
Unexpected structural complexity of thorium coordination polymers and polyoxo cluster built from simple formate ligands†
Zi-Jian
Li
 a,
Shangyao
Guo
a,
Huangjie
Lu
a,
Yongjia
Xu
a,
Zenghui
Yue
a,
Linhong
Weng
b,
Xiaofeng
Guo
c,
Jian
Lin
*a and
Jian-Qiang
Wang
a
a,
Shangyao
Guo
a,
Huangjie
Lu
a,
Yongjia
Xu
a,
Zenghui
Yue
a,
Linhong
Weng
b,
Xiaofeng
Guo
c,
Jian
Lin
*a and
Jian-Qiang
Wang
a
aCAS Key Laboratory of Interfacial Physics and Technology, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, 2019 Jia Luo Road, Shanghai 201800, China. E-mail: linjian@sinap.ac.cn
bDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, Shanghai 200433, China
cDepartment of Chemistry, Washington State University, Pullman, WA 99164-4630, USA
First published on 30th October 2019
Abstract
A simple binary synthetic approach with variable [HCOOH]/[Th(IV)] ratios and the addition of water resulted in the self-assembly of six novel thorium formate complexes with well-controlled structures ranging from 0D clusters and 2D layered networks to 3D frameworks. Specifically, small [HCOOH]/[Th(IV)] promoted the formation of the hexanuclear Th6O4(OH)4(HCOO)12 core, a secondary building unit for the construction of 3D [Th6Na(μ3-O)4(μ3-OH)4(HCOO)14]·[NH2(CH3)2]·(DMF)3(H2O)5 (Th-SINAP-1), 2D [Th6(μ3-O)4(μ3-OH)4(HCOO)12(DMF)2]·(H2O)10 (Th-SINAP-2), and 0D [Th6(μ3-O)4(μ3-OH)4(HCOO)12(H2O)6]·G (Th-SINAP-3). The large [HCOOH]/[Th(IV)] inhibited the olation/oxolation reaction, affording 3D frameworks of [Th2(HCOO)9]·[NH2(CH3)2]·(DMF)(H2O)2 (Th-SINAP-4), [Th2(HCOO)9]·[NH2(CH3)2]·(DMF)(H2O)4 (Th-SINAP-5), and [Th2(HCOO)8(H2O)(DMF)]·(H2O)3 (Th-SINAP-6) with monomeric Th(IV) cations as the metal nodes. The addition of water not only affected the hydrolysis of Th(IV) to form nucleophilic hydroxo/oxo-aquo Th species, but also influenced the overall solid-state packing via the competition between H2O, HCOO−, OH−/O2−, and DMF for coordination with Th4+ cations. This unexpected structural complexity of thorium formates opens a new avenue for the targeted design and synthesis of atypical coordination polymers, e.g. open-framework materials of actinides, for applications in diverse fields such as radionuclide adsorption.
Introduction
As the most abundant actinide metal on earth, thorium has received increasing attention owning to its potential and revolutionary use in new types of nuclear fuels. In the thorium fuel cycle, 232Th can capture neutrons and breed 233U, a fissionable fuel in molten salt reactors.1 Thus, a comprehensive understanding of the coordination chemistry of thorium can benefit the optimization of the separation processes in the nuclear fuel cycle and the prediction of the fate of thorium in the environment.2 In addition, ThIV is considered an ideal surrogate for studying the much more radiotoxic PuIV owning to its exclusive tetravalent state in nature and similarities in coordination as PuIV.3,4 Thus, the coordination chemistry of thorium can serve as a benchmark for Pu.5,6 However, thorium complexes have been significantly less investigated compared to their uranium counterparts. To date, only approximately 60 thorium coordination polymers have been reported versus the hundreds for uranium based on the Cambridge Crystallographic Data Centre (CCDC). This marginalization can be partially attributed to the relative difficulty in obtaining thorium coordination polymers.The strong Lewis acidic nature of ThIV and other tetravalent actinide ions facilitates the prevalence of their hydrolysis and condensation reactions in aqueous solutions, promoting the formation of oligomers and even colloids.7–9 However, the isolation of these polynuclear species have proven to be rather intricate and challenging, and the generated solids are often not sufficiently crystallized for single crystal X-ray diffraction characterization, especially in the case of thorium. By employing bridging ligands, such as sulphate, selenate, carboxylate, and phosphonate groups, ThIV polynulcear clusters can be stabilized and crystallized from aqueous solutions under certain conditions. However, only a handful of polynuclear ThIV complexes, including dimers,10 trimers,11 tetramers,12 hexamers,13 octamers,14 and decamers,15 have been reported. Loiseau and coworkers initiated a solvothermal synthesis method to capture tetravalent actinide poly-oxo clusters via the assistance of bridging carboxylate ligands.16–20 The extent of hydrolysis, nucleation, and condensation can be fine-tuned by the addition of water, the selection of different organic solvents, and varying the reaction temperature, which allow the isolation of a variety of clusters containing up to 38 AnIV cations.21–23 Nevertheless, thorium polyoxo clusters in association with carboxylate groups still represent an underdeveloped area, where further exploration of the chemical behaviours of thorium is necessary.
Formic acid is commonly used as a modulator to control the crystal growth and improve the crystallinity of metal organic frameworks (MOFs). Due to the strong affinity between tetravalent metal cations and dicarboxylate ligands, the poor solubility of the formed dicarboxylate MOFs often result in rapid nucleation and precipitation, forming bulk powder or even amorphous products. Monocarboxylic acid can be used to regulate the rate of framework extension, crystallization, and crystal growth.24 The competition between formate and multidentate carboxylate linkers not only allows the formation of hexanuclear metal-oxo formate clusters, but also leads to the partial replacement of bridging linkers by modulators, resulting in desirable pore sizes and defects.25–29 Thus, elucidating the role of formate in molecular assembly will help in the better design and synthesis of targeted thorium coordination polymers. Additionally, the importance of formate is highlighted by the role of carboxylate ligands in solvent extraction processes for actinide separation.30,31 As the simplest carboxylate, formate can be visualized as a model ligand to study more complicated metal–ligand complexation.32–36 Formate displays different coordination modes including bridging-bidentate, terminal-monodentate, and terminal-bidentate, giving rise to rich but arbitrary effects on the speciation and structure of actinides.25,37–40
With the aim to expand the family of thorium coordination polymers and develop a synthetic strategy to precisely control the metal–ligand molecular assembly, we systematically investigated the solvothermal reactivity of thorium nitrate with formic acid in DMF solvent. Surprisingly, five thorium formate coordination polymers and one thorium polyoxo cluster with distinct topologies were obtained by tuning the molar ratio of [HCOOH]/[Th(IV)] and adding a controlled amount of water in the reactions. This discovery highlights the potential emergence of thorium coordination polymers with unexpected structural complexity.
