DOI:
10.1039/C9QI01605H
(Research Article)
Inorg. Chem. Front., 2020,
7, 2017-2022
Ligand-centered reactivity of a pseudo-dearomatized phosphorus-nitrogen PN3P* rhodium complex towards molecular oxygen at room temperature†
Received
6th December 2019
, Accepted 20th March 2020
First published on 20th March 2020
Abstract
The ligand of an organometallic complex is typically considered as a spectator which can be modified to tune the steric and electronic properties of the coordination environment. We herein demonstarted a PN3P*Rh-CO pincer system where one of the C–H bonds of the pseudo-dearomatized pyridine ring was oxidized by O2 at room temperature to create an α,β-unsaturated carbonyl functionality on the ligand backbone. The resulting metal complex with the post-modified PN3P ligand readily reacts with thiophenol and 4-methylaniline to afford the corresponding oxidative Michael addition products.
Introduction
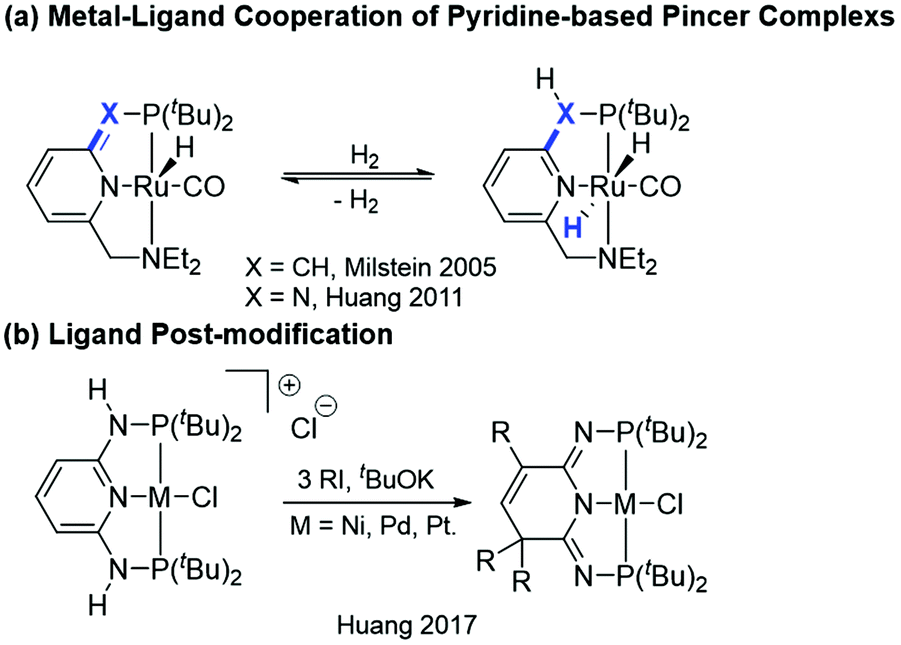
Pincer ligand platforms have become popular due to the increased stability of the corresponding metal complexes arising from the tridentate binding mode. The tunability of stereo and electronic properties of the ligands allows broad applications in catalysis.1 Early investigations on pincer complexes mainly focused on synthesis, coordination chemistry,2 and substitution reactions on the metal center.3 It was not until 2005 when Milstein and co-workers demonstrated a new type of metal–ligand cooperation (MLC, Scheme 1a, X = CH) in a pyridine-based pincer ruthenium complex for the catalytic hydrogenation of alcohols to esters.4 The MLC proceeds via the dearomatization and rearomatization of the central pyridine ring of the pincer ligand backbone with the metal oxidation state unchanged. Such a bond activation mode has received much attention in the development of pincer catalysts.5 We have also recently demonstrated that the seemingly small change, by replacing the spacer arms from the CH2 to NH group(s), has dramatically altered the thermodynamic and kinetic properties compared to those of the CH2 analogs (Scheme 1a, X = N).1c,6 