
Electron-withdrawing anion intercalation and surface sulfurization of NiFe-layered double hydroxide nanoflowers enabling superior oxygen evolution performance†
Yan-Yan
Dong‡
ab,
Dong-Dong
Ma‡
a,
Xin-Tao
Wu
 a and
Qi-Long
Zhu
*a
a and
Qi-Long
Zhu
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), Fuzhou 350002, China. E-mail: qlzhu@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 18th November 2019
Abstract
Developing earth-abundant, highly active, and durable electrocatalysts for the oxygen evolution reaction (OER) is of crucial importance for renewable energy conversion processes. Herein, we fabricated novel flower-like NiFe-layered double hydroxide nanostructures with electron-withdrawing anion intercalation and surface sulfurization via a two-step hydrothermal treatment. Benefiting from the dual-modified electronic structure of the surface active sites of NiFe-LDHs, the as-obtained catalyst showed excellent electrocatalytic activity for the OER, only demanding a low overpotential of 259 mV to achieve 10 mA cm−2 in 1.0 M KOH.
Hydrogen has been identified as one of the most viable alternative energy sources to fossil fuels, due to its high calorific value and renewability.1 Among the various hydrogen technologies, water electrolysis is a promising way to produce hydrogen in a sustainable manner.2 However, during water electrolysis, the anodic oxygen evolution reaction involves a multi-step four-electron transfer process, leading to sluggish kinetics as compared to the hydrogen evolution reduction (HER). Thus, the exploitation of high-efficiency electrocatalysts is urgently needed to enhance the OER performance and thus the overall water-splitting efficiency.3,4 Conventionally, commercial RuO2 and IrO2 show good OER performances; however, the shortage of noble metals and their high cost limit their applications, and they usually suffer from poor stability during long-term electrolysis.5 Therefore, there is an urgent need to design efficient OER catalysts based on traditional metals such as metal-phosphides,6,7 metal-sulfides,8,9 metal-oxides,10,11 metal-hydroxides,12–15 and MOF-related materials,16–19etc.
Recently, layered double hydroxides (LDHs) have attracted much attention due to their high tunability with brucite-like layers and intercalated anions.20,21 Their large interlayer distance can afford remarkably large electrochemically accessible surface areas.22 Meanwhile, the abundant and uniformly distributed metal active sites could lead to high catalytic activity and selectivity.23 Among them, NiFe-LDHs have shown great potential for the OER due to the synergistic effect between Ni2+ and Fe3+.24,25 However, the relatively poor intrinsic catalytic activity of the reactive sites of NiFe-LDHs inhibits further enhancement of their OER performance.26 Fortunately, the electronic structures and surface active sites of NiFe-LDHs can be adjusted by modification, such as anion intercalation, sulfuring, phosphating, hetero metal doping, defect introduction, etc., thus greatly improving their intrinsic properties.27–30 For example, Luo et al.28 confirmed that the interactions between the intercalated phosphorous oxoanions (H2PO2−) and the edge-sharing MO6 (M = Ni, Fe) layers can modify the surface electronic structure of the Ni2+ sites and thus enhance the OER performance. Xiang et al.31 introduced a surface sulfurization treatment to fabricate NiCo-LDH@HOS as a highly efficient and durable electrocatalyst for the OER. Yang et al.32 reported that the third metal doped into NiFe-LDHs can be incorporated into the lattices of the hybrid, thus modifying the chemical environment and boosting the electrocatalytic performance.
A careful analysis of these reports shows that the reduced electronic densities of the active sites induced by various modifications generally should be responsible for the enhancement of the activities of the NiFe-LDH based catalysts. This could be due to the fact that the higher-valent electrophilic Ni/Fe sites promote the nucleophilic addition of hydroxyls and water molecules on the catalyst surface and improve the absorption of the reaction intermediate during the OER process.33 Therefore, the introduction of strong electron-withdrawing anions could be an effective way to further improve their activities. Inspired by this, we speculate that the integration of electron-withdrawing anion intercalation and surface sulfurization could generate a significant synergistic effect for an efficient OER.
To support this hypothesis, herein we designed and fabricated molybdenum, sulphur and phosphorus oxoanion co-intercalated NiFe-LDH nanoflowers with surface sulfurization (Mo0.5PS-NiFe-LDH) using a simple two-step hydrothermal procedure. By adjusting the ratio of the dopants, the best Mo0.5PS-NiFe-LDH exhibits a remarkable activity for the OER with a low overpotential of 259 mV at 10 mA cm−2 in 1.0 M KOH, and maintains good stability during long-term electrolysis. The boosted OER electrocatalytic performance of this catalyst could be correlated to the optimal electronic structures of the active sites modulated by anion intercalation and surface functionalization.
