BioSpec 70/30 USR magnet (software version Paravision 5.1) using a Bruker volume coil (RF RES 300 1H 075/040 QSN TR). The MRI phantom consisted of 6 vials filled with a EuL solution in PBS (3 mM) and the pH ranging from 6.0 to 8.0.Z-spectra were acquired at room temperature (∼22 °C) with 139 irradiation offsets ranging from 20 to −20 ppm, saturation pulse duration of 5 s, and five different radiofrequency field strengths, B1 = 3, 4, 5, 6 and 7.5 μT.
Presaturated MR images were acquired using the rapid acquisition with relaxation enhancement (RARE) imaging protocol with the following parameters: repetition time (TR)/echo time (TE) = 15316.18/3.26 ms, field of view (FOV) = 48 × 48 mm, matrix size (MTX) = 96 × 96, slice thickness 2 mm, rare factor = 96, number of excitation (NEX) = 1, acquisition time (TA) = 35 min 28 s 948 ms.
Image analysis was performed in MATLAB (MathWorks, USA). Z-spectrum images were interpolated by splines and shifted to the centre frequency in order to remove B0 inhomogeneity artefacts.
pH evaluation using the ratiometric method was done using the inverse matrix approach, as described above in eqn (1). For generating pH maps, the pH values were plotted as a function of the corresponding MTRind ratio values. The obtained values were fitted using an exponential function according to eqn (3), yielding a monotonic function pH(MTRind ratio). This function was then applied pixel-wise to MTRind ratio maps to generate the pH maps.
| pH = c − a × e−b×MTRind ratio | (3) |
Results and discussion
Preparation
The synthesis of L started by the preparation of the 3,6,10,13-tetraaza-1,8(2,6)-dipyridinecyclotetradecaphane precursor by condensation of diformylpyridine and ethylenediamine using Ba2+ as a template, and subsequent reduction of the Schiff-base intermediate with NaBH4 (59% yield).40,50 Alkylation of the macrocycle with bromoacetamide in acetonitrile using Na2CO3 as a base provided L in 51% yield. The paramagnetic Eu3+ complex was obtained in good yield (60%) by direct reaction of the ligand with the appropriate hydrated lanthanide nitrate in methanol. The mass spectrum (ESI+) shows intense peaks due to the [Eu(L–2H)]+ entity that confirm the formation of the complex (ESI Fig. S1†).X-ray crystallography
Crystals of formula [EuL](NO3)3·3H2O were obtained by slow evaporation of aqueous solutions of the complex. They contain the [EuL]3+ cations, highly disordered nitrate anions and water molecules. While the quality of the crystallographic data of the Eu3+ complex is not very high, the overall structure of the [EuL]3+ complex and the bond distances and angles of the metal coordination environment are reasonably accurate (Fig. 2 and ESI Tables S1 and 2†). Furthermore, we obtained single crystals of the Y3+ analogue (ESI Fig. S2 and ESI Tables S1 and 2†), which assumes a very similar structure.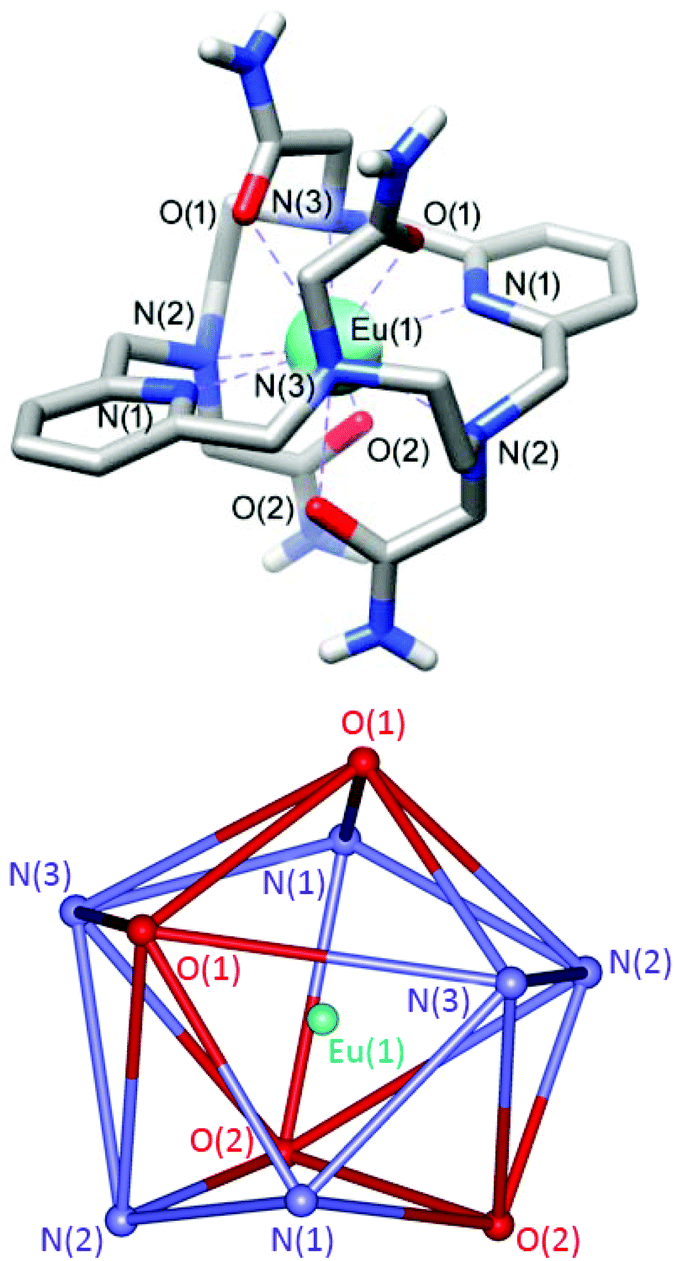 | ||
| Fig. 2 Top: View of the structure of the [EuL]3+ cation present in crystals of [EuL](NO3)3·3H2O (0.80 Å resolution). Hydrogen atoms (except those of amide groups) are omitted for simplicity. Bond distances [Å]: Eu–N(1), 2.593(15); Eu–N(2), 2.63(2); Eu–N(3), 2.644(19); Eu–O(1), 2.525(16); Eu–O(2), 2.511(15). Bottom: View of the coordination polyhedron around the metal ion. | ||
The [EuL]3+ cations present ten-coordinate metal ions that are directly bonded to the six donor atoms of the macrocyclic skeleton and the four oxygen atoms of the acetamide pendants (Fig. 2). While ten coordination is relatively uncommon for lanthanide complexes in aqueous solution, similar ten-coordinated structures were observed previously for complexes based on the same macrocyclic platform functionalized with acetate,33 methynenepyridine34 or hydroxyethyl32,35 pendant arms. The ligand L adopts a twist-wrap conformation in which the angles involving the two pyridyl nitrogen atoms and the metal ion are nearly linear (>179°). The two pyridyl units are twisted with respect to each other, so that the least-squares planes intersect at angles of ca. 19–20°. The coordination polyhedron can be best described as a sphenocorona, as confirmed by performing shape measures with the SHAPE program.51,52 A sphenocorona is a 1:5:4 polyhedron (Fig. 2) relatively common in the chemistry of the lanthanide ions.53,54 In the present case the least-squares containing 5 and 4 donor atoms present rather large mean deviations from planarity (0.27 and 0.21 Å, respectively, Fig. 2).