This new class of PN3(P) metal complexes offered intriguing properties due to the distinct pseudo-dearomatized nature,7 greatly extending the potential for different reactivities in catalysis and bond activation.8 Accordingly, metal enhanced ligand centered reactivities were realized.9 Very interesting, such properties enabled the preparation of a second generation of PN3P pincer complexes via a facile ligand post-modification to diversify the pincer platforms (Scheme 1b).10
 |
| | Scheme 1 Various reactivities in pincer metal complexes. | |
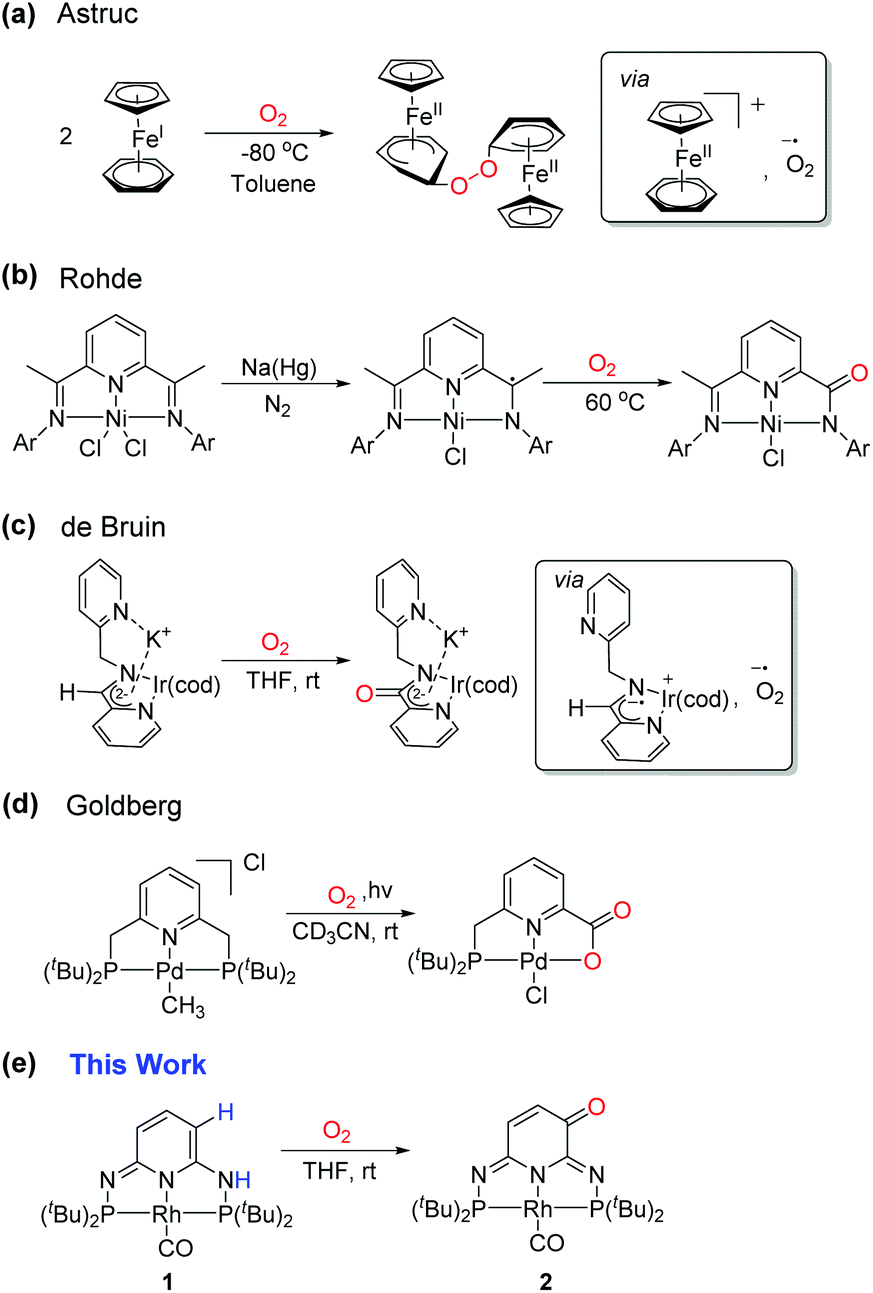
Dioxygen is a triplet diradical (3O2) in its ground state and its reaction with a singlet closed shell organometallic complex is therefore spin-forbidden and quite unexpected. Only a few examples where a ligand is oxidized by O2 could be found in the literature. For example, cobaltocene and other cobalt clusters can form peroxo-bridged dimers in the presence of O2.11 A similar observation was made in Cp(FeI)arene (Scheme 2a).12 This reaction was proposed to proceed via a single-electron transfer (SET) from the Cp(FeI)arene complex to O2 to create a superoxide radical anion, followed by the nucleophilic attack on the arene ring. The driving force for the SET was explained by the large difference of 1.0 V in redox potential between the Fe+/Fe2+ and O2/O2− couples.13 In a bis(arylimino)pyridine nickel pincer complex, a radical could be chemically introduced on the ligand and could subsequently react with O2 (Scheme 2b).14 The mono-oxygenation of a ligand backbone occurred at room temperature in a bis(picolyl)amine iridium complex via a ligand-centered radical intermediate as observed in the EPR spectroscopy (Scheme 2c).15 The PNP-ligated Pd–Me complex was found to react with O2 leading to a oxidation of the CH2 arm of the pincer ligand upon photolysis (Scheme 2d).16 Herein, we disclose yet another unique example of ligand reactivity towards O2 in our PN3P*Rh-CO complex (1).8a The subsequent reactions of the resulting α,β-unsaturated carbonyl functionality on the ligand backbone were also investigated.