The MoxPS-NiFe-LDH samples were fabricated by a two-step hydrothermal method which is schematically illustrated in Fig. 1a (for details see the Experiment section in the ESI†). The phase structures of the as-prepared samples were analysed by powder X-ray diffraction (PXRD). As shown in Fig. S1a,† all the samples exhibit the characteristic peaks of the LDH phase, consistent with the standard PDF card (PDF#40-0125). Upon the intercalation of molybdate (Mo7O246−) ions, the peak at 11.4° (labelled as the B region in Fig. S1a†) decreases due to the higher atomic scattering factor for Mo in the LDHs.34 Besides this, a broad peak appears at about 6.5° (labelled as the A region in Fig. S1a†) upon molybdate intercalation, indicating the presence of enlarged layer spacing due to the large size of the intercalated molybdate.35 The presence of Mo7O246− was further confirmed by FT-IR spectroscopy (Fig. S2†).35,36 After further introducing sulphur and phosphorus oxoanions, as well as surface sulfurization, lots of impurity peaks appear in PS-NiFe-LDH (Fig. S1b†), illustrating that the structure of NiFe-LDH can be easily disrupted, leading to other phases.37 By contrast, the PXRD patterns of MoxPS-NiFe-LDH are almost the same as those of Mox-NiFe-LDH, indicating that intercalation with molybdate can improve the structural stability of NiFe-LDH. The larger layer spacing has proven to be instrumental in mass transfer and thus will improve the electrocatalytic performance for the OER.22 Besides, the PXRD patterns of the contrast samples with different composition ratios were also recorded, which show their similar structures to the LDH phase (Fig. S1c and d†).
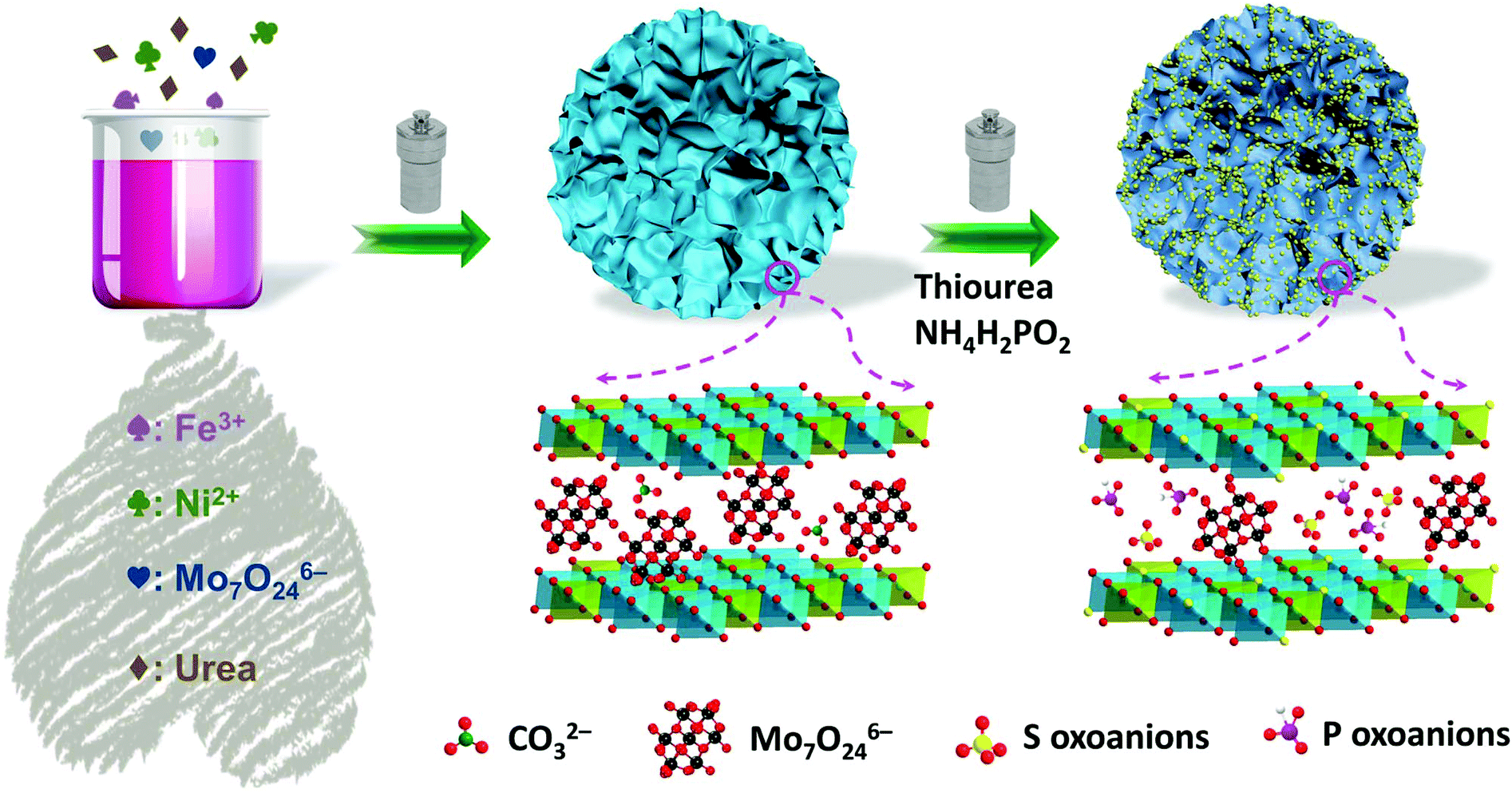 | ||
| Fig. 1 Schematic diagram for the fabrication of MoxPS-NiFe-LDH. | ||
The structure and morphology of Mo0.5PS-NiFe-LDH were examined by scanning electron microscopy (SEM), transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and energy-dispersive X-ray (EDX) spectroscopy. Although both NiFe-LDH and Mo0.5-NiFe-LDH show a flower-like shape (Fig. 2a and b), the size of the nanoflowers became smaller and the layers became thinner after the intercalation of molybdate.38 After intercalating sulfur and phosphorus oxoanions, the morphologies of both PS-NiFe-LDH and Mo0.5PS-NiFe-LDH were maintained well (Fig. 2c and d). The TEM images confirmed the nanoflower structures of Mo0.5PS-NiFe-LDH (Fig. 2e). The corresponding EDX elemental mappings demonstrate that the Ni, Fe, Mo, S, P and O elements are uniformly distributed in the nanostructure of Mo0.5PS-NiFe-LDH (Fig. 2i), suggesting the successful fabrication of the molybdenum, sulphur and phosphorus oxoanion co-intercalated NiFe-LDH materials. Notably, C mapping shows that no obvious C element was identified in the nanostructure, confirming that the CO32− anions, which are the main counter ions in pristine NiFe-LDH obtained under similar conditions,39 were almost replaced by molybdenum, sulfur and phosphorus oxoanions, which is in accordance with the decrease of the peak at 1383 cm−1, as shown in Fig. S2.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22,39 The atomic ratio of Ni, Fe, Mo, P and S elements was determined to be 4.8:1.9:1:3.7:1.5 by EDX (Fig. S3†), further confirming the successful doping of Mo, P and S based oxoanions and the majority of the phosphorus oxoanions.