Kinetic inertness
The 1H NMR spectrum of EuL recorded in a H2O solution presents 10 paramagnetically shifted signals in the range ∼20 to −20 ppm, which is in line with the D2 symmetry observed in the solid state. The spectrum recorded in 1 M HCl remains unchanged over a period of at least 25 days (ESI Fig. S3†), with no signals due to the free ligand being observed. This experiment evidences that the complex presents an astonishing inertness under very harsh conditions. For instance, the commercially available contrast agent [Gd(DOTA)]− (DOTA – 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetate) dissociates with a half-live of ∼30 hours under these conditions.55 The chemical shifts of the paramagnetically shifted signals could be easily assigned by comparison with those of the analogue bearing hydroxyethyl pendant arms instead of acetamides.35 The similarity of the 1H NMR spectra of these two complexes indicates that they present similar magnetic anisotropies, and thus similar coordination environments in solution (ESI Table S3†).Additional experiments were carried out to assess the inertness of EuL in phosphate buffer at pH 7.0 upon addition of one equivalent of Zn2+. These conditions were proposed to study the stability of Gd-based contrast agents.56 The 1H NMR spectra of EuL (5 mM, 25 °C) recorded in phosphate buffer (67 mM) before and after addition of one equivalent of Zn2+ are identical, and confirm the stability of the complex under these conditions. The spectra did not show signals due to diamagnetic species and the solution did not show visible presence of precipitate due to insoluble EuPO4. Moreover, the 1H NMR spectrum remained unchanged upon incubation of the mixture at 40 °C for 72 h. 1H NMR experiments were also recorded at variable temperatures of up to 73 °C (ESI Fig. S4†). Again no dissociation of the complex was observed, nor signs of fluxional behaviour, which shows that the complex presents a very rigid structure in solution. The only noticeable change observed upon increasing temperature is the decrease of the observed paramagnetic shifts, as expected due to the 1/T and 1/T2 dependencies of the contact and pseudocontact contributions, respectively.57 The 1H NMR spectrum also remains unchanged upon increasing the pH to 10.0 (ESI Fig. S5†). Finally, the EuL complex was challenged with 10 equivalent of the DTPA chelator (DTPA – diethylenetriamine pentaacetic acid) in phosphate buffer at pH 7.0. The luminescence steady-state emission spectrum of the solution did not experience any noticeable changes upon addition of DTPA over a period of 72 h (ESI Fig. S6†). Furthermore, the luminescence emission decay curves recorded before and after addition of DTPA are virtually superimposable, demonstrating that the EuL complex remains intact under these conditions (ESI Fig. S7†). These experiments confirm the astonishing kinetic inertness of the complex, which appears to be related to the efficient protection from the environment of the metal ion in the ten-coordinate structure.
UV/Vis and luminescence properties
The UV absorption and luminescence emission experiments on the EuL complex gave further insights on its coordination and photophysical properties in solution (Fig. 3). The absorption spectrum presents a maximum at 268 nm (ε = 11000 M−1 cm−1) characteristic of the pyridyl group,58 and this band can be used to sensitized Eu3+ luminescence emission. The absorption and excitation spectra are almost identical, which indicates that Eu3+ sensitization occurs via energy transfer from excited states of the ligand to Eu3+ excited states. The luminescence emission spectrum consists of the 5D0 → 7FJ bands typical for Eu3+. The integrated and corrected relative intensities of the 5D0 → 7FJ transitions represent 0.21, 15.8, 25.7, 5.5, 39.5, 2.5 and 10.8% of the overall emission intensity for J = 0, 1, 2, 3, 4, 5 and 6, respectively. Thus, the overall emission intensity is dominated by the 5D0 → 7F4 transition, while the intensities of the 5D0 → 7F1 and 5D0 → 7F2 transitions are rather similar.
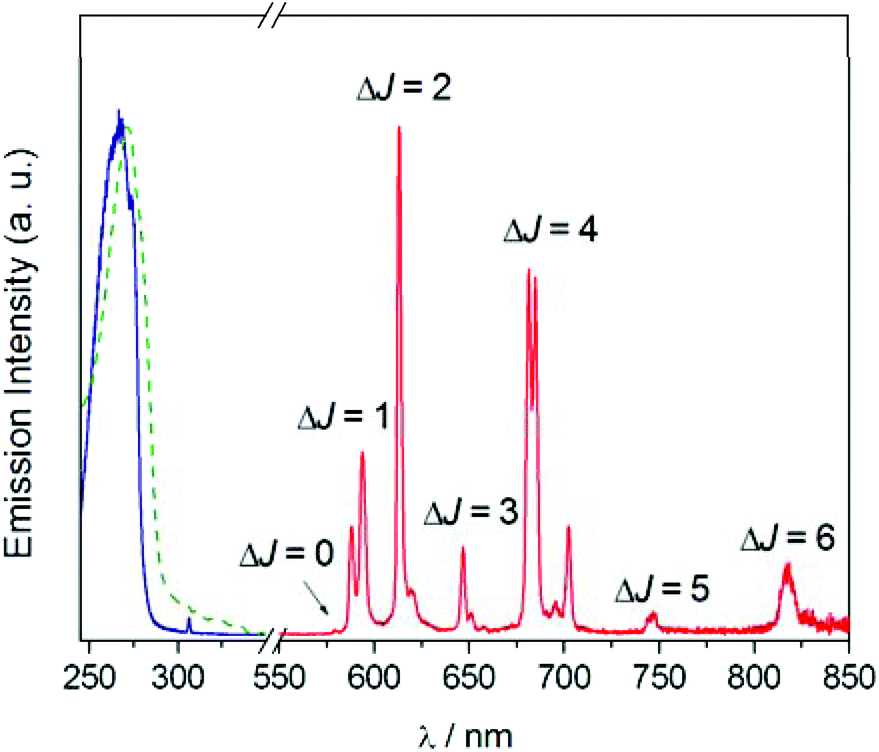 | ||
| Fig. 3 UV-Vis absorption (dashed green line), excitation (solid blue line) and emission (solid red line) spectra of EuL recorded in water (10−5 M). λexc = 268 nm, λem = 613 nm. | ||
Furthermore, the 5D0 → 7F0 transition is very weak and the 5D0 → 7F1 transition displays only two of the maximum 2J + 1 components. This emission pattern is characteristic of a rather symmetric crystal field around the metal coordination environment,59 which is attributed to the D2 symmetry observed both in the solid state and in solution. The emission spectrum exhibits unusually intense ΔJ = 5 and ΔJ = 6 transitions, a rare feature that is characteristic of this type of coordination.34 Further investigations indicated that the excited 5D0 state presents a monoexponential decay with a lifetime of 0.914 ms and a modest luminescence quantum yield of ΦEu = 0.11, which can be attributed to the presence of amide NH oscillators and possibly water molecules in the second-hydration sphere that provide a rather efficient vibrational deactivation of the 5D0 excited state of Eu3+.60 The emission lifetime determined in D2O solution amounts to 1.902 ms, which results in a hydration number of q = 0 by using the method proposed by Beeby including the correction associated to the presence of four amide NH oscillators.60
CEST experiments
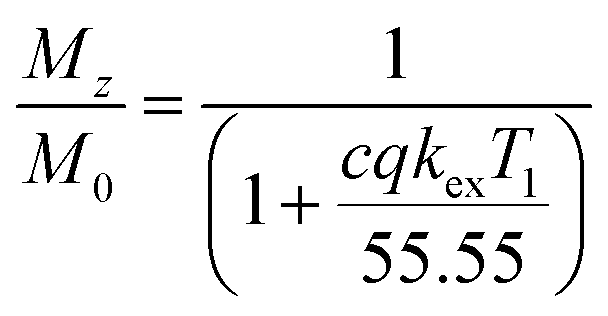 | (4) |
The Z-spectra of EuL were recorded at 25 and 37 °C and a magnetic field of 7 T (Fig. 4). Already at the chelate concentration of 5 mM and low saturation pulse power (5 μT), the advantageous CEST properties of this complex became obvious. Namely, two paramagnetically shifted and well resolved resonances at 15 and 9.5 ppm from bulk water exhibited CEST effect of ∼25% at 25 °C, which corresponds to a proton transfer enhancement (PTE) of 5550.61 Upon heating to 37 °C, the intensity of CEST effect remained similar, while the shift of the resonances decreased by ∼1 ppm along with a broadening of the peak closer to bulk water, suggesting an increase in the exchange rates due to higher temperature. For comparison, we recorded the Z-spectra of iopamidol (Isovue™ or Solutrast™) under the same conditions (5 mM, pH 7.4, 25 and 37 °C).