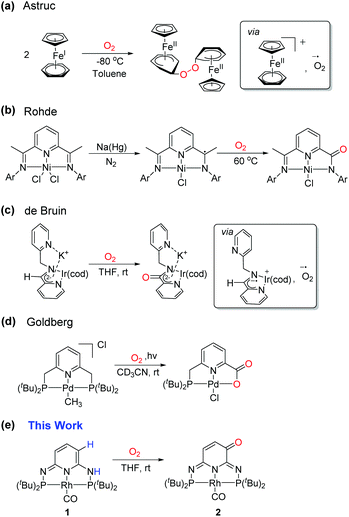 |
| | Scheme 2 Reactions between organometallic complex and O2. | |
Results and discussion
Reaction of 1 with O2
The reaction with O2 and 1 proceeded with a rapid color change from green to red-brown in 2 hours at room temperature. 2 and 3 were isolated by flash chromatography as the major products in 40% and 2% yields, respectively, with the formation of a trace amount of 4 (Scheme 3). The 31P{1H} NMR spectrum of 2 displays an AB system with two sets of doublet of doublet at δ 140.8 ppm (2JPP = 252.5 Hz, 1JRhP = 127.3 Hz) and δ 139.5 ppm (2JPP = 252.5 Hz, 1JRhP = 127.3 Hz), consistent with the non-equivalent phosphine groups. The 1H NMR spectrum shows two different signals, a doublet at δ 6.91 ppm and multiplet at δ 6.10 ppm, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, for two protons on the ligand backbone. The rhodium carbonyl carbon of 2 can be observed as a doublet of triplet at δ 199.80 ppm (1JRhC = 68.6 Hz, 2JPC = 10.8 Hz) in the 13C NMR, while the carbonyl carbon on the ligand backbone appears at δ 176.46 ppm. Similarly, 3 also shows two sets of doublet of doublet at δ 141.4 ppm (2JPP = 253.2 Hz, 1JRhP = 127.3 Hz) and δ 138.7 ppm (2JPP = 253.2 Hz, 1JRhP = 127.3 Hz). The 1H NMR spectrum exhibits a broad signal at δ 6.50 ppm, in agreement with only one proton remaining on the backbone.
:1 ratio, for two protons on the ligand backbone. The rhodium carbonyl carbon of 2 can be observed as a doublet of triplet at δ 199.80 ppm (1JRhC = 68.6 Hz, 2JPC = 10.8 Hz) in the 13C NMR, while the carbonyl carbon on the ligand backbone appears at δ 176.46 ppm. Similarly, 3 also shows two sets of doublet of doublet at δ 141.4 ppm (2JPP = 253.2 Hz, 1JRhP = 127.3 Hz) and δ 138.7 ppm (2JPP = 253.2 Hz, 1JRhP = 127.3 Hz). The 1H NMR spectrum exhibits a broad signal at δ 6.50 ppm, in agreement with only one proton remaining on the backbone.
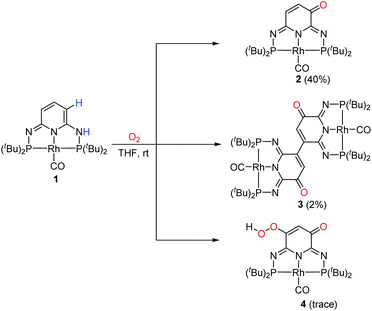 |
| | Scheme 3 Reaction of 1 with O2 at room temperature. | |
Crystals suitable for X-ray analysis of 2 (Fig. 1), 3 (Fig. 2) and 4 (Fig. S14†) were obtained from their respective benzene solutions. 2 exhibits a distorted square-planar geometry around the tetracoordinated Rh(I) center with angles of P1–Rh1–P2 and N1–Rh1–C6 as 159.33 (2)° and 178.65(13)°, respectively. The C1–N3 (1.285 Å) and C5–N2 (1.291 Å) bond distances strongly indicate the double bond characters, in good agreement with the solution NMR data of 2. 3 displays a dimeric structure resulting from the oxidative coupling of the backbone of 2 linked by formation of a C–C bond (C2–C23: 1.492 Å), with a distorted square-planar geometry around two Rh(I) centers (P1–Rh1–P2: 159.05(17), C43–Rh1–N1: 178.3(7), P4–Rh2–P3: 160.3(3) and C44–Rh2–N4: 178.2(7)). 4 shows a similar structure with the only significant difference being the presence of an –OOH moiety connecting to the C5 atom of the oxidized pyridine ring. The crystallography analysis also confirmed the tetracoordinated environment for the Rh(I) center (P2–Rh2–P3: 159.19(5)° and N2–Rh1–C6: 179.6(2)°).