22,39 The atomic ratio of Ni, Fe, Mo, P and S elements was determined to be 4.8:1.9:1:3.7:1.5 by EDX (Fig. S3†), further confirming the successful doping of Mo, P and S based oxoanions and the majority of the phosphorus oxoanions.
 | ||
| Fig. 2 SEM images of (a) NiFe-LDH, (b) Mo0.5-NiFe-LDH, (c) PS-NiFe-LDH and (d) Mo0.5PS-NiFe-LDH. (e, f) TEM, (g) HRTEM and (h) HAADF-STEM images, and (i) EDX elemental mappings of Mo0.5PS-NiFe-LDH. Scale bars: (a, b, c, d) 1 μm; (e) 500 nm; (f) 200 nm; (g) 5 nm; (h) 100 nm; and (i) 500 nm. | ||
To reveal the elemental composition, chemical status and particularly the change of electronic structure upon functionalization, X-ray photoelectron spectroscopic (XPS) analyses were conducted for the catalysts (Fig. 3). The deconvoluted spectra of Ni 2p, as shown in Fig. 3b, show two spin–orbit doublets, which are the characteristic peaks of Ni2+ and Ni3+, and satellites. The observed two main peaks for Mo0.5PS-NiFe-LDH at binding energy (BE) values of 856.07 and 873.45 eV with a spin–orbit splitting of 17.38 eV are ascribed to Ni 2p3/2 and Ni 2p1/2 with +2 oxidation state, respectively, while another pair of weak peaks at 859.87 and 875.42 eV suggests the existence of a small amount of Ni3+.40,41 The deconvoluted spectrum of Fe 2p for Mo0.5PS-NiFe-LDH (Fig. 3c) shows two peaks at BE values of 712.23 (Fe 2p3/2) and 725.49 eV (Fe 2p1/2), which signifies that iron exists in the Fe3+ state.34,42 Compared to those of NiFe-LDH, the Ni 2p and Fe 2p peaks of both Mo0.5-NiFe-LDH and Mo0.5PS-NiFe-LDH are shifted to the more positive energies, manifesting the strong interactions between the intercalated Mo, P and S based oxoanions and the Ni and Fe sites, which reduces the surface electron densities of the active sites.43,44 The Mo 3d spectrum of Mo0.5PS-NiFe-LDH splits into two main peaks of 2p5/2 and 2p3/2 at 232.24 and 235.29 eV, which are attributed to Mo6+ (Fig. 3d).45,46 After further intercalating the sulfur and phosphorus oxoanions, an obvious positive-shift of Mo6+ can be observed due to electron loss, indicating stronger electron-withdrawing properties of the sulfur and/or phosphorus oxoanions.47 In the S 2p spectrum of Mo0.5PS-NiFe-LDH (Fig. 3e), the BE values at 162.42 and 164.12 eV ascribed to S 2p3/2 and S 2p1/2 correspond to the metal–sulfide (M–S) bonding,48,49 while the peaks at 168.10 and 169.20 eV are assigned to the sulphur oxoanions.50,51 The results illustrate that some of the S atoms were bonded with metals by replacing the O atoms in the edge-sharing MO6 units to form surface metal sulfides and the rest of the S atoms were oxidized and then replaced the interlaminar CO32− ions, which is consistent with the EDX mapping results. As presented in Fig. 3f, the only peak of P 2p at 132.9 eV is associated with the oxidized P species.52,53 The XPS results reveal the successful intercalation of molybdenum, sulfur and phosphorus oxoanions and surface sulfurization in Mo0.5PS-NiFe-LDH, which generate a strong synergistic effect and modulate the chemical environments and electronic structures of the active Ni and Fe sites for electrocatalysis.