Results and discussion
Structure description
Crystals of thorium formate coordination polymers and polyoxo cluster were obtained via the solvothermal reaction of thorium nitrate and formic acid in DMF with the addition of a controlled amount of water. The crystal structures of the isolated products are surprisingly rich even though they were synthesized from a simple binary reaction system. By systematically controlling the [HCOOH]/[Th(IV)] molar ratio and addition of water, six different phases were obtained, including [Th6Na(μ3-O)4(μ3-OH)4(HCOO)14]·[NH2(CH3)2]·(DMF)3(H2O)5(Th-SINAP-1), [Th6(μ3-O)4(μ3-OH)4(HCOO)12(DMF)2]·(H2O)10 (Th-SINAP-2), Th6(O)4(OH)4(HCOO)12(H2O)6·G (Th-SINAP-3) [Th2(HCOO)9]·[NH2(CH3)2]·(DMF)(H2O)2 (Th-SINAP-4), [Th2(HCOO)9]·[NH2(CH3)2]·(DMF)(H2O)4 (Th-SINAP-5), and [Th2(HCOO)8(H2O)(DMF)]·(H2O)3 (Th-SINAP-6). Single-crystal and powder X-ray diffraction measurements were used to characterize their structures and phase purities, respectively. Each of the prepared complexes can be classified as thorium hexamer- or monomer-containing coordination polymers or polyoxo cluster, depending on the nuclearity of the Th4+ cations. Th-SINAP-1, Th-SINAP-2, and Th-SINAP-3 are constructed from the assembly of common Th6(μ3-O)4(μ3-OH)4 secondary building units (SBUs) and HCOO− anions, but their overall topologies vary from a three-dimensional (3D) framework and two-dimensional (2D) layer to zero-dimensional (0D) discrete molecular cluster, respectively. Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6 are built by packing less condensed Th4+ monomers and HCOO− anions in different manners, which leads to diverse structures. The resulting versatile phases are rather uncommon, although a handful of coordination polymers combining tetravalent actinide cations with bicarboxylate, and tricarboxylate ligands having been identified in diverse topologies and nuclearity.41–44The single-crystal X-ray diffraction (SCXRD) studies revealed that Th-SINAP-1 exhibits a 3D framework and crystallizes in the tetragonal space group P4/nmm (Table S1†). Its asymmetric unit is composed of 1/8 Th6(μ3-O)4(μ3-OH)4 hexamer, 14/8 HCOO− ligands, 1/8 Na+ cation, and 1/8 μ2-H2O molecule. Each Th6(μ3-O)4(μ3-OH)4 cluster comprises three crystallographically independent Th4+ cations, all of which are nine-fold coordinated with a monocapped square anti-prismatic geometry. Th(1) and Th(3) are bonded by four μ2-HCOO− anions, two μ3-O2−, two μ3-OH−, and one μ2-H2O, whereas the coordination sphere of Th(2) is provided by five μ2-HCOO− anions, two μ3-O2−, and two μ3-OH− (Fig. 1a). The Th–O2−/OH−/HCOO− distances range from 2.299(14) to 2.519(15) Å, while the Th–Ow distances are 2.745(17) and 2.858(17) Å, both of which are in good agreement with the previously reported values (Table S2†).6 The Th6(μ3-O)4(μ3-OH)4 hexanuclear unit is decorated by 16 μ2-HCOO− anions, 12 of which bridge Th4+ ions within the cluster and four join the intermolecular hexamers into a 2D lamellar structure along the ab plane (Fig. 1b). The μ2-H2O groups further link these layers along the c axis with an AA stacking mode (Fig. 1c), generating a porous 3D network with two-fold interpenetration (Fig. 1d and e). It is interesting to note that one crystallographically unique Na+ cation was identified in the channels of the framework (Fig. S1†), which did not originate from the reagent solution. Na+ may have unexpectedly dissolved from the glass vials since the synthesis conducted in a Teflon-lined container under comparable conditions did not yield Th-SINAP-1. This was also confirmed by performing rational synthesis with the addition of NaNO3 in the solution, from which an identical product was isolated. Considering the anionic nature of the framework, positive charge is required to ensure the electroneutrality of Th-SINAP-1. Accordingly, the highly disordered NH2(CH3)2+ cations generated from decomposition of DMF can be considered as the counter cation species residing in the pores of the framework. Although NH2(CH3)2+ cations could not be accurately located on the electron density map due to their highly disordered nature in the voids and the high structural symmetry of Th-SINAP-1, the presence of NH2(CH3)2+ in the voids of MOFs isolated from DMF solution is frequently observed. Similarly, dimethylamine cations have been regarded as the charge-neutralizing species in Sr(HCOO)2:Eu2+/Eu3+,37 [Tb(BPDC)2]·(CH3)2NH2,45 [(CH3)2NH2]2[ZnNa2(μ2-H2O)2(TATAT)]·2DMF,46etc.
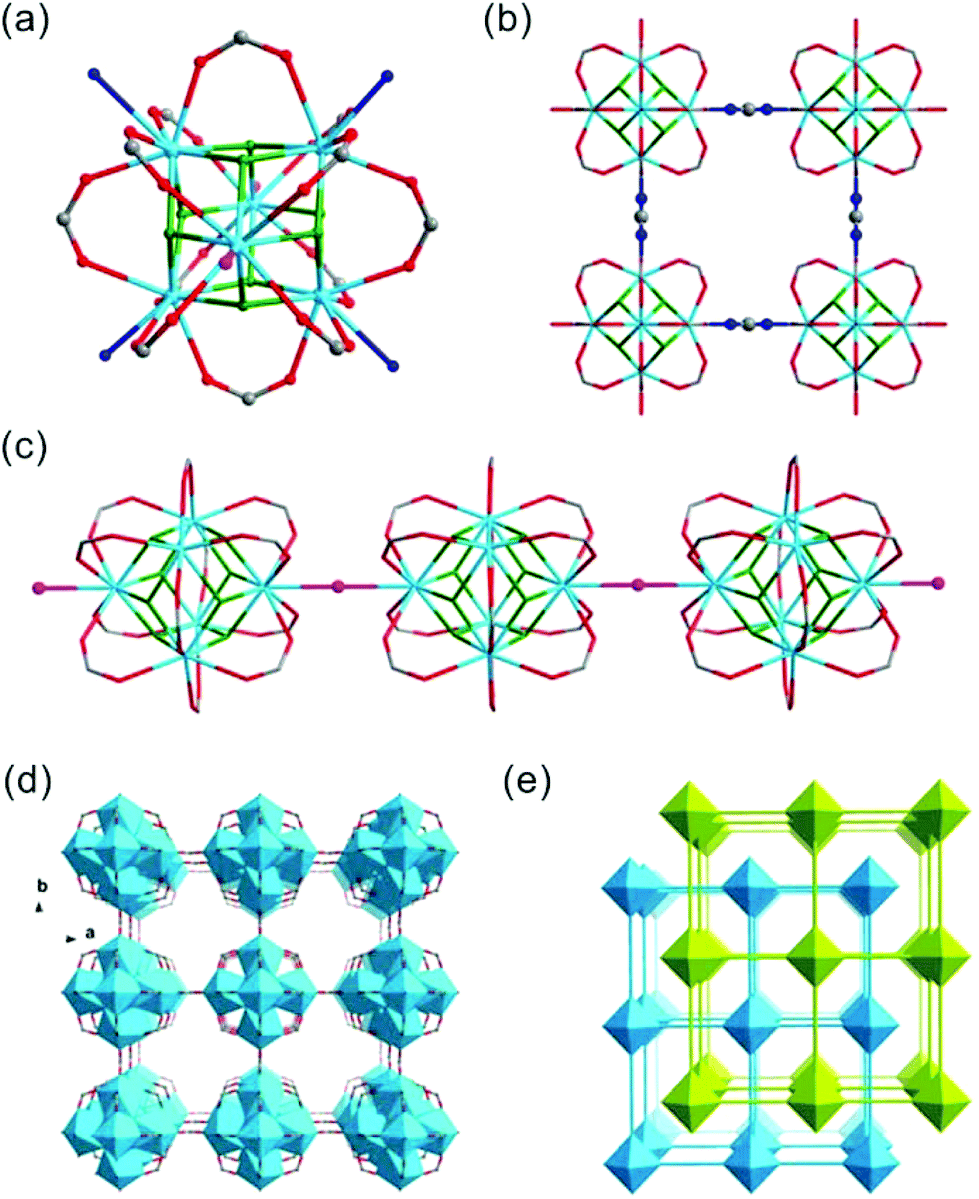 | ||
| Fig. 1 (a) Th6(μ3-O)4(μ3-OH)4(μ2-H2O)2(HCOO)14 cluster of Th-SINAP-1. The O atoms from μ3-OH/-O, μ2-H2O, bridging HCOO−, and chelating HCOO− groups are shown in green, pink, blue, and red, respectively. The Th atoms are shown in sky blue. (b) View of the 2D lamellar fragment of Th-SINAP-1 built from Th6 clusters and formate groups. (c) Additional bridging of Th6 clusters by μ2-H2O groups to create a 3D framework. (d) One independent network of Th-SINAP-1. (e) Simplified topological representation of Th-SINAP-1 showing the 2-fold interpenetration. The Na+ ions are omitted for clarity. | ||
Th-SINAP-2 possesses a 2D layered network and crystallizes in the orthorhombic space group Fmmm (Table S1†). Its asymmetric unit consists of 1/8 Th6(μ3-O)4(μ3-OH)4 hexamer, decorated or bridged by 1.5 HCOO− ligands and 1/4 DMF molecule. Three crystallographically independent Th4+ ions and three unique HCOO− ligands can be identified in the structure. Th(1) and Th(2) are coordinated with eight O atoms (HCOO− × 4, μ3-O2− × 2, and μ3-OH− × 2), while Th(3) is nine-coordinated (HCOO− × 4, μ3-O2− × 2, μ3-OH− × 2, and η1-DMF × 1) with an additional O atom donated from the terminal DMF molecule, forming square anti-prismatic and monocapped square anti-prismatic geometries, respectively (Fig. 2a). All of the Th–O distances are in the range of 2.266(4) to 2.558(5) Å, which are typical in thorium complexes (Table S2†).44,47 Eight C(1) and C(3) formate anions chelate with adjacent Th4+ ions in the Th6(μ3-O)4(μ3-OH)4 clusters (Fig. 2a), whereas the C(2) formates act as bidentate bridging ligands, which interconnect the neighboring Th6(μ3-O)4(μ3-OH)4 clusters and form layered grids extending along the ac plane (Fig. 2b). These grids stack on top of each other in a staggered arrangement with an ABAB mode, generating a pseudo-3D framework with approximately 22% void calculated by PLATON (Fig. 2c).48 The network of Th-SINAP-2 can be simplified as a 4-connected net of sql topology (Fig. 2d).