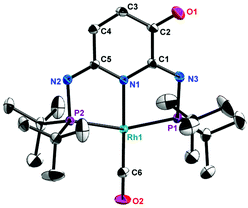 |
| | Fig. 1 Molecular structure of 2. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths [Å] and bond angles [°]: C2–O1, 1.233(4); C3–C4, 1.358(4); C6–O2, 1.150(4); C1–N3, 1.285(3); C5–N2, 1.291(3); Rh1–N1, 2.059(2); Rh1–C6, 1.824(3); Rh1–P1, 2.2921(6); Rh1–P2, 2.2918(7); P2–Rh1–P1, 159.33(2); N1–Rh1–C6, 178.65(13); P2–Rh1–N1, 79.70(6); N1–Rh1–P1, 79.63(6); P2–Rh1–C6, 100.76(10); C6–Rh1–P1, 99.91(10). | |
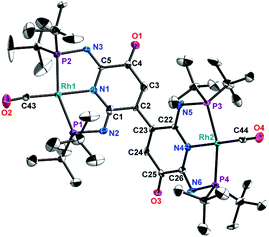 |
| | Fig. 2 Molecular structure of 3. Thermal ellipsoids are shown at the 50% probability level. Selected bond lengths [Å] and bond angles [°]: C4–O1, 1.23(2); C2–C3, 1.30(3); C43–O2, 1.147(19); C5–N3, 1.28(2); C1–N2, 1.28(2); Rh1–N1, 2.040(13); Rh1–C43, 1.834(16); Rh1–P1, 2.289(5); Rh1–P2, 2.292(5); C2–C23, 1.49(2); C25–O3, 1.23(2); C23–C24, 1.35(3); C44–O4, 1.17(2); C26–N6, 1.32(2); C22–N5, 1.28(2); Rh2–N4, 2.054(12); Rh2–C44, 1.82(2); Rh2–P3, 2.278(11); Rh2–P4, 2.276(5); N1–Rh1–C43, 178.3(7); P1–Rh1–P2, 159.05(17); N1–Rh1–P1, 79.8(4); P1–Rh1–C43, 101.5(6); C43–Rh1–P2, 99.4(6); P2–Rh1–N1, 79.3(4); N4–Rh2–C44, 178.2(7); P3–Rh2–P4, 160.3(3); N4–Rh2–P3, 80.8(5); P3–Rh2–C44, 98.5(6); C44–Rh2–P4, 100.7(6); P4-Rh2-N4, 79.9(4). | |
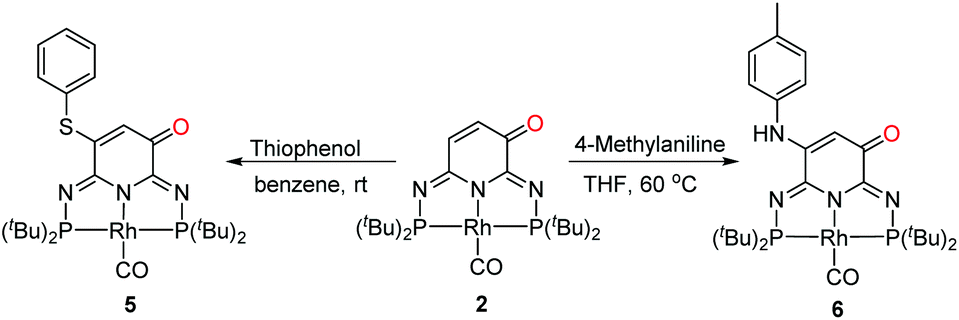
Oxidative Michael addition of 2
The ligand backbone of 2 has an α,β-unsaturated carbonyl functionality which may react as a Michael acceptor. The reactions of thiophenol and 4-methylaniline were performed under air to produce 5 and 6 in 95% and 60% yields, respectively (Scheme 4). The 31P{1H} NMR spectra of 5 and 6 both show an AB pattern with two sets of doublet of doublet at δ 138.7 ppm (2JPP = 252.7 Hz, 1JRhP = 128.0 Hz) and δ 137.0 ppm (2JPP = 252.7 Hz, 1JRhP = 128.0 Hz), and δ 141.8 ppm (2JPP = 254.2 Hz, 1JRhP = 127.3 Hz) and δ 135.6 ppm (2JPP = 254.2 Hz, 1JRhP = 127.3 Hz), respectively. The 1H NMR spectra exhibit a broad signal at δ 5.72 ppm and δ 6.38 ppm for 5 and 6, respectively. These observations implied that 2 underwent an oxidative Michael addition. The molecular structures of 5 and 6 were finally confirmed by single crystal X-ray diffraction analysis (Fig. 3), revealing similar geometries to that of 4.