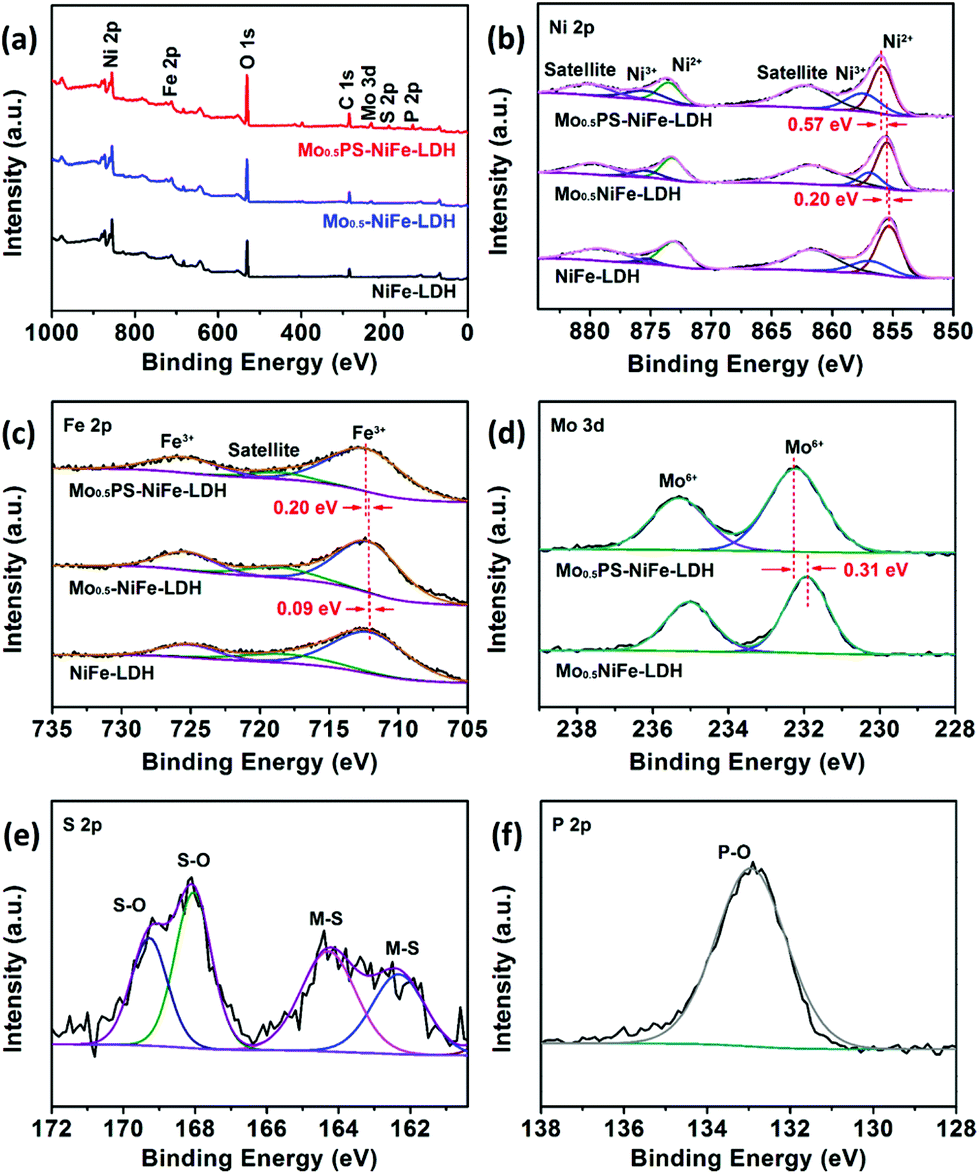 | ||
| Fig. 3 (a) XPS survey spectra and high-resolution XPS spectra of (b) Ni 2p and (c) Fe 2p of NiFe-LDH, Mo0.5-NiFe-LDH, and Mo0.5PS-NiFe-LDH; high-resolution XPS spectra of (d) Mo 3d of Mo0.5-NiFe-LDH and Mo0.5PS-NiFe-LDH; high-resolution XPS spectra of (e) S 2p and (f) P 2p of Mo0.5PS-NiFe-LDH. | ||
The electrocatalytic OER performances of MoxPS-NiFe-LDH (the molar ratio of Ni:Fe of the samples was 3:1 except for special instructions) were examined in 1.0 M KOH using a typical three-electrode system (for details see the ESI†, Electrochemical measurements). For comparison, NiFe-LDH, PS-NiFe-LDH, Mo0.5-NiFe-LDH and the commercial benchmark RuO2 catalyst were measured under the same conditions. According to the linear sweep voltammetry (LSV) polarization curves shown in Fig. 4a, the OER activities of the catalysts with different functionalizations were apparently improved as indicated by the considerable negative-shift of the polarization curves (Fig. 4a, Mo0.5PS-NiFe-LDH vs. PS-NiFe-LDH, Mo0.5-NiFe-LDH vs. NiFe-LDH), which can be ascribed to the larger layer spacing, electron interactions and synergistic effect between the intercalated oxoanions and the electrochemically active sites of the LDH.54,55 Besides, surface sulfurization may also enhance the OER properties due to the modification of the electronic structure (Fig. 4a).33,56 Meanwhile, the Ni2+/Ni3+ oxidative peaks of Mo0.5PS-NiFe-LDH at ca. 1.4 V (vs. RHE) were significantly decreased, which may originate from the reduction of ammonium hypophosphite and further implies the modification of the electronic structure of the active sites upon the intercalation of the phosphorus oxoanion.28,57 In comparison, the Mo0.5PS-NiFe-LDH catalyst delivered higher current densities and more negative OER onset potentials than NiFe-LDH, PS-NiFe-LDH, Mo0.5-NiFe-LDH and RuO2. Particularly, Mo0.5PS-NiFe-LDH showed the lowest overpotential of only 259 mV to achieve a current density of 10 mA cm−2 (Fig. 4b). To further investigate the influences of the molar quantity of the Mo element, reaction time, Ni/Fe ratio and functionalization on the catalytic activity, a series of other comparative samples were synthesized and tested (Fig. S4†). It was obvious that Mo0.5PS-NiFe-LDH showed a much better catalytic performance than those of the contrast samples.