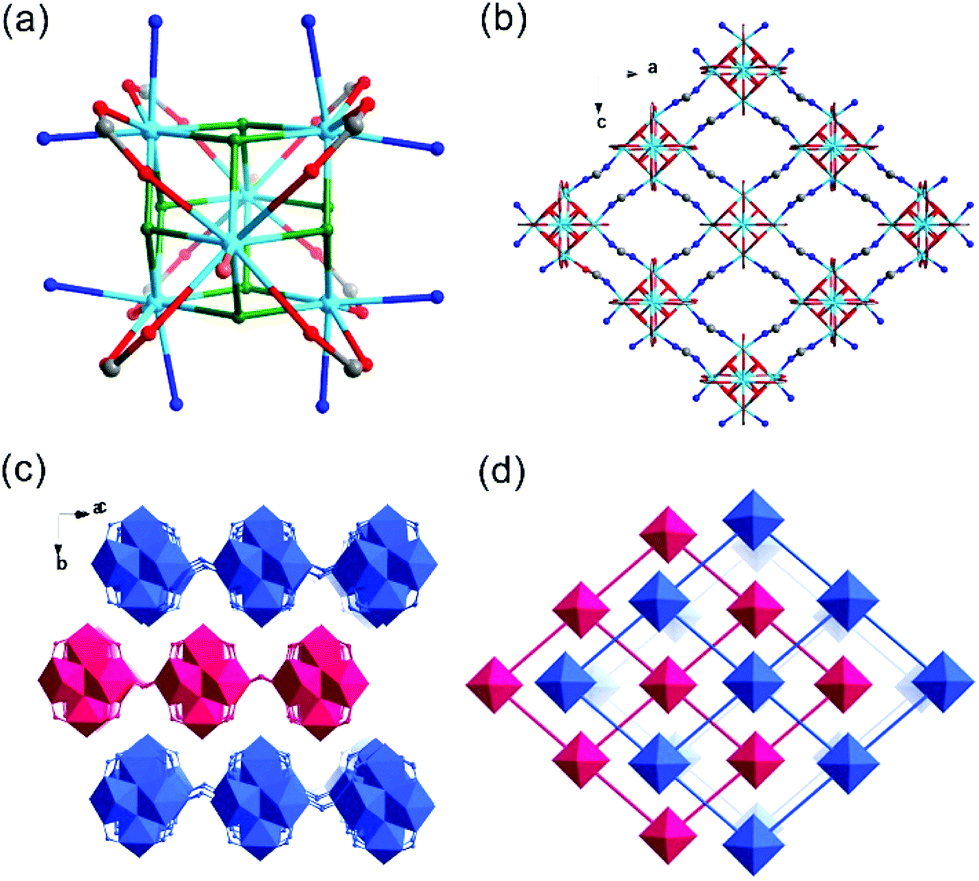 | ||
| Fig. 2 (a) Th6(μ3-O)4(μ3-OH)4(DMF)2(HCOO)8(HCOO)4 cluster of Th-SINAP-2. The O atoms from μ3-OH/-O, DMF, bridging HCOO−, and chelating HCOO− groups coordinated to the Th6 cluster are shown in green, orange, blue, and red, respectively. The Th atoms are shown in sky blue. Only the O atoms of DMF molecules are shown for clarity. (b) View of the 2D layered structure of Th-SINAP-2 extending along the ac plane. (c) Pseudo-3D structure formed by stacking 2D grids with an ABAB mode. (d) Simplified topology of Th-SINAP-2. | ||
Th-SINAP-3 crystallizes in the cubic space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c and its asymmetric unit consists of 1/4 of Th6O4(OH)4(HCOO)12(H2O)6 hexanuclear core and an HCOO− linker (Table S1†). The Th6 hexameric core is composed of six thorium ions bridged by four μ3-OH and four μ3-O groups, and further decorated by twelve HCOO− anions, as shown in Fig. 3a and b. Each Th(IV) center is nine-coordinated with four μ3-O or μ3-OH oxygen atoms, four HCOO− anions, and one water molecule, with the Th–O distances of 2.348(8)–2.397(8) Å, 2.471(7)–2.564(6) Å, and 2.679(6) Å, respectively. Th-SINAP-3 contains identical cluster moieties as the Th(IV) formate hexamer reported by Soderholm et al., which both feature dense 0D structures without any obvious intermolecular space (Fig. 3c).6 However, the synthesis and the overall packing of these two complexes are essentially different, where Th-SINAP-3 has much larger unit cell dimensions and higher symmetry.
c and its asymmetric unit consists of 1/4 of Th6O4(OH)4(HCOO)12(H2O)6 hexanuclear core and an HCOO− linker (Table S1†). The Th6 hexameric core is composed of six thorium ions bridged by four μ3-OH and four μ3-O groups, and further decorated by twelve HCOO− anions, as shown in Fig. 3a and b. Each Th(IV) center is nine-coordinated with four μ3-O or μ3-OH oxygen atoms, four HCOO− anions, and one water molecule, with the Th–O distances of 2.348(8)–2.397(8) Å, 2.471(7)–2.564(6) Å, and 2.679(6) Å, respectively. Th-SINAP-3 contains identical cluster moieties as the Th(IV) formate hexamer reported by Soderholm et al., which both feature dense 0D structures without any obvious intermolecular space (Fig. 3c).6 However, the synthesis and the overall packing of these two complexes are essentially different, where Th-SINAP-3 has much larger unit cell dimensions and higher symmetry.