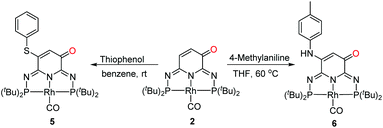 |
| | Scheme 4 Oxidative Michael addition of 2. | |
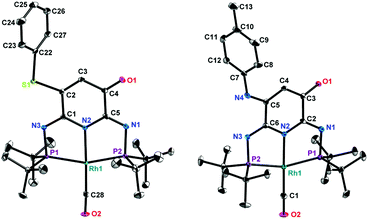 |
| | Fig. 3 Molecular structures of 5 (left) and 6 (right). Thermal ellipsoids are shown at the 50% probability level; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: for 5: C4–O1, 1.220(3); C2–C3, 1.346(4); C2–S1, 1.753(3); C28–O2, 1.151(4); C1–N3, 1.295(4); C5–N1, 1.294(4); Rh1–N2, 2.061(2); Rh1–P1, 2.2849(7); Rh1–C28, 1.824(3); Rh1–P2, 2.2978(7); N2–Rh1–C28, 177.65(10); P1–Rh1–P2, 159.49(2); N2–Rh1–P1, 80.19(6); P1–Rh1–C28, 98.42(8); C28–Rh1–P2, 101.94(8); P2–Rh1–N2, 79.38(6). For 6: C3–O1, 1.235(6); C4–C5, 1.362(7); C5–N4, 1.351(7); C1–O2, 1.156(7); C6–N3, 1.295(7); C2–N1, 1.291(8); Rh1–N2, 2.049(4); Rh1–C1, 1.810(6); Rh1–P2, 2.3054(15); Rh1–P1, 2.2894(15); N2–Rh1–C1, 179.0(3); P2–Rh1–P1, 158.56(6); N2–Rh1–P2, 79.85(13); P2–Rh1–C1, 100.85(19); C1–Rh1–P1, 99.78(19); P1–Rh1–N2, 79.46(13). | |
Mechanistic insights for the reaction of 1 with O2
In order to gain insight into the mechanism of the oxidation reaction between 1 and O2, multiple spectroscopic investigations were performed. From the similar reactions between metal complexes and O2 shown in Scheme 2, a radical mechanism is highly plausible. Therefore, the reaction was investigated using the EPR spectroscopy. A toluene solution of 1 was exposed to O2 and frozen in liquid nitrogen. The X-band EPR data at 10 K show signals originating from two paramagnetic species which grow together (Fig. 4A). One species has a g value of 2.06 and 1.99, showing typical characteristics of superoxide species,17 and the other has a sharp signal at g = 2.004, suggesting that the unpaired electron of these species is delocalized on the pyridine ligand. After being left at room temperature for 2 hours, the EPR signal gradually disappeared.
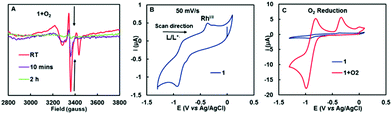 |
| | Fig. 4 (A) Detection of reaction intermediate in the reaction between 1 and O2 with EPR spectroscopy. Conditions: T = 10 K, freq. = 9.3931 GHz, att. = 20 dB. (B) Cyclic voltammogram of 1 in THF (1.0 mM of complex 1, scan rate: 50 mV s−1, Glassy carbon electrode (GC, diameter 3 mm) as a working electrode, Pt-wire as a counter electrode, Ag/AgCl/1 M KCl as a reference electrode, 0.1 M nBu4NPF6 as a supporting electrolyte. (C) Electrochemical investigation of 1 in presence of O2, with clear indication of reduction current (enhancement of current) (scan rate: 50 mV s−1). | |
With the presence of a ligand-centered radical intermediate established, cyclic voltammetry was performed to gain quantitative information about the charge transfer properties of 1. Ideally, it should be possible to identify the possible redox couples of 1, especially the ligand-centered redox couple 1/1˙+. The hypothesis is to observe a ligand-centered redox couple that is more negative than that of the O2/O2˙− couple, thereby explaining the driving force behind the electron transfer from the pincer ligand to O2.