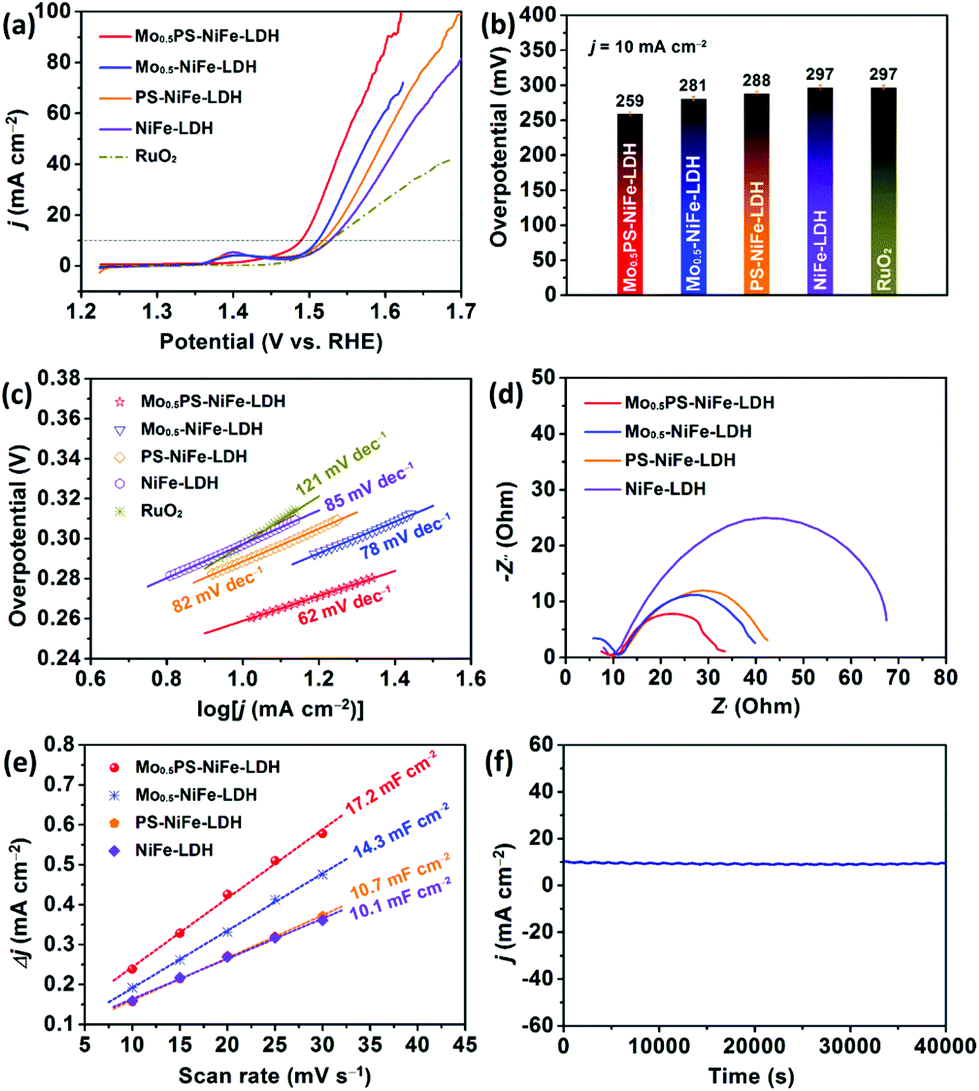 | ||
| Fig. 4 Electrochemical performances of the catalysts in 1.0 M KOH: (a) polarization curves at 5 mV s−1, (b) required overpotentials at the current density of 10 mA cm−2, (c) Tafel plots, (d) Nyquist plots, (e) current density variation at a non-redox region plotted against scan rates to estimate Cdl, and (f) amperometric i–t curve of Mo0.5PS-NiFe-LDH. | ||
The catalytic kinetics of the catalysts were evaluated using the Tafel slope and electrochemical impedance spectroscopy (EIS). As shown in Fig. 4c, Mo0.5PS-NiFe-LDH presents the smallest Tafel value (62 mV dec−1), compared to RuO2 (121 mV dec−1), NiFe-LDH (85 mV dec−1), PS-NiFe-LDH (82 mV dec−1), and Mo0.5-NiFe-LDH (78 mV dec−1), indicating the fastest OER kinetics. The semicircles shown in Fig. 4d are Nyquist plots obtained at the overpotential of 274 mV. The semicircles in the EIS curves correspond to the charge transfer resistance (Rct), which is related to the OER kinetics occurring at the electrode/electrolyte interface. Apparently, Mo0.5PS-NiFe-LDH shows the smallest Rct value, illustrating that it has relatively low charge transfer resistance and superior charge transport kinetics, owing to its nanoflower morphology and structural funactionalization.58,59 The results prove that Mo0.5PS-NiFe-LDH showed better kinetics than the other catalysts.