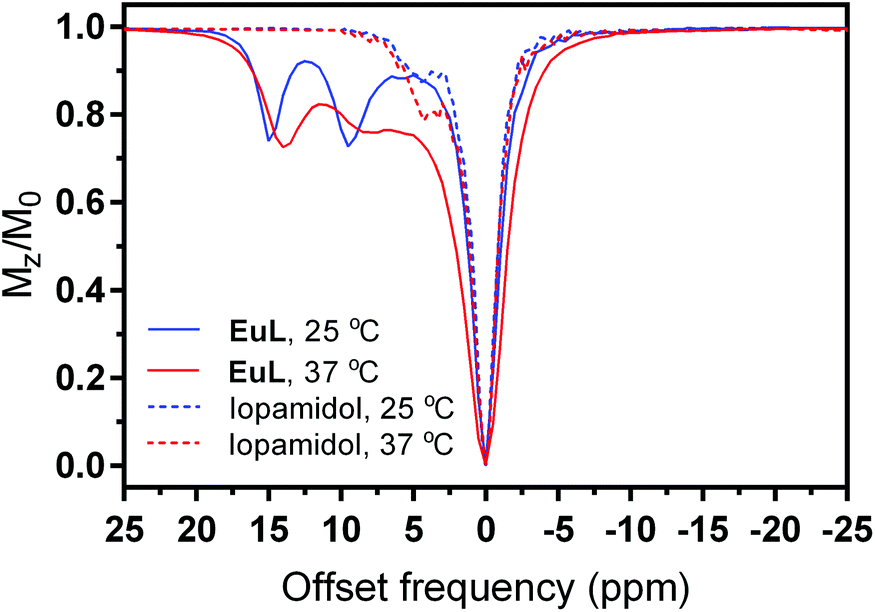 | ||
| Fig. 4 Z-spectra of EuL (lines) and iopamidol (dashes) at 25 °C (blue) and 37 °C (red) at 5 mM concentration, B1 = 5 μT and saturation time 5 s (PBS, pH 7.4). | ||
In addition to the existence of diamagnetic CEST peaks closer to the resonance of bulk water, their intensity was weaker than in EuL, reaching a maximum CEST effect of 20% (Fig. 4). We have applied quantitative CEST (qCEST) analysis to determine exchange rate values (kex),47 using the Bloch–McConnell (BM) equations and assuming a 3-pool model (bulk water and two paramagnetically-shifted exchanging pools). The qCEST procedure employing data of 7 different saturation powers resulted in kex values of 1029 ± 63 Hz and 1914 ± 78 Hz for 25 °C and 2652 ± 132 Hz and 4699 ± 229 Hz at 37 °C, with the strongly shifted peak (14–15 ppm) having an exchange rate which was roughly twice as slow as that of the peak with smaller paramagnetic shift. These kex values are just slightly higher than those reported for iopamidol also at the physiological pH and fall into the optimal region for reaching the maximal α value at the low B1 fields applied for preclinical and clinical use (see below and description of CEST experiments in Experimental section);62,63 hence, we proceeded with the characterization of the EuL complex as a potential pH-sensitive paraCEST agent. Namely, the exchange rates are expected to change significantly with variations in pH, which would give rise to different CEST effects. We therefore recorded a series of Z-spectra at variable pH values from 6.0 to 8.0, using pulses with different saturation powers ranging from 1 to 15 μT (ESI Fig. S8†); subsequently, we performed the qCEST as well as the inverse difference of opposite frequencies (asymmetry analysis) to obtain the kex values and the inverse MT ratio difference (MTRind), respectively.46
The multi-B1 experiments at variable pH values revealed advantageous exchange properties of EuL for pH sensing. The exchange rates remained roughly in 2:1 ratio throughout the studies for the peak with smaller shift (Table 1). CEST effects of at least 5% were recorded already using B1 = 2.5 μT at all pH values above 6.4; we note that this lies within the saturation power limits permitted for use at clinical settings, ensuring low SAR levels. Moreover, the increase of exchange rates along with the increase in pH additionally affected the CEST effects. When MTRind were plotted as a function of pH, the CEST signals exhibited pH dependency until pH ∼ 7.4 (ESI Fig. S9†). Above this pH the MTRind dropped, due to a decreased labelling efficiency at the given B1 level and the faster exchange.
The concentration independent ratio of different MTRind was calculated either by comparing the effects at different frequencies and same saturation power, or using the so-called ratio of RF power mismatch method.15 When the obtained ratios were plotted as a function of pH, the pH-dependent effect became even more obvious (Fig. 5 and ESI Fig. S10†). The ratio of MTRind at B1 = 7.5 μT and 2.5 μT at either frequencies exhibited significant changes in the region from slightly acidic to physiological pH (6.4–7.4, Fig. 5a), as previously observed in the analysis and behaviour of iobitrol.15 Similar results were obtained when the ratio of MTRind at B1 = 7.5 μT and 14 ppm versus the signal ratio at B1 = 2.5 μT and 8.5 ppm was calculated, or when just the ratios of the signals of two peaks at B1 = 5 μT was used (Fig. 5b), as previously demonstrated for the behaviour of iopamidol.64 We stress these are very advantageous properties, highly desirable in the application of responsive probes: all results reported here were achieved with EuL at low saturation powers and probe concentrations. Next, the ratiometric analysis yields results that are independent of probe concentration.12 Finally, the existence of CEST effects at two frequencies that – for a fixed power – changes proportionally with pH due to a virtually constant ratio of kex values (Table 1 and ESI Fig. S11†) allows for a simplified analysis in which the results obtained with one experiment can be cross-checked with those at the other frequency, i.e. one CEST signal can serve as an internal reference to another one.
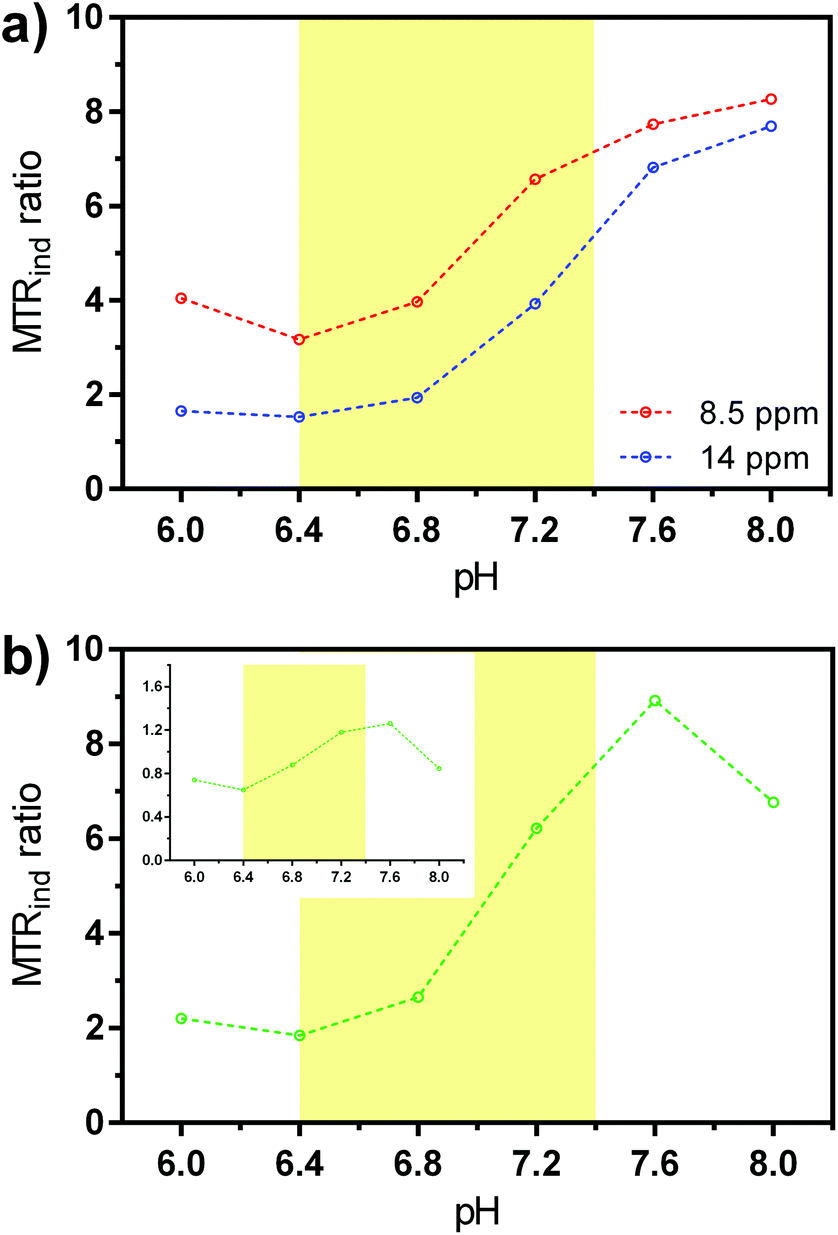 | ||
| Fig. 5 Ratio of MTRind for EuL (5 mM in PBS, 37 °C) at varying pH values. (a) Ratio of signals at 7.5 μT and 2.5 μT at 8.5 ppm (red) and 14 ppm (blue). (b) Ratio of signal obtained with 7.5 μT at 14 ppm with the signal obtained with 2.5 μT at 8.5 ppm; inset: the ratio of signals at 14 and 8.5 ppm and B1 = 5 μT. The pH region 6.4–7.4 is highlighted with yellow color. | ||
Thus, we attempted to combine the ample knowledge and experience collected in the field of CEST MRI in order to foresee the paraCEST agents that comprise the most promising properties and suggest the best direction for their further improvement. We summarized the majority of existing paraCEST agents by grouping them into the four groups (A to D, respectively), according to the similarities either by the exchange rate or paramagnetic shift (ESI Table S4†). We then performed the initial set of simulations to emphasize the importance of a particular physical parameter in correlation with other standard parameters and values. Finally, we combined the particular set of physical values valid for each one of the groups and made their direct comparison. In all cases, we considered two types of outputs, i.e. parameters that suit clinical or preclinical conditions and measurements.