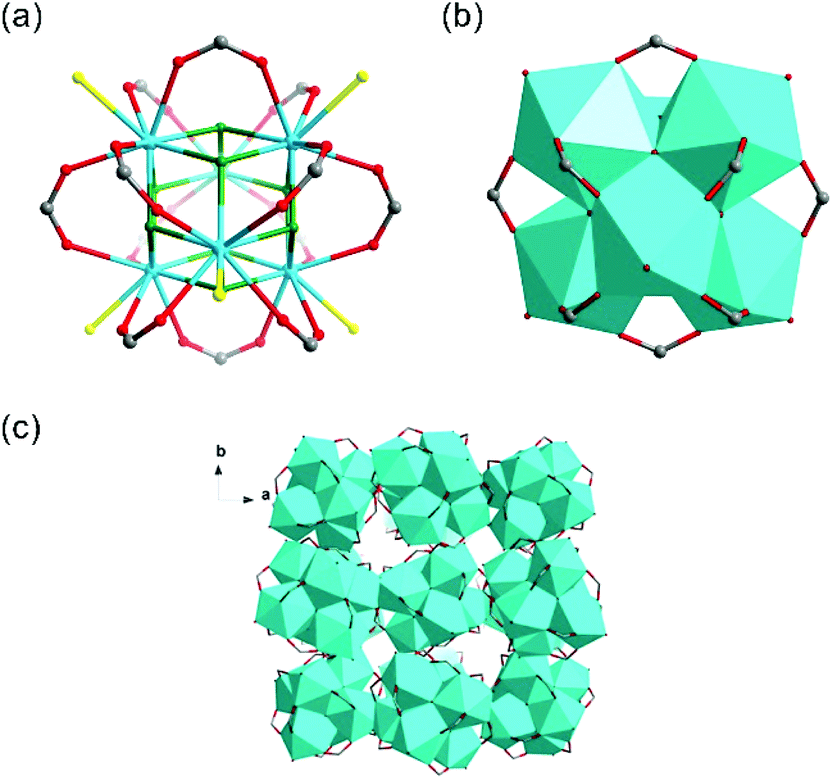 | ||
| Fig. 3 (a) Th6(μ3-O)4(μ3-OH)4(HCOO)12(H2O)6 cluster of Th-SINAP-3. The O atoms from μ3-OH/-O, H2O, chelating –HCOO groups coordinated to one Th6 cluster are shown in green, yellow, and red, respectively. The Th atoms are shown in sky blue. (b) Polyhedral representation of the Th6(μ3-O)4(μ3-OH)4(HCOO)12(H2O)6 cluster. (c) Polyhedral representation of the overall packing of Th-SINAP-3. | ||
Th-SINAP-4 crystallizes in the cubic space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m and adopts a 3D microporous structure (channel size of ca. 3.7 Å × 3.7 Å) (Table S1†). The asymmetric unit of Th-SINAP-4 contains 1/6 crystallographically independent Th4+ monomeric ion, and 1/2 and 1/4 HCOO− ions. The Th4+ ion is coordinated with nine O atoms, which are exclusively contributed from nine HCOO− anions (Th–O = 2.344(18)–2.478(19) Å) forming a tricapped trigonal prismatic geometry (Fig. 4a). To better understand the assembly of the 3D framework, a tetrahedral motif constructed from four Th4+ nodes bridged by six C(1) HCOO− anions may serve as the SBU (Fig. 4b). A 3D open-framework with a pcu topology is formed by the assembly of C(2) HCOO− anions with Th4+ nodes (Fig. 4c). These SBUs are further embedded in the open-framework to create an anionic network, as shown in Fig. 4d. Considering the electroneutrality of the structure, the anionic framework is charged balanced by dimethylammonium cations derived from the decomposition of the DMF solvent. The porosity of Th-SINAP-4 was calculated using PLATON, which indicated that the solvent-accessible volume is 14.1%.48
3m and adopts a 3D microporous structure (channel size of ca. 3.7 Å × 3.7 Å) (Table S1†). The asymmetric unit of Th-SINAP-4 contains 1/6 crystallographically independent Th4+ monomeric ion, and 1/2 and 1/4 HCOO− ions. The Th4+ ion is coordinated with nine O atoms, which are exclusively contributed from nine HCOO− anions (Th–O = 2.344(18)–2.478(19) Å) forming a tricapped trigonal prismatic geometry (Fig. 4a). To better understand the assembly of the 3D framework, a tetrahedral motif constructed from four Th4+ nodes bridged by six C(1) HCOO− anions may serve as the SBU (Fig. 4b). A 3D open-framework with a pcu topology is formed by the assembly of C(2) HCOO− anions with Th4+ nodes (Fig. 4c). These SBUs are further embedded in the open-framework to create an anionic network, as shown in Fig. 4d. Considering the electroneutrality of the structure, the anionic framework is charged balanced by dimethylammonium cations derived from the decomposition of the DMF solvent. The porosity of Th-SINAP-4 was calculated using PLATON, which indicated that the solvent-accessible volume is 14.1%.48
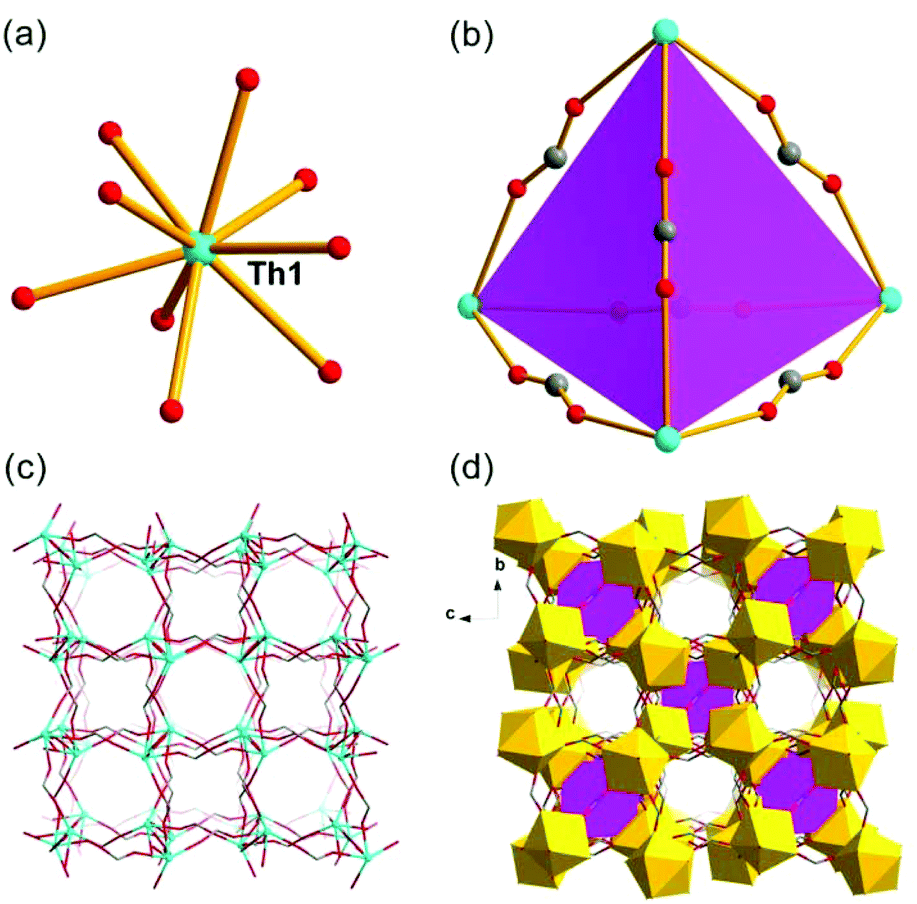 | ||
| Fig. 4 (a) Coordination environment of the Th(IV) ion in Th-SINAP-4. (b) Illustration of the tetrahedral SBU created by four Th(IV) ions and six C(1) HCOO− anions. (c) Assembly of Th(IV) ions and C(2) HCOO− anions into a 3D open-framework with a pcu topology. (d) Polyhedral representation of the 3D porous structure of Th-SINAP-4 with the tetrahedral SBU shown in purple. | ||