To reveal the high electrochemical performance of Mo0.5PS-NiFe-LDH for the OER, the electrochemically active surface area (ECSA) was estimated from the double-layer capacitance (Cdl) by carrying out CV tests in the non-faradaic region (Fig. S5†). As shown in Fig. 4e, the Cdl value of Mo0.5PS-NiFe-LDH was calculated to be 17.2 mF cm−2, which is apparently larger than those of NiFe-LDH (10.1 mF cm−2), PS-NiFe-LDH (10.7 mF cm−2), and Mo0.5-NiFe-LDH (14.3 mF cm−2). This result indicates that Mo0.5PS-NiFe-LDH is capable of providing more active sites to enhance the electrocatalytic OER activity.30,35,60
In addition to the improved catalytic activity, Mo0.5PS-NiFe-LDH showed high stability during the long-term OER reaction. As shown in Fig. S6,† three parallel Mo0.5PS-NiFe-LDH samples were tested and they showed good repeatability. After successive CV scanning for 1000 cycles, the overpotential increased only 12 mV at the current density of 10 mA cm−2. Meanwhile, chronoamperometry of Mo0.5PS-NiFe-LDH was further performed under electromagnetic stirring, and the curve shows that the catalytic current density was well maintained over 40000 s (Fig. 4f). These results demonstrate the excellent electrocatalytic stability of the Mo0.5PS-NiFe-LDH catalyst. For better understanding the catalytic activity and durability, the morphology and composition of Mo0.5PS-NiFe-LDH after the OER test were further characterized by PXRD, SEM and XPS. As shown in Fig. S7,† the characteristic diffraction peaks of the sample were maintained, illustrating that no new crystalline phase was formed during the test and the LDH structure remained unchanged. The XPS measurement for Mo0.5PS-NiFe-LDH after the stability test shows no obvious change in the high-resolution Ni 2p, Fe 2p, Mo 3d and P 2p spectra (Fig. S8†). However, the valence state of S was changed and the strength of M–S bonds was decreased, revealing that the S-based species could be reconstructed to form surface metal hydroxysulfides and thus influenced the surface electronic structure during the OER test. The SEM image showed that the sample presented a flower-like but thicker lamellar structure than that before the OER test (Fig. S9†), which may result from the surface reconstruction and the residual Nafion membrane covering the sample surface. The above results indicate that the surface electronic structure was further adjusted during the OER process to probably obtain abundant Ni2+/3+/Fe3+ sites and surface metal hydroxysulfides as the active species, thus leading to high catalytic performance.5,30,31,50,53,61–64
Conclusions
In this work, we have introduced a facile strategy with the combination of electron-withdrawing anion intercalation and surface sulfurization to hydrothermally construct NiFe-LDH based nanoflowers with dual-modified electronic structures as highly efficient and durable electrocatalysts for the OER. Owing to the intercalation of the electron-withdrawing molybdenum, sulfur and phosphorus oxoanions and surface sulfurization, the surface chemical states and electronic structures of the active sites in the resulting Mo0.5PS-NiFe-LDH catalyst were modulated, which greatly enhance their intrinsic activity for the OER. As a result, Mo0.5PS-NiFe-LDH showed a much superior OER performance compared to pristine NiFe-LDH and benchmark RuO2. This work provides a new way to fabricate efficient non-noble catalysts for the OER and other electrocatalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the One Thousand Young Talents Program under the Recruitment Program of Global Experts, the National Natural Science Foundation of China (NSFC) (21901246, 21905278 and 21771179), and the Natural Science Foundation of Fujian Province (2019J05158 and 2019J01133).Notes and references
- J. H. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- X. S. Wang, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Adv. Mater., 2018, 31, 1803625 CrossRef PubMed.
- W. P. Yang, X. X. Li, Y. Li, R. M. Zhu and H. Pang, Adv. Mater., 2019, 31, 1804740 Search PubMed.
- F. L. Lyu, Q. F. Wang, S. M. Choi and Y. D. Yin, Small, 2018, 15, 1804201 CrossRef PubMed.
- X. T. Han, C. Yu, H. W. Huang, W. Guo, C. T. Zhao, H. L. Huang, S. F. Li, Z. B. Liu, X. Y. Tan, Z. M. Gao, J. H. Yu and J. S. Qiu, Nano Energy, 2019, 62, 136–143 CrossRef CAS.
- J. Wang and F. Ciucci, Appl. Catal., B, 2019, 254, 292–299 CrossRef CAS.
- Y. J. Tang, A. M. Zhang, H. J. Zhu, L. Z. Dong, X. L. Wang, S. L. Li, M. Han, X. X. Xu and Y. Q. Lan, Nanoscale, 2018, 10, 8404–8412 RSC.
- Y. Yang, H. Q. Yao, Z. H. Yu, S. M. Islam, H. Y. He, M. W. Yuan, Y. H. Yue, K. Xu, W. C. Hao, G. B. Sun, H. F. Li, S. L. Ma, P. Zapol and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS.
- D. C. Zhong, L. M. Cao, Y. W. Hu and T. B. Lu, ChemSusChem, 2018, 11, 3708–3713 CrossRef.
- A. L. Strickler, M. E. Escribano and T. F. Jaramillo, Nano Lett., 2017, 17, 6040–6046 CrossRef CAS.