For paraCEST probes, we used the following grouping principles (definitions of slow, intermediate and fast exchange rates were simplified for the purpose of presenting these results; otherwise they depend on the NMR time scale of the applied magnetic field):
– Group A: Paramagnetic transition-metal complexes (Fe2+, Ni2+, Co2+), having slow exchange rates (kex = 200–400 Hz), high paramagnetic shift (>50 ppm) and high number of exchanging amide protons (up to 4);17,18,65–67
– Group B: Paramagnetic EuL complex from this work, characterized by intermediate exchange rates (kex = 1.3–2.6 kHz at pH 7.2), fairly small paramagnetic shift (8–15 ppm) and high number of exchanging amide protons (4);
– Group C: Paramagnetic Tm3+ complex characterized with an intermediate exchange rate (kex = 3 kHz), high paramagnetic shift (∼−50 ppm) and high number of exchanging amide protons (4).21 Here, we considered also the Yb3+ complex, which is also based on a DOTA chelator and should have similar CEST properties. This complex exhibits smaller paramagnetic shift (similar to that of group B); however, we estimate its performance to belong to this group according to the kex values reported for 25 °C only.68
– Group D: Paramagnetic Eu3+ and Tb3+ complexes characterized with fast exchange rates (kex = 10 kHz and above), high paramagnetic shift (∼50 and −550 ppm) and low number of exchanging water protons (2).20,69,70 The term fast exchange should not be related to the actual exchange regime on the NMR time scale, but rather how the exchange rate compared with those of groups A–C.
For the simulations, we used the computational method that incorporates in vivo-like water and semi-solid MT properties.71 Namely, by defining a set of different parameters for the abstract cases or existing paraCEST probes, we estimated CEST effects,72 which can be expected in the brain tissue (grey matter) at 7 T magnetic field. Also, we assumed the use of a pulse with lower SAR (B1 = 5 μT, saturation time = 0.5 s) to mimic the clinical CEST MRI limits, whereas the pulses with higher power and longer duration and thus higher SAR can be tolerated for the preclinical cases without having significant impact for welfare of the animal subject (B1 = 10 μT, saturation time = 5 s). These two conditions are important restrictions, if performance is compared without restriction for SAR, faster exchanging sites will always win. All these studies assumed cases with same concentration of the exchanging protons (10 mM); otherwise, the obtained results can be scaled by the appropriate factor, if lower or higher concentrations are used. We first performed calculations for the abstract cases, aiming to show variations of the particular physical parameter while varying only one of these parameters (Fig. 6). If fixing the exchange rate to 200, 2000 and 10000 Hz (slow to fast exchange rates), respectively, the results indicate that probes with the intermediate kex (up to a few kHz, see below) are the most suitable for the clinical settings, irrespective of the frequency shift of the CEST resonance (Fig. 6a). On the other hand, generally faster exchange rates are preferred for the preclinical settings, while the frequency shift of the paraCEST agent shows the following trend: a shift of at least 50 ppm is preferred, whereas at >100 pm the negative effect of the semi-solid MT is completely eliminated (Fig. 6b). We note that larger paramagnetic shifts are preferred at scanners with the lower magnetic field (e.g. at 3 T) to reach beyond the semi-solid MT, while this effect is reducing with the increase of the magnetic fields (i.e. semi-solid MT covers smaller frequency region at higher magnetic fields; hence lower paramagnetic shifts of the CEST effect on the paraCEST agent can be tolerated).73
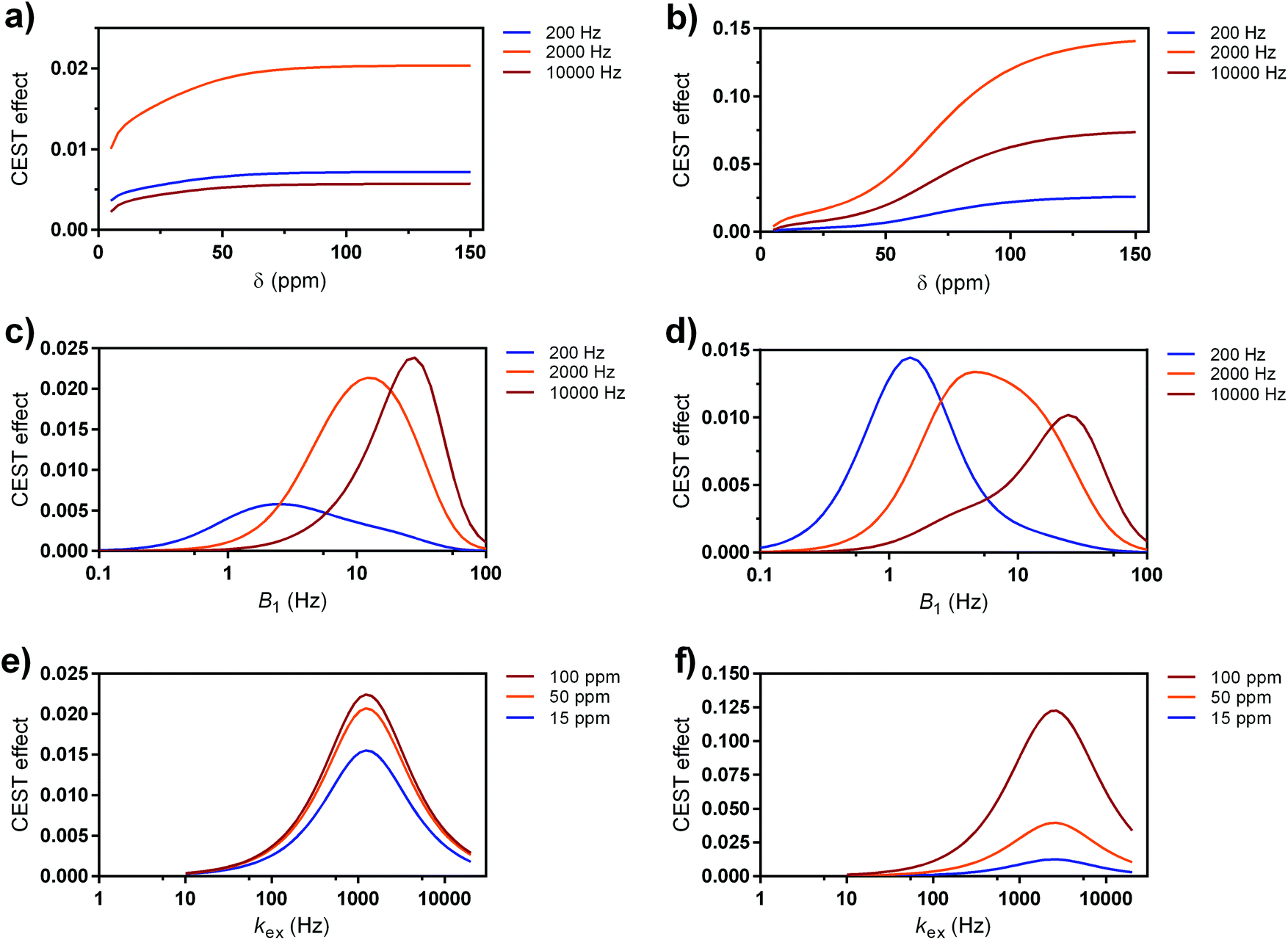 | ||
| Fig. 6 Simulated CEST effects of paraCEST probes at 7 T magnetic field at (a, c and e) clinical (B1 = 5 μT, sat. time = 0.5 s) and (b, d and f) preclinical settings (B1 = 10 μT, sat. time = 5 s), respectively. (a and b) Dependence at three different kex regimes as a function of paramagnetic shift of CEST resonance. (c and d) Dependence at three different kex regimes as a function applied saturation power at δ = 15 ppm. (e and f) Dependence at three different resonance shifts as a function of kex value. | ||
Next, we checked the optimal B1 fields (saturation powers) for the maximal labelling efficiency for these three types of exchange rates (slow, intermediate and fast), at the fixed resonance frequency that corresponds to EuL (δB = 15 ppm). At both the clinical and preclinical settings, intermediate exchange rate (groups B–C) has the maximal α in suitable regions of B1 that match the requirements (B1 < 5 or 10 μT, respectively). For groups A and D (low and high kex, respectively) it is obvious that labelling exists; however, it is less efficient (Fig. 6c and d).