Th-SINAP-5 crystallizes in the orthorhombic space group Cmcm and its asymmetric unit contains two 1/2 crystallographically independent monomeric Th(IV) ions, three and 1/2 HCOO− ions, and four 1/4 HCOO− ions (Table S1†). Both of the Th4+ ions are nine-coordinated with O atoms from HCOO− anions with Th–O distances ranging from 2.338(12) to 2.504(10) Å, forming a tricapped trigonal prismatic geometry (Fig. 5a and Table S2†). The coordination from μ2-HCOO− anions function as bringing units and link ThO9 units into 2D layers. More explicitly, Th(1) nodes are bridged by the C(1) and C(3) HCOO− anions, and Th(2) nodes are connected by the C(6), C(7) and C(8) HCOO− anions, forming a honeycomb lattice, as shown in Fig. 5b. These layers are further connected by the C(2), C(4) and C(5) HCOO− anions, generating a 3D anionic network (Fig. 5c). Similar to Th-SINAP-2 and Th-SINAP-4, the negatively charged framework can be neutralized by NH2(CH3)2+ cations derived from the decomposition of DMF solvent. Another key feature of Th-SINAP-5 is its system of channels with dimensions of 5.0 Å × 4.2 Å extending along the b axis (Fig. 5d). Th4+ cations and HCOO− anions do not fill all the space in the framework, and consequently large free voids with approximately 22% void were identified by PLATON.48
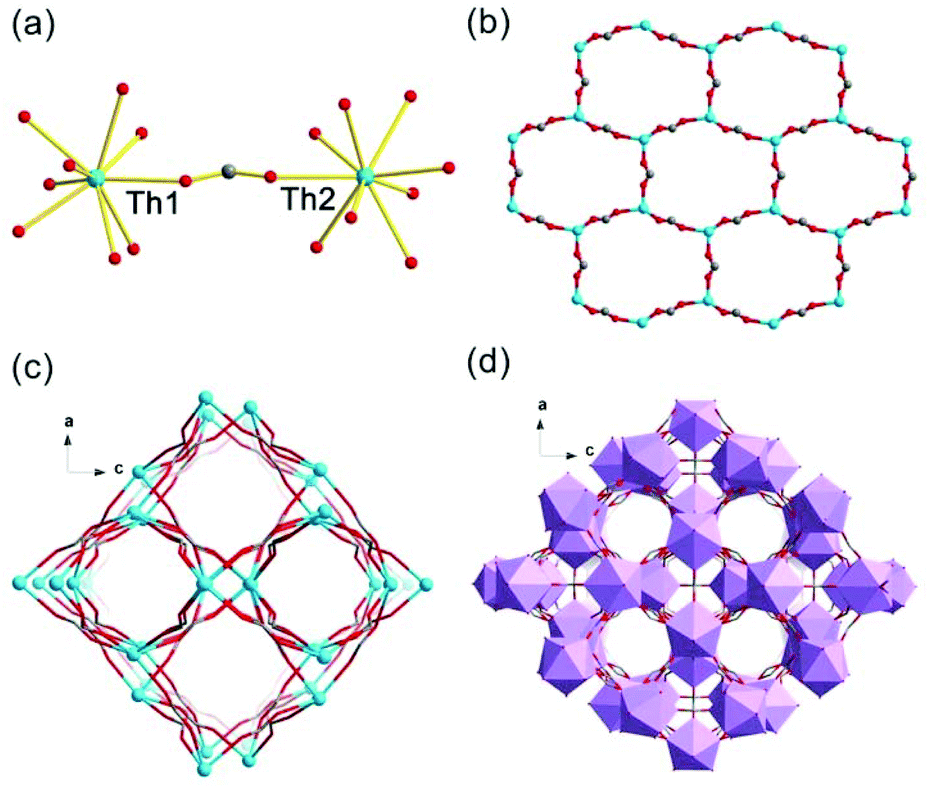 | ||
| Fig. 5 (a) Coordination environment of Th(IV) ions in Th-SINAP-5. (b) One 2D layer formed by Th(1) ions bridged by C(1) and C(3) HCOO− anions or Th(2) ions bridged by C(6), C(7), and C(8) HCOO− anions. (c) 3D network formed by the connection of Th1 and Th2 ions through C(2), C(4) and C(5) HCOO− anions. (d) Polyhedral representation of the 3D porous structure. | ||
The SCXRD study showed that Th-SINAP-6 features a 3D topology and it crystallizes in the chiral orthorhombic space group P212121 (Table S1†). The absolute chiral configuration of Th-SINAP-6 was confirmed by circular dichroism (CD) spectroscopy, as shown in Fig. S2.† Both of the crystallographically independent Th4+ cations are nine-coordinated and adopt a monocapped square anti-prismatic geometry, within which eight O atoms are provided by HCOO− anions. The ninth O atom can be assigned to the coordinating DMF and water molecule for Th(1) and Th(2), respectively (Fig. 6a). The Th–O (HCOO−) distances range from 2.368(5) to 2.547(5) Å, while the Th–O distances are 2.470(6) and 2.535(6) Å for the coordinating DMF and H2O, respectively. A closer structural examination on the origin of the chirality demonstrated that the neighbouring Th centres are bridged by HCOO− units, forming one left-handed and one right-handed helical chain for Th(1) and Th(2) (Fig. 6b and c), respectively. These two types of chains are further linked by the C(1), C(4), C(5), and C(6) formate anions to generate the final 3D chiral structure without a large void (Fig. 6d). Another significant difference between Th-SINAP-6 and other Th(IV) formate complexes is that the DMF solvent molecules participate in the coordination with the Th centers in Th-SINAP-6, besides their involvement in the overall packing.