Electrochemical analysis of 1 showed a quasi-reversible wave in the cylclic voltammogram with E1/2 = −0.89 V (ΔEp = 60 mV) vs. Ag/AgCl/1 M KCl in THF, corresponding to the ligand+/ligand redox couple (Fig. 4B). One broad redox couple was also observed at −0.56 V (Epc = −0.77 V; Epa = −0.35 V), corresponding to RhII/I. The CVs corresponding to two redox processes were obtained at different scan rates (Fig. S13A†) and showed a linear relationship, in agreement with a diffusion-controlled process (Fig. S13B and C†). The redox potential of O2/O2˙− is −0.50 V vs. NHE (i.e. −0.70 V vs. Ag/AgCl),18 suggesting that the outer-sphere electron transfer (ET) from the pseudo-dearomatized pyridine ligand to O2 should be exergonic by 4.3 kcal mol−1. Thus, ET to O2 is thermodynamically favorable, and a concerted process may be operative. Furthermore, electrochemical investigation of 1 in the presence of O2, showed a clear indication of oxygen reduction current (enhancement of current; Fig. 4B). A loss of electrochemical reversibility of the ligand+/ligand redox couple upon addition of O2 also implies the outer sphere electron transfer to O2.
Conclusions
In summary, we have shown that the pseudo-dearomatized ligand backbone of PN3P*Rh-CO (1) underwent the C–H activation with O2 to form 2 with a α,β-unsaturated carbonyl moiety at room temperature, showcasing an unprecedented ligand-centered reactivity with the central metal Rh(I) remaining untouched. 2 can function as a Michael acceptor to readily react with thiol and amino groups, such as thiophenol and 4-methylaniline, to generate the ligand post-modified 5 and 6, respectively, demonstrating its potential for bio-conjugation. Further investigations on their bio-synthetic applications are ongoing and will be reported in due course.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
We acknowledge the financial support from the King Abdullah University of Science and Technology (KAUST).
Notes and references
-
(a)
G. van Koten, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, p. 1 Search PubMed;
(b) C. Gunanathan and D. Milstein, Bond Activation and Catalysis by Ruthenium Pincer Complexes, Chem. Rev., 2014, 114, 12024 CrossRef CAS PubMed;
(c) H. Li, B. Zheng and K.-W. Huang, A New Class of PN3-Pincer Ligands for Metal–Ligand Cooperative Catalysis, Coord. Chem. Rev., 2015, 293–294, 116 CrossRef CAS;
(d) J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236 CrossRef CAS PubMed;
(e) M. E. van der Boom and D. Milstein, Cyclometalated Phosphine-Based Pincer Complexes: Mechanistic Insight in Catalysis, Coordination, and Bond Activation, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed.
-
(a) W. S. J. Kelly, G. H. Ford and S. M. Nelson, Studies on the Magnetic Crossover in Five-Co-ordinate Complexes of Iron(II), Cobalt(II), and Nickel(II). Part I, J. Chem. Soc. A, 1971, 388 RSC;
(b) C. J. Moulton and B. L. Shaw, Transition Metal–Carbon Bonds. Part XLII. Complexes of Nickel, Palladium, Platinum, Rhodium and Iridium with the Tridentate Ligand 2,6-Bis[(di-t-butylphosphino)methyl]phenyl, J. Chem. Soc., Dalton Trans., 1976, 1020 RSC;
(c) W. Schirmer, U. Flörke and H.-J. Haupt, Darstellung, Eigenschaften und Molekülstrukturen von Komplexen des versteiften dreizähnigen Chelatliganden N, N’-Bis(diphenylphosphino)-2, 6-diaminopyridin mit MII - und M0-Übergangsmetallen [MII = Ni, Pd, Pt; M0 = Cr, Mo, W], Z. Anorg. Allg. Chem., 1987, 545, 83 CrossRef CAS.