Finally, when the value for CEST resonance frequency is fixed against the variable kex values, one concludes that an intermediate exchange rate of just a few kHz is optimal for the best labelling; furthermore, the large paramagnetic shift of the CEST resonance(s) is advantageous, although the differences are not significant at the clinical settings (Fig. 6e). However, highly shifted CEST resonances are very advantageous for preclinical conditions (Fig. 6f).
Another set of simulations was performed using the specific set of parameters valid for each of the A–D groups of molecules (ESI Table S4†). The most important matter in these calculations was including the difference in the abundance of exchanging protons that cause CEST effect. Namely, the calculations assumed same concentrations of molecules (5 mM), in which the clear advantage went in direction of those having higher number of exchanging protons per molecule (groups A–C).
Furthermore, we included two examples for each group A and D, aiming to provide better comparison between structurally and behaviourally different systems. In specific, we included a macrocyclic Fe2+ complex,65 an acyclic Co2+ complex,18 macrocyclic Eu3+ and Tb3+ complexes20,69,70 as group A1, A2, D1 and D2 probes, respectively (ESI Table S4†). In this calculation, we did not consider faster T1 relaxation noticed for the group A and C probes; this effect will reduce the maximal CEST effects that is based on their other physical properties presented here (kex, paramagnetic shift, number of exchanging protons).
The obtained results were expressed either in the form of simulated Z-spectra (Fig. 7a and b) or CEST effects (Fig. 7c and d), again for clinical and preclinical settings (Fig. 7a, c and b, d, respectively). They again showed the advantageous properties of the paramagnetic EuL platform.74 Although the other three groups of probes possess CEST effects that are well paramagnetically shifted, their other features result in similar magnitude of the signal as EuL. Indeed, EuL has excellent properties for the clinical settings: it is not only that its effects are comparable to those of group A and C probes – indeed, only EuL together with group A2 probe exhibit two strong CEST signals (Fig. 7c) suitable for ratiometric investigations. Also, while other compounds show increased CEST effects for preclinical settings, EuL shows almost the same effect size in both settings.
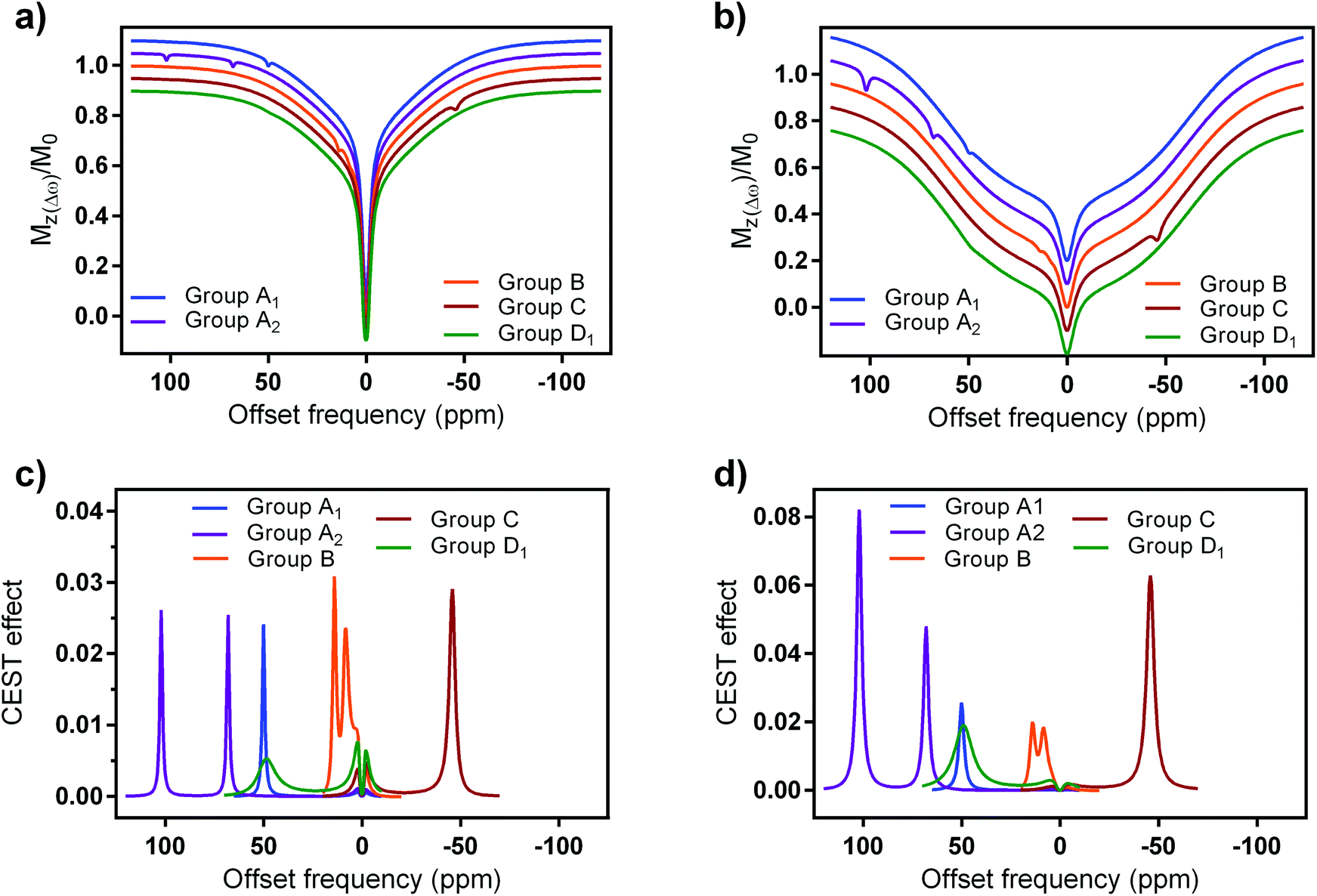 | ||
| Fig. 7 Simulated (a and b) z-spectra and (c and d) CEST effects of selected paraCEST probes at 7 T magnetic field at a, (c) clinical (B1 = 5 μT, sat. time = 0.5 s) and (b and d) preclinical settings (B1 = 10 μT, sat. time = 5 s), respectively. Water and semisolid MT parameters were chosen to mimic grey brain matter, in specific: water relaxation times, T1w = 1.67 s and T2w = 0.043 s; fractional concentration, transverse relaxation time, exchange rate and chemical shift of the MT pool: χMT = 0.05, T2MT = 9.1 μs, kex(MT) = 40 Hz, δMT = 0 ppm; Super-Lorentzian lineshape.74 | ||
The results presented here show some further aspects that should be considered in future design and application of paraCEST probes. Group A2 and C probes also display strong signals at both clinical/preclinical settings, mainly due to possession of high number of exchanging protons, optimal or slightly slower exchange rates and high paramagnetic shift (Fig. 7d). Finally, the group D probes show limitations for further applications under clinical settings. Here, lower number of exchanging protons clearly has a major impact on the final CEST effect, while fairly fast exchange rates make them suitable for possible investigations at preclinical settings (Fig. 7d and ESI Fig. S12†).