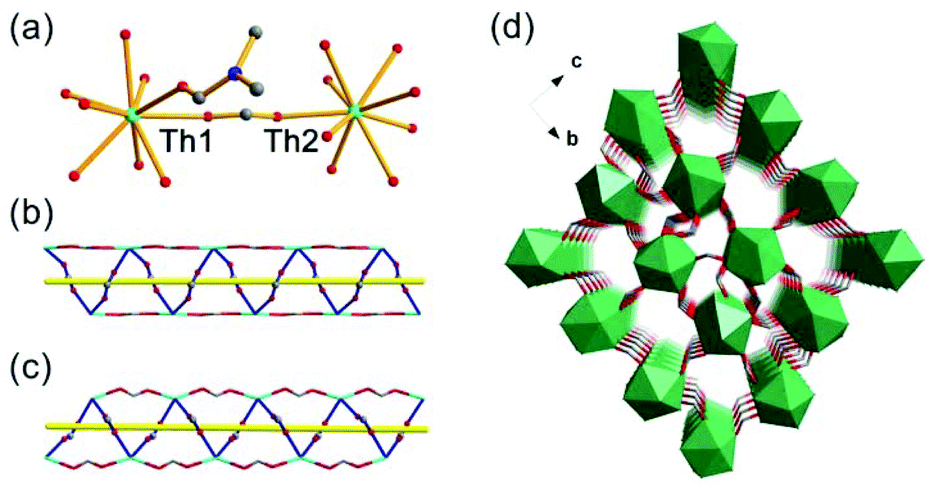 | ||
| Fig. 6 (a) Coordination environments of Th(IV) ions in Th-SINAP-6. (b) Left-handed helical chain composed of Th(1) cations and formate anions. (c) Right-handed helical chain composed of Th(2) cations and formate anions. (d) Polyhedral representation of the 3D structure of Th-SINAP-6. The C and N atoms of the coordinating DMF molecules are omitted for clarity. | ||
FTIR spectroscopy
FTIR spectra were recorded for Th-SINAP-1, Th-SINAP-2, Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6, but not for Th-SINAP-3 owning to our inability to obtain its pure phase (Fig. 7). The solvent species including water and DMF molecules can be confirmed by the broad O–H stretching bands of water ranging from 3000 to 3700 cm−1 and C–H vibrations of DMF varying from 2800 to 2900 cm−1.49 All the complexes exhibit two strong peaks centered at ∼1550 and ∼1350 cm−1, corresponding to the νasym(COO) and νsym(COO) vibrations of the carboxylate groups, respectively.50,51 The crystal-field effects and correlation field splitting within the crystalline compounds result in a split and shift in the carboxylate functions.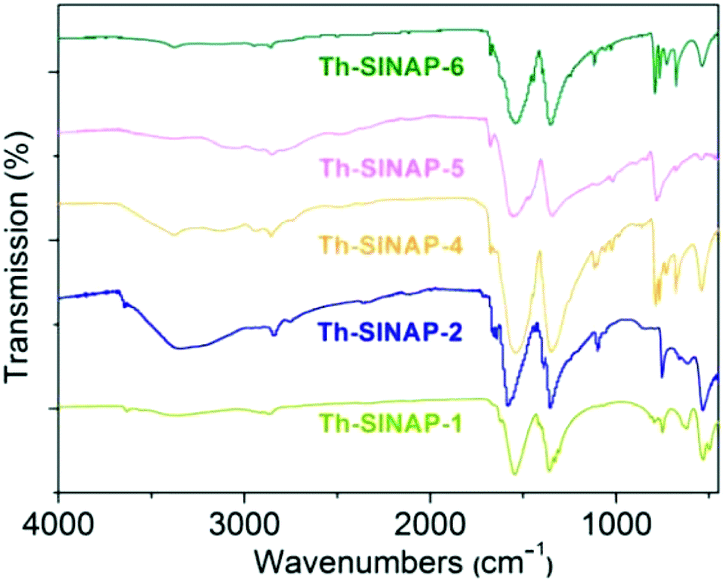 | ||
| Fig. 7 FTIR spectra of Th-SINAP-1, Th-SINAP-2, Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6. | ||
Thermal properties
Thermogravimetric analyses (TGA) were performed to confirm the thermal stabilities of the obtained thorium complexes. As shown in Fig. S3,† the initial weight loss from 32 °C to approximately 250 °C for Th-SINAP-1, Th-SINAP-2, Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6 was 11.6%, 13.8%, 11.0%, 15.2%, and 12.9%, respectively, corresponding to the departure of DMF, lattice, and coordinating water molecules (calcd 12.2%, 13.6%, 10.6%, 13.7%, and 13.4%, respectively). Sharp weight loss occurred from 250 °C to 330 °C as the second stage, which may originate from the departure of intercalated molecules.52,53 This event can result in the formation of thorium oxide for all the complexes.Iodine adsorption
The porous frameworks of Th-SINAP-1, Th-SINAP-4, and Th-SINAP-5 suggest their potential application for the adsorption of iodine (129I and 131I), one of the major fission products in the nuclear waste stream. Iodine has attracted significant concern due to its high mobility, volatility, and radiotoxicity as well as its important role in the metabolic processes in human beings.54,55 The stable 127I isotope was used as a surrogate for 129I and 131I due to their identical chemical properties. Owing to their relatively high yields, the sorption capacities of Th-SINAP-1 and Th-SINAP-4 were tested by examining iodine vapour uptake in a sealed vessel at 353 K. The colour of Th-SINAP-1 and Th-SINAP-4 kept deepening continuously from white to the final black. The SEM-EDS analysis further confirmed the adsorption of iodine by Th-SINAP-1 and Th-SINAP-4 (Fig. S4†). Gravimetric measurements were performed, which showed that the adsorption equilibrium of Th-SINAP-1 can be reached within 3 h with excellent adsorption capacity of 124 wt% (Fig. 8), which is comparable to that of other reported MOFs, including MFM-300(Sc) and MFM-300(Fe).56 In contrast, Th-SINAP-4 exhibited a moderate iodine uptake capacity of 34 wt%.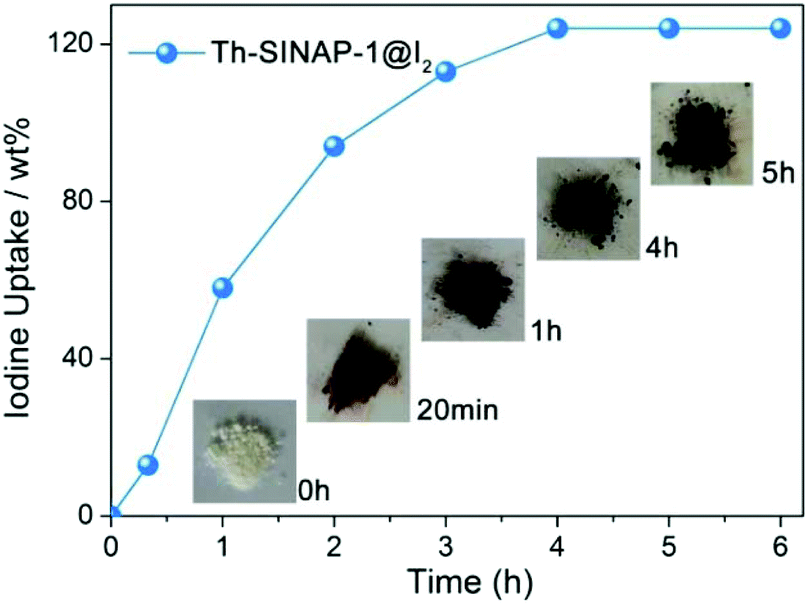 | ||
| Fig. 8 I2 vapour uptake curves and colour changes of Th-SINAP-1 at 353 K. | ||
Discussion
A simple solvothermal synthesis route by systematically controlling the molar ratio of thorium nitrate and formic acid and by adding water to the reaction system was developed, which produced six distinct thorium formate phases. By increasing the [HCOOH]/[Th(IV)] molar ratio, one of the most direct impacts is the increase in H+ concentration and proportionally the decrease in hydroxo/oxo concentration, which are involved in the olation/oxolation mechanisms of Th(IV) species.13,14 In addition, the presence of hydrating water molecules in Th(NO3)4·6H2O and the addition of water in the reagents allow the controlled hydrolysis of Th(IV) to form nucleophilic hydroxo/oxo-aquo Th species.16,18,43 The formation of theses phases was characterized via powder X-ray diffraction (PXRD) (Fig. S5 and S6†) and the reaction scheme is shown in Fig. 9.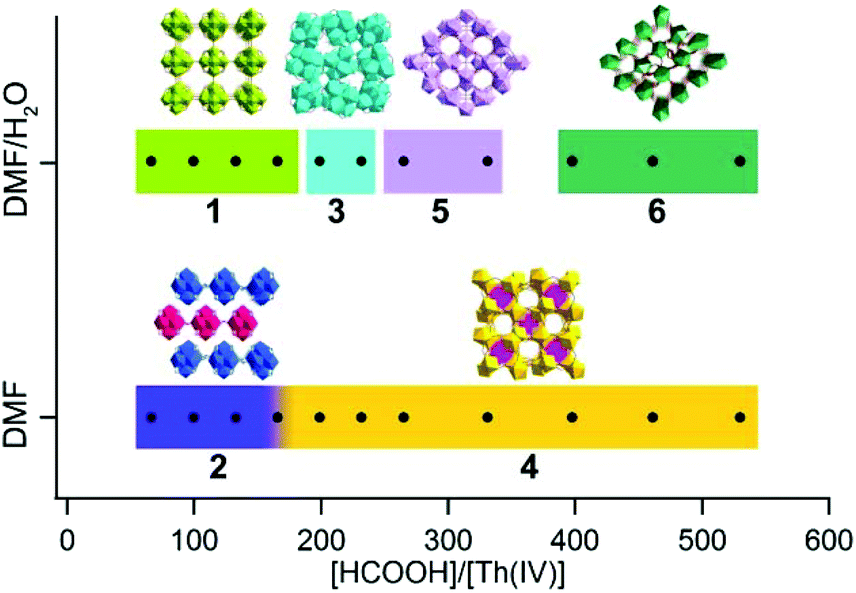 | ||
| Fig. 9 Composition diagram of the Th(IV)-formate system as function of [HCOOH]/[Th(IV)] and the addition of water to the system. | ||
At relatively small [HCOOH]/[Th(IV)] (≤232) ratios, hexanuclear Th6O4(OH)4(HCOO)12 clusters emerged, based on which prismatic, rhombohedral, and octahedral crystals of Th-SINAP-1, Th-SINAP-2, and Th-SINAP-3 were crystallized and isolated, respectively (Fig. S7†). In contrast, the further addition of HCOOH (265 ≤ [HCOOH]/[Th(IV)] ≤ 530) inhibited hydrolysis and nucleation, affording monomeric Th(IV) species, including tablet crystals of Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6 (Fig. S7†). These observations led to the initial speculation that acidity plays an essential role in influencing the nucleation of the metal centres since high acidity limits the deprotonation of water and subsequently inhibits the larger assembly of nucleophilic hydroxo/oxo thorium clusters. The effect of acidity was further studied independently of formate using hydrochloric acid as a pH regulator (Table S4†). Only Th-SINAP-2 could be isolated under comparable conditions and other synthesis resulted in the formation of amorphous products or clear solutions. This result suggests that the relative concentration of the ligand plays a central role also in influencing competition for ligation and directing the structure.