- L. Qian, Z. Y. Lu, T. H. Xu, X. C. Wu, Y. Tian, Y. P. Li, Z. Y. Huo, X. M. Sun and X. Duan, Adv. Energy Mater., 2015, 5, 1500245 CrossRef.
- X. F. Zhang, B. Y. Zhang, S. S. Liu, H. W. Kang, W. Q. Kong, S. R. Zhang, Y. Shen and B. C. Yang, Appl. Surf. Sci., 2018, 436, 974–980 CrossRef CAS.
- Y. Zhang, Q. Shao, Y. C. Pi, J. Guo and X. Q. Huang, Small, 2017, 13, 1700355 CrossRef.
- D. D. Zhao, K. Z. Jiang, Y. C. Pi and X. Q. Huang, ChemCatChem, 2017, 9, 84–88 CrossRef CAS.
- W. Zhou, D. D. Huang, Y. P. Wu, J. Zhao, T. Wu, J. Zhang, D. S. Li, C. H. Sun, P. Y. Feng and X. H. Bu, Angew. Chem., Int. Ed., 2019, 58, 4227–4231 CrossRef CAS PubMed.
- Y. P. Wu, J. W. Tian, S. Liu, B. Li, J. Zhao, L. F. Ma, D. S. Li, Y. Q. Lan and X. H. Bu, Angew. Chem., Int. Ed., 2019, 58, 12185–12189 CrossRef CAS PubMed.
- Z. B. Liang, R. Zhao, T. J. Qiu, R. Q. Zou and Q. Xu, Energy Chem., 2019, 1, 100001 CrossRef.
- J. W. Tian, Y. P. Wu, Y. S. Li, J. H. Wei, J. W. Yi, S. Li, J. Zhao and D. S. Li, Inorg. Chem., 2019, 58, 5837–5843 CrossRef CAS.
- M. Gong and H. J. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- C. Q. Dong, L. L. Han, C. Zhang and Z. H. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 16096–16104 CrossRef CAS.
- L. N. Dang, H. F. Liang, J. Q. Zhuo, B. K. Lamb, H. Y. Sheng, Y. Yang and S. Jin, Chem. Mater., 2018, 30, 4321–4330 CrossRef CAS.
- G. L. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- Z. H. Li, M. F. Shao, H. L. An, Z. X. Wang, S. M. Xu, M. Wei, D. G. Evans and X. Duan, Chem. Sci., 2015, 6, 6624–6631 RSC.
- X. M. Li, X. G. Hao, Z. D. Wang, A. Abudula and G. Q. Guan, J. Power Sources, 2017, 347, 193–200 CrossRef CAS.
- Y. F. Zeng, L. J. Chen, R. Chen, Y. Y. Wang, C. Xie, L. Tao, L. L. Huang and S. Y. Wang, J. Mater. Chem. A, 2018, 6, 24311–24316 RSC.
- M. Luo, Z. Cai, C. Wang, Y. M. Bi, L. Qian, Y. C. Hao, L. Li, Y. Kuang, Y. P. Li, X. D. Lei, Z. Y. Huo, W. Liu, H. L. Wang, X. M. Sun and X. Duan, Nano Res., 2017, 10, 1732–1739 CrossRef CAS.
- K. Nejati, S. Davari, A. Akbari, K. A. Zeynali and Z. Rezvani, Int. J. Hydrogen Energy, 2019, 44, 14842–14852 CrossRef CAS.
- Y. Y. Wang, M. Qiao, Y. F. Li and S. Y. Wang, Small, 2018, 14, 1800136 CrossRef PubMed.
- K. Xiang, J. Guo, J. Xu, T. T. Qu, Y. Zhang, S. Y. Chen, P. P. Hao, M. H. Li, M. J. Xie, X. F. Guo and W. P. Ding, ACS Appl. Energy Mater., 2018, 1, 4040–4049 CrossRef CAS.
- Y. Yang, L. N. Dang, M. J. Shearer, H. Y. Sheng, W. J. Li, J. Chen, P. Xiao, Y. H. Zhang, R. J. Hamers and S. Jin, Adv. Energy Mater., 2018, 8(15), 1703189 CrossRef.
- L. Z. Fan, P. L. Zhang, Q. Daniel, B. J. J. Timmer, F. G. Zhang and L. C. Sun, ACS Energy Lett., 2018, 3, 2865–2874 CrossRef CAS.
- Y. Zhang, P. H. Yu, J. J. Wu, F. Chen, Y. D. Li, Y. L. Zhang, Y. Zuo and Y. A. L. Qi, J. Coat. Technol. Res., 2018, 15, 303–313 CrossRef CAS.
- X. Y. Chen, Z. M. Wang, Y. Z. Wei, X. Zhang, Q. H. Zhang, L. Gu, L. J. Zhang, N. L. Yang and R. B. Yu, Angew. Chem., Int. Ed. DOI:10.1002/anie.201909879.
- B. Courcot and A. J. Bridgeman, J. Phys. Chem. A, 2009, 113, 10540–10548 CrossRef CAS.
- X. Long, G. X. Li, Z. L. Wang, H. Y. Zhu, T. Zhang, S. Xiao, W. Y. Guo and S. H. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS.