 | ||
| Fig. 8 CEST MRI on tube phantoms with EuL (3 mM in PBS, RT). pH maps were generated from the ratio of experiments done at B1 = 7.5 and 4 μT, saturation time 5 s and with the peak at 9.7 ppm. | ||
Conclusions
In summary, we have reported on a paramagnetic platform that forms kinetically highly inert complexes and has an excellent outlook for the development of entirely new class of paraCEST probes. The investigated Eu3+ chelate displays very affirmative CEST features due to two paramagnetically shifted protons that are in slow to intermediate exchange with bulk water. The variations in pH reflect on the intensity of the obtained CEST effect, which exhibits pH-dependent behaviour either at a single resonance or from the ratio of applied saturation powers and different resonance peaks. Moreover, the high intensity of the CEST effect allows for applications of small quantities of this paraCEST probe, as well as low saturation powers that match clinically approved protocols. Indeed, the theoretical calculations predict that this platform has one of the best set of cumulative properties for CEST applications, which should become the landmark for development of novel classes of paraCEST probes.We want to highlight that this promising platform likely requires additional chemical modifications to be used for in vivo applications. For instance, it has been shown that tri-cationic DOTA-tetraamide complexes are toxic, while negatively charged analogues are well tolerated at much higher doses.77 Nevertheless, the chemical nature of this platform envisions an immense number of potential chemical transformations and the development of a great variety of structures that, combined with a number of chelated paramagnetic metals, can further improve on the already excellent features to result in probes sensitive to different types of targets. For instance, functionalization of the acetamide pendants with negatively charged groups at the α-carbon would result in negatively charged complexes that should maintain the D2 symmetry of the complex and two resonances for the amide groups. Ultimately, this macrocyclic platform could pave the way to many exciting advances in the field of responsive paraCEST agents with high potential for clinical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Rolf Pohmann and Dr Dario Longo for helpful discussions. T. G. thanks the German Academic Exchange Service (DAAD) for the Ph.D. fellowship. D. E.-G. and C. P.-I. thank Ministerio de Economía y Competitividad (CTQ2016-76756-P) and Xunta de Galicia (ED431B 2017/59 and ED431D 2017/01) for generous financial support. M. Z. thanks the research support of the German Research Foundation (DFG, grant ZA 814/2-1) and European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 667510). Open Access funding provided by the Max Planck Society.Notes and references
- A. E. Merbach, L. Helm and É. Tóth, The chemistry of contrast agents in medical magnetic resonance imaging, Wiley, Chichester, 2013 Search PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS.
- R. A. Moats, S. E. Fraser and T. J. Meade, A “smart” magnetic resonance imaging agent that reports on specific enzymatic activity, Angew. Chem., Int. Ed. Engl., 1997, 36, 726–728 CrossRef CAS.
- A. Y. Louie, M. M. Huber, E. T. Ahrens, U. Rothbacher, R. Moats, R. E. Jacobs, S. E. Fraser and T. J. Meade, In vivo visualization of gene expression using magnetic resonance imaging, Nat. Biotechnol., 2000, 18, 321–325 CrossRef CAS.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Lanthanide Probes for Bioresponsive Imaging, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS.
- J. Lux and A. D. Sherry, Advances in gadolinium-based MRI contrast agent designs for monitoring biological processes in vivo, Curr. Opin. Chem. Biol., 2018, 45, 121–130 CrossRef CAS PubMed.
- K. Rajamaki, T. Nordstrom, K. Nurmi, K. E. O. Akerman, P. T. Kovanen, K. Oorni and K. K. Eklund, Extracellular Acidosis Is a Novel Danger Signal Alerting Innate Immunity via the NLRP3 Inflammasome, J. Biol. Chem., 2013, 288, 13410–13419 CrossRef CAS.
- Y. Kato, S. Ozawa, C. Miyamoto, Y. Maehata, A. Suzuki, T. Maeda and Y. Baba, Acidic extracellular microenvironment and cancer, Cancer Cell Int., 2013, 13, 89 CrossRef CAS PubMed.
- R. J. Gillies, N. Raghunand, M. L. Garcia-Martin and R. A. Gatenby, pH imaging, IEEE Eng. Med. Biol. Mag., 2004, 23, 57–64 CrossRef.
- F. Kallinowski, K. H. Schlenger, S. Runkel, M. Kloes, M. Stohrer, P. Okunieff and P. Vaupel, Blood-Flow, Metabolism, Cellular Microenvironment, and Growth-Rate of Human-Tumor Xenografts, Cancer Res., 1989, 49, 3759–3764 CAS.
- M. T. McMahon, A. A. Gilad, J. W. M. Bulte and P. C. M. van Zijl, Chemical Exchange Saturation Transfer Imaging: Advances and Applications, Pan Stanford Publishing Pte. Ltd, Singapore, 2017 Search PubMed.
- L. A. Ekanger and M. J. Allen, Overcoming the concentration-dependence of responsive probes for magnetic resonance imaging, Metallomics, 2015, 7, 405–421 RSC.
- D. V. Hingorani, A. S. Bernstein and M. D. Pagel, A review of responsive MRI contrast agents: 2005–2014, Contrast Media Mol. Imaging, 2015, 10, 245–265 CrossRef CAS PubMed.
- L. Q. Chen, C. M. Howison, J. J. Jeffery, I. F. Robey, P. H. Kuo and M. D. Pagel, Evaluations of Extracellular pH within In Vivo Tumors Using acidoCEST MRI, Magn. Reson. Med., 2014, 72, 1408–1417 CrossRef PubMed.
- D. L. Longo, P. Z. Sun, L. Consolino, F. C. Michelotti, F. Uggeri and S. Aime, A General MRI-CEST Ratiometric Approach for pH Imaging: Demonstration of in Vivo pH Mapping with Iobitridol, J. Am. Chem. Soc., 2014, 136, 14333–14336 CrossRef CAS PubMed.
- D. Delli Castelli, E. Terreno and S. Aime, Yb-III-HPDO3A: A Dual pH- and Temperature-Responsive CEST Agent, Angew. Chem., Int. Ed., 2011, 50, 1798–1800 CrossRef CAS PubMed.
- S. J. Dorazio, A. O. Olatunde, J. A. Spernyak and J. R. Morrow, CoCEST: cobalt(II) amide-appended paraCEST MRI contrast agents, Chem. Commun., 2013, 49, 10025–10027 RSC.
- A. E. Thorarinsdottir, K. Du, J. H. P. Collins and T. D. Harris, Ratiometric pH Imaging with a CoII2 MRI Probe via CEST Effects of Opposing pH Dependences, J. Am. Chem. Soc., 2017, 139, 15836–15847 CrossRef CAS PubMed.
- A. Bar-Shir, J. W. M. Bulte and A. A. Gilad, Molecular Engineering of Nonmetallic Biosensors for CEST MRI, ACS Chem. Biol., 2015, 10, 1160–1170 CrossRef CAS PubMed.