-
(a) S. Nemeh, C. Jensen, E. Binamira-Soriaga and W. C. Kaska, Interaction of {2,6-bis[(di-tert-butylphosphino)methyl]phenyl}rhodium(I) with Hydrocarbons. X-ray Molecular Structure of {2,6-bis[(di-tert-butylphosphino)methyl]phenyl}chlorohydridorhodium(III), Organometallics, 1983, 2, 1442 CrossRef CAS;
(b) O. V. Ozerov, C. Guo, L. Fan and B. M. Foxman, Oxidative Addition of N−C and N−H Bonds to Zerovalent Nickel, Palladium, and Platinum, Organometallics, 2004, 23, 5573 CrossRef CAS;
(c) R. Çelenligil-Çetin, L. A. Watson, C. Guo, B. M. Foxman and O. V. Ozerov, Decarbonylation of Acetone and Carbonate at a Pincer-Ligated Ru Center, Organometallics, 2005, 24, 186 CrossRef;
(d) C. C. Comanescu and V. M. Iluc, Synthesis and Reactivity of a Nucleophilic Palladium(II) Carbene, Organometallics, 2014, 33, 6059 CrossRef CAS;
(e) B. J. Barrett and V. M. Iluc, Metal-ligand Cooperation between Palladium and a Diphosphine Ligand with an Olefinic Backbone, Inorg. Chim. Acta, 2017, 460, 35 CrossRef CAS;
(f) E. A. LaPierre, W. E. Piers and C. Gendy, Redox-State Dependent Activation of Silanes and Ammonia with Reverse Polarity (PCcarbeneP)Ni Complexes: Electrophilic vs. Nucleophilic carbenes, Dalton Trans., 2018, 47, 16789 RSC;
(g) L. Nurdin, D. M. Spasyuk, L. Fairburn, W. E. Piers and L. Maron, Oxygen–Oxygen Bond Cleavage and Formation in Co(II)-Mediated Stoichiometric O2 Reduction via the Potential Intermediacy of a Co(IV) Oxyl Radical, J. Am. Chem. Soc., 2018, 140, 16094 CrossRef CAS PubMed;
(h) J. R. Logan, W. E. Piers, J. Borau-Garcia and D. M. Spasyuk, Ligand Attachment Chemistry in the Preparation of PCsp3P and PCsp2P Complexes of Rhodium, Organometallics, 2016, 35, 1279 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Facile Conversion of Alcohols into Esters and Dihydrogen Catalyzed by New Ruthenium Complexes, J. Am. Chem. Soc., 2005, 127, 10840 CrossRef CAS PubMed.
-
(a) J. I. van der Vlugt and J. N. H. Reek, Neutral Tridentate PNP Ligands and Their Hybrid Analogues: Versatile Non-Innocent Scaffolds for Homogeneous Catalysis, Angew. Chem., Int. Ed., 2009, 48, 8832 CrossRef CAS PubMed;
(b) D. Milstein, Discovery of Environmentally Benign Catalytic Reactions of Alcohols Catalyzed by Pyridine-Based Pincer Ru Complexes, Based on Metal–Ligand Cooperation, Top. Catal., 2010, 53, 915 CrossRef CAS.
-
(a)
K.-W. Huang, T. Chen, L. He, D. Gong, W. Jia and L. Yao, Phospho-Amino Pincer-Type Ligands and Catalytic Metal Complexes
Thereof, U.S. Patent8598351B2, 2013 Search PubMed;
(b) L.-P. He, T. Chen, D. Gong, Z. Lai and K.-W. Huang, Enhanced Reactivities toward Amines by Introducing an Imine Arm to the Pincer Ligand: Direct Coupling of Two Amines To Form an Imine Without Oxidant, Organometallics, 2012, 31, 5208 CrossRef CAS.
- T. P. Gonçalves and K.-W. Huang, Metal–Ligand Cooperative Reactivity in the (Pseudo)-Dearomatized PNx(P) Systems: The Influence of the Zwitterionic Form in Dearomatized Pincer Complexes, J. Am. Chem. Soc., 2017, 139, 13442 CrossRef PubMed.
-
(a) Y. Wang, B. Zheng, Y. Pan, C. Pan, L. He and K.-W. Huang, C-H and H-H Bond Activation via Ligand Dearomatization/Rearomatization of a PN3P-Rhodium(I) Complex, Dalton Trans., 2015, 44, 15111 RSC;
(b) L.-P. He, T. Chen, D.-X. Xue, M. Eddaoudi and K.-W. Huang, Efficient Transfer Hydrogenation Reaction Catalyzed by a Dearomatized PN3P Ruthenium Pincer Complex under Base-free Conditions, J. Organomet. Chem., 2012, 700, 202 CrossRef CAS;
(c) C. Zhou, J. Hu, Y. Wang, C. Yao, P. Chakraborty, H. Li, C. Guan, M.-H. Huang and K.-W. Huang, Selective Carbonylation of Benzene to Benzaldehyde Using a Phosphorus–Nitrogen PN3P–Rhodium(I) Complex, Org. Chem. Front., 2019, 6, 721 RSC;
(d) Y. Pan, C.-L. Pan, Y. Zhang, H. Li, S. Min, X. Guo, B. Zheng, H. Chen, A. Anders, Z. Lai, J. Zheng and K.-W. Huang, Selective Hydrogen Generation from Formic Acid with Well-Defined Complexes of Ruthenium and Phosphorus–Nitrogen PN3-Pincer Ligand, Chem. – Asian J., 2016, 11, 1357 CrossRef CAS PubMed;
(e) H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, PN3(P)-Pincer Complexes: Cooperative Catalysis and Beyond, ACS Catal., 2019, 9, 1619 CrossRef CAS.