On the other hand, the presence of water molecules is directly related to the formation of [Th6O4(OH)4]12+ cores and the overall packing of these moieties. Although Th-SINAP-2 was synthesized from a water-free organic solvent system, the inclusion of hydrating water (0.08 mmol) in the thorium source induced the occurrence of the olation/oxolation reaction, forming similar hydroxo/oxo Th hexamers as observed in Th-SINAP-1 under a comparable [HCOOH]/[Th(IV)] ratio. An excess of water (24.44 mmol) for the synthesis of Th-SINAP-1 led directly to additional μ2-H2O bridging between the adjacent Th6 hexamers, facilitating the formation of a two-fold interpenetrating 3D framework. Due to the lack of this bridging unit in Th-SINAP-2, a terminal η1-DMF takes the bridging role of μ2-H2O, which terminates the linkage of layered grids and results in a 2D lamellar structure. Moreover, with comparable [HCOOH]/[Th(IV)] ratios, crystallization of the 0D discrete molecular cluster Th-SINAP-3 was favoured upon the addition of a small amount (Vwater/VDMF = 0.44 mL/1.99 mL) of water, whereas the 3D framework of Th-SINAP-4 composed of monomeric ThIV ions was obtained in the absence of water. Thus, we postulate that under the synthetic conditions of Th-SINAP-3 and Th-SINAP-4, thermodynamic competition may exist between HCOO−, OH−, O2−, H2O, and DMF for the coordination with Th4+ cations, which results in different SBUs.57 Further addition of formic acid to some extent prevented the olation/oxolation reaction of thorium, and thus mononuclear Th(IV) ions directly coordinated with the HCOO− ligands to form the 3D framework of Th-SINAP-4. The addition of a controlled amount of water allowed higher concentration of OH− or O2− species and the production of the Th6 hexameric cluster Th-SINAP-3.
It is noteworthy that both Th-SINAP-4 and Th-SINAP-5 feature open-frameworks with the same molecular formula but their structures are essentially different. This observation suggests that the crystal chemistry of the final products could be drastically changed by the addition of a small amount of water during the solvothermal synthesis. This also implies that the energetic difference between these two phases is small, and that tuning the DMF/H2O ratio can trigger phase transition. Microporous thorium coordination polymers are inclined to be assembled from highly-charged cations or metal–oxo/hydroxo clusters linked by coordinationally flexible main group oxoanions, including borates and sulfates, as well as by multidentate-carboxylate groups in MOFs.13,58,59 O'Hare reported the very first series of thorium carboxylate open-frameworks, namely TOF-1, TOF-2, and TOF-3.60–62 Th6-based MOFs including Th6O4(OH)4(H2O)6(bdc)6·6DMF·12H2O and [Th6(μ3-O)2(HCOO)4(H2O)6(TCPP)4] were further synthesized by Thierry and Farha, respectively.34,44 Recently, Wang et al. synthesized the most porous thorium-organic framework SCU-8 with a surface area of 1360 m2 g−1.63Th-SINAP-4 and Th-SINAP-5 are built from the assembly of Th4+ cations and simple μ2-chelating HCOO− anions, and they represent another unusual case of thorium bearing open-frameworks without adding structure-directing templates. The details of their synthesis and structures will help to address the questions surrounding how best to make new micro- and mesoporous materials without resorting to templating agents that must subsequently be removed before use.
Reactions with [HCOOH]/[Th(IV)] ≥ 398 and addition of water produced Th-SINAP-6, giving a rare example of chiral thorium coordination polymers built from achiral moieties. To the best of our knowledge, only two chiral thorium MOFs, [C9H17N2][Th(TPO)Cl2]·18H2O (TPO = tris-(4-carboxylphenyl)phosphineoxide) and [Th8(O)(L)6(HL)6(H2O)5]·0.5H2O (L = o-sulfophenylarsonic acid), have been reported to date.64,65
Conclusions
An easy synthetic route using thorium nitrate and formic acid in DMF led to the crystallization of five new thorium coordination polymers and one rare thorium polyoxo cluster, with diverse dimensions of structures ranging from 0D molecular cluster and 2D layered network to 3D frameworks. The assemblies and structural variations can be tuned via the [HCOOH]/[Th(IV)] molar ratio and the addition of water, both of which control the hydrolysis, nucleation, and complexation. At a low [HCOOH]/[Th(IV)] ratio, three hexanuclear Th6(O)4(OH)4(HCOO)12 clusters-based complexes, Th-SINAP-1, Th-SINAP-2, and Th-SINAP-3, were isolated, and they exhibit 3D, 2D, and 0D topologies, respectively. An increase in the [HCOOH]/[Th(IV)] ratio limits the olation/oxolation reactions during the synthesis and enables monomeric Th(IV) cations as the dominating SBUs of three 3D frameworks (Th-SINAP-4, Th-SINAP-5, and Th-SINAP-6), including the unexpected chiral network Th-SINAP-6. The effect of water can be best illustrated by comparing the structures Th-SINAP-3 and Th-SINAP-4. Under anhydrous conditions, the system favoured the crystallization of monomeric species Th-SINAP-4, while the addition of water to the same reaction system allowed the condensation of Th(IV) and afforded a 0D hexanuclear cluster SINAP-3. The role of water (and solvent) during crystallization will be revealed in future studies, where we will focus on the thermodynamic driving forces (e.g. metastability, solvent–host interaction) for the formation of these versatile Th complexes, which can facilitate the understanding of the underlying formation mechanism.The coordination versatility of HCOO− anions renders them ideal components for the construction of open-frameworks. The combination of large Th(IV) cations with small bridging HCOO− anions enabled the creation of open-frameworks, as exemplified by Th-SINAP-4 and Th-SINAP-5. In addition, the chelating role of the HCOO− anions in molecular assemblies helps stabilize the metal-hydro/oxo clusters and increase the size of the building blocks. HCOO− anions can act as both chelating ligands and bidentate linkers to connect the thorium clusters to generate a 3D open-framework, e.g.Th-SINAP-1. Importantly, this strategy allows for increasing the porosity of the frameworks without introducing structure-directing templates, which makes Th-SINAP-1 and Th-SINAP-4 potential materials for application in the capture of radionuclides (129I and 131I). These results give predictive insights into the formation and assembly of open-frameworks and open the potential of developing these materials for actinides. Investigations are underway to prepare the other thorium MOFs for the utility of radionuclide remediation.