- Y. Wu, S. Zhang, T. C. Soesbe, J. Yu, E. Vinogradov, R. E. Lenkinski and A. D. Sherry, pH imaging of mouse kidneys in vivo using a frequency-dependent paraCEST agent, Magn. Reson. Med., 2016, 75, 2432–2441 CrossRef PubMed.
- N. McVicar, A. X. Li, M. Suchy, R. H. E. Hudson, R. S. Menon and R. Bartha, Simultaneous In Vivo pH and Temperature Mapping Using a PARACEST-MRI Contrast Agent, Magn. Reson. Med., 2013, 70, 1016–1025 CrossRef CAS PubMed.
- T. Chauvin, P. Durand, M. Bernier, H. Meudal, B. T. Doan, F. Noury, B. Badet, J. C. Beloeil and E. Toth, Detection of enzymatic activity by PARACEST MRI: A general approach to target a large variety of enzymes, Angew. Chem., Int. Ed., 2008, 47, 4370–4372 CrossRef CAS PubMed.
- R. Trokowski, J. M. Ren, F. K. Kalman and A. D. Sherry, Selective sensing of zinc ions with a PARACEST contrast agent, Angew. Chem., Int. Ed., 2005, 44, 6920–6923 CrossRef CAS PubMed.
- K. Du, A. E. Thorarinsdottir and T. D. Harris, Selective Binding and Quantitation of Calcium with a Cobalt-Based Magnetic Resonance Probe, J. Am. Chem. Soc., 2019, 141, 7163–7172 CrossRef CAS PubMed.
- D. V. Hingorani, E. A. Randtke and M. D. Pagel, A CatalyCEST MRI Contrast Agent That Detects the Enzyme-Catalyzed Creation of a Covalent Bond, J. Am. Chem. Soc., 2013, 135, 6396–6398 CrossRef CAS PubMed.
- Y. Wu, I. Y. Zhou, T. Igarashi, D. L. Longo, S. Aime and P. Z. Sun, A generalized ratiometric chemical exchange saturation transfer (CEST) MRI approach for mapping renal pH using iopamidol, Magn. Reson. Med., 2018, 79, 1553–1558 CrossRef CAS PubMed.
- P. A. Bottomley, Turning Up the Heat on MRI, J. Am. Coll. Radiol., 2008, 5, 853–855 CrossRef PubMed.
- M. J. P. van Osch and A. G. Webb, Safety of Ultra-High Field MRI: What are the Specific Risks?, Curr. Radiol. Rep., 2014, 2, 61 CrossRef.
- S. J. Ratnakar, M. Woods, A. J. M. Lubag, Z. Kovacst and A. D. Sherry, Modulation of water exchange in europium(III) DOTA-tetraamide complexes via electronic substituent effects, J. Am. Chem. Soc., 2008, 130, 6–7 CrossRef CAS PubMed.
- T. Mani, G. Tircso, O. Togao, P. Zhao, T. C. Soesbe, M. Takahashi and A. D. Sherry, Modulation of water exchange in Eu(III) DOTA-tetraamide complexes: considerations for in vivo imaging of PARACEST agents, Contrast Media Mol. Imaging, 2009, 4, 183–191 CrossRef CAS PubMed.
- W. S. Fernando, A. F. Martins, P. Zhao, Y. Wu, G. E. Kiefer, C. Platas-Iglesias and A. D. Sherry, Breaking the Barrier to Slow Water Exchange Rates for Optimal Magnetic Resonance Detection of paraCEST Agents, Inorg. Chem., 2016, 55, 3007–3014 CrossRef CAS PubMed.
- G. Castro, M. Regueiro-Figueroa, D. Esteban-Gomez, R. Bastida, A. Macias, P. Perez-Lourido, C. Platas-Iglesias and L. Valencia, Exceptionally Inert Lanthanide(III) PARACEST MRI Contrast Agents Based on an 18-Membered Macrocyclic Platform, Chem. – Eur. J., 2015, 21, 18662–18670 CrossRef CAS PubMed.
- L. Valencia, J. Martinez, A. Macias, R. Bastida, R. A. Carvalho and C. F. G. C. Geraldes, X-ray diffraction and 1H NMR in solution: Structural determination of lanthanide complexes of a Py2N6Ac4 ligand, Inorg. Chem., 2002, 41, 5300–5312 CrossRef CAS PubMed.
- A. Nonat, D. Esteban-Gomez, L. Valencia, P. Perez-Lourido, J. L. Barriada, L. J. Charbonniere and C. Platas-Iglesias, The role of ligand to metal charge-transfer states on the luminescence of Europium complexes with 18-membered macrocyclic ligands, Dalton Trans., 2019, 48, 4035–4045 RSC.
- G. Castro, M. Regueiro-Figueroa, D. Esteban-Gomez, P. Perez-Lourido, C. Platas-Iglesias and L. Valencia, Magnetic Anisotropies in Rhombic Lanthanide(III) Complexes Do Not Conform to Bleaney's Theory, Inorg. Chem., 2016, 55, 3490–3497 CrossRef CAS PubMed.
- S. J. Ratnakar, T. C. Soesbe, L. L. Lumata, Q. N. Do, S. Viswanathan, C. Y. Lin, A. D. Sherry and Z. Kovacs, Modulation of CEST Images in Vivo by T1 Relaxation: A New Approach in the Design of Responsive PARACEST Agents, J. Am. Chem. Soc., 2013, 135, 14904–14907 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- A. S. Chauvin, F. Gumy, D. Imbert and J. C. G. Bunzli, Europium and terbium tris(dipicolinates) as secondary standards for quantum yield determination, Spectrosc. Lett., 2004, 37, 517–532 CrossRef CAS.
- P. A. Brayshaw, J. C. G. Bunzli, P. Froidevaux, J. M. Harrowfield, Y. Kim and A. N. Sobolev, Synthetic, Structural, and Spectroscopic Studies on Solids Containing Tris(Dipicolinato) Rare-Earth Anions and Transition or Main-Group Metal-Cations, Inorg. Chem., 1995, 34, 2068–2076 CrossRef CAS.
- G. L. Rothermel, L. Miao, A. L. Hill and S. C. Jackels, Macrocyclic Ligands with 18-Membered Rings Containing Pyridine or Furan Groups - Preparation, Protonation, and Complexation by Metal-Ions, Inorg. Chem., 1992, 31, 4854–4859 CrossRef CAS.
- SAINT, ver, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- SADABS, Program for Empirical Absorption Correction of Area Detector Data, ver, University of Götingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- P. Vandersluis and A. L. Spek, Bypass - an Effective Method for the Refinement of Crystal-Structures Containing Disordered Solvent Regions, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- L. J. Farrugia, ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI), J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- M. Zaiss, J. Z. Xu, S. Goerke, I. S. Khan, R. J. Singer, J. C. Gore, D. F. Gochberg and P. Bachert, Inverse Z-spectrum analysis for spillover-, MT-, and T1-corrected steady-state pulsed CEST-MRI - application to pH-weighted MRI of acute stroke, NMR Biomed., 2014, 27, 240–252 CrossRef CAS PubMed.
- M. Zaiss, G. Angelovski, E. Demetriou, M. T. McMahon, X. Golay and K. Scheffler, QUESP and QUEST revisited – fast and accurate quantitative CEST experiments, Magn. Reson. Med., 2018, 79, 1708–1721 CrossRef CAS PubMed.
- M. Zaiss, Bloch-McConnell Simulation & Fit, http://cest-sources.org/doku.php?id=bm_sim_fit.
- M. Zaiss, A. Anemone, S. Goerke, D. L. Longo, K. Herz, R. Pohmann, S. Aime, M. Rivlin, G. Navon, X. Golay and K. Scheffler, Quantification of hydroxyl exchange of D-Glucose at physiological conditions for optimization of glucoCEST MRI at 3, 7 and 9.4 Tesla, NMR Biomed., 2019, 32, e4113 CrossRef.