- K. Nejati, A. R. Akbari, S. Davari, K. A. Zeynali and Z. Rezvani, New J. Chem., 2018, 42, 2889–2895 RSC.
- Z. Y. Lu, L. Qian, W. W. Xu, Y. Tian, M. Jiang, Y. P. Li, X. M. Sun and X. Duan, Nano Res., 2016, 9, 3152–3161 CrossRef CAS.
- K. Q. Qin, L. P. Wang, S. W. Wen, L. C. Diao, P. Liu, J. J. Li, L. Y. Ma, C. S. Shi, C. Zhong, W. B. Hu, E. Z. Liu and N. Q. Zhao, J. Mater. Chem. A, 2018, 6, 8109–8119 RSC.
- G. Rajeshkhanna, T. I. Singh, N. H. Kim and J. H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 42453–42468 CrossRef CAS.
- Y. Q. Zheng, B. Cheng, W. You, J. G. Yu and W. K. Ho, J. Hazard. Mater., 2019, 369, 214–225 CrossRef CAS.
- T. T. Zhou, Z. Cao, P. Zhang, H. Y. Ma, Z. Gao, H. Wang, Y. Lu, J. He and Y. F. Zhao, Sci. Rep., 2017, 7, 46154 CrossRef CAS PubMed.
- S. Nayak, G. Swain and K. Parida, ACS Appl. Mater. Interfaces, 2019, 11, 20923–20942 CrossRef CAS PubMed.
- H. Q. Chu, D. Zhang, B. W. Jin and M. Yang, Appl. Catal., B, 2019, 255, 117744 CrossRef CAS.
- J. Guo, X. J. Yang, S. L. Bai, X. Xiang, R. X. Luo, J. He and A. F. Chen, J. Colloid Interface Sci., 2019, 540, 9–19 CrossRef CAS.
- A. P. Wu, Y. Gu, Y. Xie, C. G. Tian, H. J. Yan, D. X. Wang, X. M. Zhang, Z. C. Cai and H. G. Fu, ACS Appl. Mater. Interfaces, 2019, 11, 25986–25995 CrossRef CAS.
- Q. J. Yang, Y. Liu, M. Yan, Y. Lei and W. D. Shi, Chem. Eng. J., 2019, 370, 666–676 CrossRef CAS.
- Z. Zhang, Y. P. Deng, Z. Y. Xing, D. Luo, S. Sy, Z. P. Cano, G. H. Liu, Y. Jiang and Z. W. Chen, ACS Nano, 2019, 13, 7062–7072 CrossRef CAS PubMed.
- G. G. Zhang, J. Y. Yuan, Y. Liu, W. Lu, N. Q. Fu, W. F. Li and H. T. Huang, J. Mater. Chem. A, 2018, 6, 10253–10263 RSC.
- Q. L. Zhu, W. Xia, T. Akita, R. Q. Zou and Q. Xu, Adv. Mater., 2016, 28, 6391–6398 CrossRef CAS.
- Q. Qin, H. Jang, L. L. Chen, G. Nam, X. E. Liu and J. Cho, Adv. Energy Mater., 2018, 9, 1801478 CrossRef.
- X. Zhang, Y. H. Wu, Y. F. Sun, Q. Y. Liu, L. Tang and J. X. Guo, Inorg. Chem. Front., 2019, 6, 604–611 RSC.
- N. Han, F. P. Zhao and Y. G. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 RSC.
- S. Y. Hao, L. C. Chen, C. L. Yu, B. Yang, Z. J. Li, Y. Hou, L. C. Lei and X. W. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- C. Y. Liu, H. Ma, M. W. Yuan, Z. H. Yu, J. Li, K. Shi, Z. P. Liang, Y. Yang, T. J. Zhu, G. B. Sun, H. F. Li and S. L. Ma, Electrochim. Acta, 2018, 286, 195–204 CrossRef CAS.
- D. D. Ma, C. S. Cao, X. F. Li, J. T. Cheng, L. L. Zhou, X. T. Wu and Q. L. Zhu, Electrochim. Acta, 2019, 321, 134679 CrossRef CAS.
- S. Y. Hao, L. C. Chen, C. L. Yu, B. Yang, Z. J. Li, Y. Hou, L. C. Lei and X. W. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- J. Bao, Z. L. Wang, J. F. Xie, L. Xu, F. C. Lei, M. L. Guan, Y. Zhao, Y. P. Huang and H. M. Li, Chem. Commun., 2019, 55, 3521–3524 RSC.
- Y. Duan, Z. Y. Yu, S. J. Hu, X. S. Zheng, C. T. Zhang, H. H. Ding, B. C. Hu, Q. Q. Fu, Z. L. Yu, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS.
- Z. Z. Liu, X. Shang, B. Dong and Y. M. Chai, J. Catal., 2018, 361, 204–213 CrossRef CAS.
- S. Zhang, X. Y. Zhang, J. Li and E. K. Wang, J. Mater. Chem. A, 2017, 5, 20588–20593 RSC.
- X. Q. Du, C. R. Huang and X. S. Zhang, Int. J. Hydrogen Energy, 2019, 44, 19595–19602 CrossRef CAS.
- B. L. Deng, L. S. Zhou, Z. Q. Jiang and Z. J. Jiang, J. Catal., 2019, 373, 81–92 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi01367a |
| ‡ Y.-Y. D. and D.-D. M. contributed equally. |
| This journal is © the Partner Organisations 2020 |