-
(a) H. Li, T. P. Gonçalves, J. Hu, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, A Pseudodearomatized PN3P*Ni–H Complex as a Ligand and σ-Nucleophilic Catalyst, J. Org. Chem., 2018, 83, 14969 CrossRef CAS PubMed;
(b) H. Li, T. P. Gonçalves, Q. Zhao, D. Gong, Z. Lai, Z. Wang, J. Zheng and K.-W. Huang, Diverse Catalytic Reactivity of a Dearomatized PN3P*–Nickel Hydride Pincer Complex towards CO2 Reduction, Chem. Commun., 2018, 54, 11395 RSC.
-
(a) X. Wang, L. Yao, Y. Pan and K.-W. Huang, Synthesis of Group 10 Metal Complexes with a New Unsymmetrical PN3P-Pincer Ligand through Ligand Post-Modification: Structure and Reactivity, J. Organomet. Chem., 2017, 845, 25 CrossRef CAS;
(b) C. Yao, X. Wang and K.-W. Huang, Nitrogen Atom Transfer Mediated by a new PN3P-Pincer Nickel Core via a Putative Nitrido Nickel Intermediate, Chem. Commun., 2018, 54, 3940 RSC;
(c) C. Yao, P. Chakraborty, E. Aresu, H. Li, C. Guan, C. Zhou, L.-C. Liang and K.-W. Huang, Monomeric Nickel Hydroxide Stabilized by a Sterically Demanding Phosphorus–Nitrogen PN3P-Pincer Ligand: Synthesis, Reactivity and Catalysis, Dalton Trans., 2018, 47, 16057 RSC;
(d) C. Yao, T. Zhang, C. Zhou and K.-W. Huang, Interrogating the Steric Outcome During H2 Heterolysis: In-plane Steric Effects in the Regioselective Protonation of the PN3P-Pincer Ligand, Dalton Trans., 2019, 48, 12817 RSC.
-
(a) H. Kojima, S. Takahashi and N. Hagihara, An Oxygen Adduct of Cobaltocene, J. Chem. Soc., Chem. Commun., 1973, 230 RSC;
(b) J. H. Kaldis, M. A. Brook and M. J. McGlinchey, Solvent-Mediated Generation of Cobalt-Cluster-Stabilised Propargyl Cations and Radicals: Allyl Migration versus Peroxide Formation, Chem. – Eur. J., 2008, 14, 10074 CrossRef CAS PubMed.
- J. R. Hamon and D. Astruc, How Dioxygen Activates Carbon-Hydrogen Bonds of Simple Arenes in Unstable CpFe(I) Arene Complexes and the Versatile Reactivity of Superoxide Anion Generated from Dioxygen and Organoiron “Electron Eeservoirs”, Organometallics, 1988, 7, 1036 CrossRef CAS.
- D. Astruc, Electron-reservoir Complexes and Other Redox-robust Reagents: Functions and Applications, New J. Chem., 2009, 33, 1191 RSC.
- T. D. Manuel and J.-U. Rohde, Reaction of a Redox-Active Ligand Complex of Nickel with Dioxygen Probes Ligand-Radical Character, J. Am. Chem. Soc., 2009, 131, 15582 CrossRef CAS PubMed.
- C. Tejel, M. P. del Río, M. A. Ciriano, E. J. Reijerse, F. Hartl, S. Záliš, D. G. H. Hetterscheid, N. T. i Spithas and B. de Bruin, Ligand-Centred Reactivity of Bis(picolyl)amine Iridium: Sequential Deprotonation, Oxidation and Oxygenation of a “Non-Innocent” Ligand, Chem. – Eur. J., 2009, 15, 11878 CrossRef CAS PubMed.
- K. A. Smoll, W. Kaminsky and K. I. Goldberg, Photolysis of Pincer-Ligated PdII–Me Complexes in the Presence of Molecular Oxygen, Organometallics, 2017, 36, 1213 CrossRef CAS.
- T. Ozawa, A. Hanaki and H. Yamamoto, On a Spectrally Well-Defined and Stable Source of Superoxide ion, O2−, FEBS Lett., 1977, 74, 99 CrossRef CAS.
- D. T. Sawyer and M. J. Gibian, The Chemistry of Superoxide Ion, Tetrahedron, 1979, 35, 1471 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed descriptions of the preparation and characterization of compound 2–6. CCDC 1967061, 1967062, 1967064, 1967065 and 1987739. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01605h |
| ‡ These authors contributed equally. |
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*