- C. Nunez, R. Bastida, A. Macias, M. Mato-Iglesias, C. Platas-Iglesias and L. Valencia, A hexaaza macrocyclic ligand containing acetohydrazide pendants for Ln(III) complexation in aqueous solution. Solid-state and solution structures and DFT calculations, Dalton Trans., 2008, 3841–3850 RSC.
- M. Pinsky and D. Avnir, Continuous symmetry measures. 5. The classical polyhedra, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed.
- D. Casanova, J. Cirera, M. Llunell, P. Alemany, D. Avnir and S. Alvarez, Minimal distortion pathways in polyhedral rearrangements, J. Am. Chem. Soc., 2004, 126, 1755–1763 CrossRef CAS PubMed.
- Y. Q. Sun, M. Liang, W. Dong, G. M. Yang, D. Z. Liao, Z. H. Jiang, S. P. Yan and P. Cheng, Hydrothermal syntheses, crystal structures, magnetism and fluorescence quenching of oxamidato-bridged pentanuclear Cu(4)(II)Ln(III) complexes containing macrocyclic ligands (Ln=Eu, Tb) and the crystal structure of a hexanuclear (Ni5SmIII)-Sm-II complex, Eur. J. Inorg. Chem., 2004, 1514–1521 CrossRef CAS.
- A. Ruiz-Martinez and S. Alvarez, Stereochemistry of Compounds with Coordination Number Ten, Chem. – Eur. J., 2009, 15, 7470–7480 CrossRef CAS PubMed.
- E. Toth, E. Brucher, I. Lazar and I. Toth, Kinetics of Formation and Dissociation of Lanthanide(III)-DOTA Complexes, Inorg. Chem., 1994, 33, 4070–4076 CrossRef CAS.
- S. Laurent, L. Vander Elst, C. Henoumont and R. N. Muller, How to measure the transmetallation of a gadolinium complex, Contrast Media Mol. Imaging, 2010, 5, 305–308 CrossRef CAS PubMed.
- J. A. Peters, J. Huskens and D. J. Raber, Lanthanide induced shifts and relaxation rate enhancements, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 28, 283–350 CrossRef CAS.
- G. Castro, R. Bastida, A. Macias, P. Perez-Lourido, C. Platas-Iglesias and L. Valencia, Lanthanide(III) Complexation with an Amide Derived Pyridinophane, Inorg. Chem., 2015, 54, 1671–1683 CrossRef CAS PubMed.
- K. Binnemans, Interpretation of europium(III) spectra, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, Non-radiative deactivation of the excited states of europium, terbium and ytterbium complexes by proximate energy-matched OH, NH and CH oscillators: an improved luminescence method for establishing solution hydration states, J. Chem. Soc., Perkin Trans. 2, 1999, 493–503 RSC.
- J. Y. Zhou and P. C. M. van Zijl, Chemical exchange saturation transfer imaging and spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 48, 109–136 CrossRef CAS.
- S. Aime, L. Calabi, L. Biondi, M. De Miranda, S. Ghelli, L. Paleari, C. Rebaudengo and E. Terreno, Iopamidol: Exploring the potential use of a well-established X-ray contrast agent for MRI, Magn. Reson. Med., 2005, 53, 830–834 CrossRef CAS.
- P. Z. Sun, D. L. Longo, W. Hu, G. Xiao and R. H. Wu, Quantification of iopamidol multi-site chemical exchange properties for ratiometric chemical exchange saturation transfer (CEST) imaging of pH, Phys. Med. Biol., 2014, 59, 4493–4504 CrossRef CAS PubMed.
- D. L. Longo, W. Dastru, G. Digilio, J. Keupp, S. Langereis, S. Lanzardo, S. Prestigio, O. Steinbach, E. Terreno, F. Uggeri and S. Aime, Iopamidol as a Responsive MRI-Chemical Exchange Saturation Transfer Contrast Agent for pH Mapping of Kidneys: In Vivo Studies in Mice at 7 T, Magn. Reson. Med., 2011, 65, 202–211 CrossRef CAS PubMed.
- S. J. Dorazio and J. R. Morrow, Iron(II) Complexes Containing Octadentate Tetraazamacrocycles as ParaCEST Magnetic Resonance Imaging Contrast Agents, Inorg. Chem., 2012, 51, 7448–7450 CrossRef CAS.
- S. J. Dorazio, P. B. Tsitovich, K. E. Siters, J. A. Spernyak and J. R. Morrow, Iron(II) PARACEST MRI Contrast Agents, J. Am. Chem. Soc., 2011, 133, 14154–14156 CrossRef CAS PubMed.
- A. O. Olatunde, S. J. Dorazio, J. A. Spernyak and J. R. Morrow, The NiCEST Approach: Nickel(II) ParaCEST MRI Contrast Agents, J. Am. Chem. Soc., 2012, 134, 18503–18505 CrossRef CAS PubMed.
- S. R. Zhang, L. Michaudet, S. Burgess and A. D. Sherry, The Amide Protons of an Ytterbium(III) dota Tetraamide Complex Act as Efficient Antennae for Transfer of Magnetization to Bulk Water, Angew. Chem., Int. Ed., 2002, 41, 1919–1921 CrossRef CAS PubMed.
- Y. K. Wu, T. C. Soesbe, G. E. Kiefer, P. Y. Zhao and A. D. Sherry, A Responsive Europium(III) Chelate That Provides a Direct Readout of pH by MRI, J. Am. Chem. Soc., 2010, 132, 14002–14003 CrossRef CAS PubMed.
- X. J. Wang, Y. K. Wu, T. C. Soesbe, J. Yu, P. Y. Zhao, G. E. Kiefer and A. D. Sherry, A pH-Responsive MRI Agent that Can Be Activated Beyond the Tissue Magnetization Transfer Window, Angew. Chem., Int. Ed., 2015, 54, 8662–8664 CrossRef CAS PubMed.
- M. Zaiss, A. Anemone, S. Goerke, D. L. Longo, K. Herz, R. Pohmann, S. Aime, M. Rivlin, G. Navon, X. Golay and K. Scheffler, Quantification of hydroxyl exchange of D-Glucose at physiological conditions for optimization of glucoCEST MRI at 3, 7 and 9.4 T, NMR Biomed., 2019, 32, e4113 CrossRef PubMed.
- P. C. M. van Zijl and N. N. Yadav, Chemical Exchange Saturation Transfer (CEST): What is in a Name and What Isn't?, Magn. Reson. Med., 2011, 65, 927–948 CrossRef CAS PubMed.
- R. M. Henkelman, G. J. Stanisz and S. J. Graham, Magnetization transfer in MRI: a review, NMR Biomed., 2001, 14, 57–64 CrossRef CAS PubMed.
- G. J. Stanisz, E. E. Odrobina, J. Pun, M. Escaravage, S. J. Graham, M. J. Bronskill and R. M. Henkelman, T1, T2 relaxation and magnetization transfer in tissue at 3T, Magn. Reson. Med., 2005, 54, 507–512 CrossRef PubMed.
- G. Helmlinger, F. Yuan, M. Dellian and R. K. Jain, Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation, Nat. Med., 1997, 3, 177–182 CrossRef CAS PubMed.
- P. Z. Sun, E. F. Wang and J. S. Cheung, Imaging acute ischemic tissue acidosis with pH-sensitive endogenous amide proton transfer (APT) MRI-Correction of tissue relaxation and concomitant RF irradiation effects toward mapping quantitative cerebral tissue pH, Neuroimage, 2012, 60, 1–6 CrossRef CAS PubMed.
- M. Woods, P. Caravan, C. F. G. C. Geraldes, M. T. Greenfield, G. E. Kiefer, M. Lin, K. McMillan, M. I. M. Prata, A. C. Santos, X. K. Sun, J. F. Wang, S. R. Zhang, P. Y. Zhao and A. D. Sherry, The Effect of the Amide Substituent on the Biodistribution and Tolerance of Lanthanide(III) DOTA-Tetraamide Derivatives, Invest. Radiol., 2008, 43, 861–870 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrum, additional crystal structures, kinetic inertness studies, further details of CEST simulations and experiments. CCDC 1886847 and 1886848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01612k |
| This journal is © the Partner Organisations 2020 |