Experimental
Synthesis
Caution! 232Th used in this study is an α emitter with the daughter of radioactive Ra-228. All thorium compounds used and investigated were operated in an authorized laboratory designed for actinide element studies. Standard precautions for handling radioactive materials should be followed.Materials
Th(NO3)4·6H2O (99%, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences), formic acid (≥99.5%, Fisher), and N,N′-dimethylformamide (DMF, water ≤50 ppm, Adamas) were used as received.Solutions containing Th(NO3)4·6H2O, HCOOH, DMF, and H2O were mixed in a capped glass vial. Six phases of thorium formates were crystalized after heating for a few days. The pH values before and after crystallization were recorded, as shown in Table S3.†
Th-SINAP-1
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.2, 0.3, 0.4, or 0.5 mL; 5.30, 7.95, 10.60, or 13.25 mmol, respectively), DMF (1.99 mL), and H2O (0.44 mL) in a capped glass vial was heated at 120 °C for 48 h. The colourless prismatic crystals were filtered, washed with MeOH and Et2O, and dried at room temperature. Yield: ca. 47% based on Th(NO3)4·6H2O. Alternatively, this compound could be prepared using the following procedure. A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.2 mL, 5.3 mmol), DMF (1.45 mL), NaNO3 (3.4 mg, 0.04 mmol), and H2O (0.44 mL) in a capped vial was heated at 120 °C for only 18 h, after which colourless needle-like crystals crystallized from the solution. Yield: ca. 40% based on Th(NO3)4·6H2O. Anal. Calcd for, [Th6Na(μ3-O)4(μ3-OH)4(HCOO)14]·[NH2(CH3)2]·(DMF)3(H2O)5, C25H57N4NaTh6O44, C, 11.85%; H, 2.27%; N, 2.21%. Found: C, 11.71%; H, 2.42%; N, 2.29%. IR: 3623 (w), 2867 (w), 1546 (vs), 1360 (vs), 795 (w), 751 (m), 620 (w), 531 (w), 498 (w) cm−1.Th-SINAP-2
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.2, 0.3, 0.4 mL; 5.30, 7.95 or 10.60 mmol, respectively), and DMF (4 mL) in a capped vial was heated at 120 °C for 24 h. Colourless rhombohedral crystals were filtered, washed with MeOH and Et2O, and dried at room temperature. Yield, 0.018 g, 33% based on Th(NO3)4·6H2O. Anal. Calcd for [Th6(μ3-O)4(μ3-OH)4(HCOO)12(DMF)2]·(H2O)10, C18H50N2Th6O44, C, 9.04%; H, 2.11%; N, 1.17%. Found: C, 8.61%; H, 2.47%; N, 0.89%. IR: 3650 (w), 2840 (w), 1580 (vs), 1350 (vs), 1100 (w), 753 (m), 617 (w), 533 (w) cm−1.Th-SINAP-3
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.6 or 0.7 mL, 15.90 or 18.55 mmol, respectively), DMF (1.99 mL), and H2O (0.44 mL) in a capped vial was heated at 120 °C for 72 h. Colourless octahedral crystals appeared in an extremely low yield (ca. <10%) with many unidentified amorphous solids always present. Although many attempts were made to improve the yield by changing the reaction parameters such as temperature, time, and concentration, no obvious enhancement in yield was achieved. Therefore, conventional characterization such as EA, IR and PXRD was not possible.Th-SINAP-4
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.6, 0.7, 0.8, 1, 1.2, 1.4, and 1.6 mL; 15.90, 18.55, 21.20, 26.50, 31.80, 34.45, and 42.40 mmol, respectively), and DMF (4 mL) in a capped vial was heated at 120 °C for 48 h. Colourless tablet crystals were filtered, washed with MeOH and Et2O, and dried at room temperature. Yield: ca. 36% based on Th(NO3)4·6H2O. Anal. Calcd for [Th2(HCOO)9][NH2(CH3)2]·(DMF)(H2O)2, C14H28N2Th2O21, C, 16.41%; H, 2.75%; N, 2.73%. Found: C, 16.26%; H, 2.94%; N, 2.85%. IR: 3377 (w), 2944 (w), 2857 (w), 1674 (w), 1541 (vs), 1348 (vs), 1118 (w), 1061 (w), 1023 (w), 786 (s), 765 (m), 729 (w), 677 (m), 539 (s) cm−1.Th-SINAP-5
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (0.8 or 1.0 mL; 21.20 or 26.50 mmol, respectively), DMF (1.99 mL), H2O (0.44 mL) in a capped vial was heated at 120 °C for 48 h. Then the clear solution was left to stand in air for a further 3 days, after which colourless tablet crystal crystallized from the solution. The colourless crystals were filtered, washed with MeOH and Et2O, and dried at room temperature. Yield: 21% based on Th(NO3)4·6H2O. Anal. Calcd for [Th2(HCOO)9]·[NH2(CH3)2]·(DMF)(H2O)4, C14H32N2Th2O23, C, 15.86%; H, 3.04%; N, 2.64%. Found: C, 15.49%; H, 3.15%; N, 2.44%. IR: 3410 (w), 2918 (w), 1675 (w), 1556 (vs), 1344(vs), 1015 (w), 782(s), 677(w), 541 (w) cm−1.Th-SINAP-6
A mixture of Th(NO3)4·6H2O (47 mg, 0.08 mmol), HCOOH (1.2, 1.4, or 1.6 mL, 31.80, 34.45, or 42.40 mmol, respectively), DMF (1.99 mL), and H2O (0.44 mL) in a capped vial was heated at 120 °C for 48 h. Then the clear solution was left to stand in air for a further 3 days, after which colourless tablet crystals crystallized from the solution. The crystals were filtered, washed with MeOH and Et2O, and dried at room temperature. Yield: 40% based on Th(NO3)4·6H2O. Anal. Calcd for [Th2(HCOO)8(DMF)]·(H2O)3, C11H21NTh2O20, C, 13.89%; H, 2.22%; N, 1.47%. Found: C, 14.14%; H, 2.3; N, 1.58%. IR: 3389 (w), 2866 (w), 1545 (vs), 1351 (vs), 1124 (m), 1032 (w), 791 (s), 765 (m), 729 (w), 677 (s), 536 (m) cm−1.Crystallographic studies
Single-crystal X-ray diffraction data for all complexes was collected on a Bruker D8-Venture single-crystal X-ray diffractometer equipped with a Turbo X-ray source (Mo Kα radiation, λ = 0.71073 Å) adopting the direct-drive rotating-anode technique and a CMOS detector. The data frames were collected using the APEX3 program and processed using the SAINT routine. The empirical absorption correction was applied using the SADABS program.66 The structure was solved by Intrinsic Phasing with ShelXT and refined with ShelXL using OLEX2.67–69 All non-H atoms were subjected to anisotropic refinement by full-matrix program. Hydrogen atoms of the μ3-hydroxyls on the faces of the clusters were not located during the refinement. Hydrogen atoms of the formate groups were placed in calculated positions. Contributions to scattering due to these highly disordered solvent molecules were removed using the SQUEEZE routine of PLATON.48 The structures were then refined again using the data generated. Crystallographic data and details of the data collection are given in Table S1.†Powder X-ray diffraction (PXRD)
PXRD patterns of Th-SINAP-1, Th-SINAP-2, Th-SINAP-3, Th-SINAP-4, and Th-SINAP-6, were collected in the 2θ range of 5° to 50° with a step of 0.02° using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å) equipped with a Lynxeye one-dimensional detector.Fourier transform infrared (FTIR) spectroscopy
FTIR spectra in the range of 400–4000 cm−1 were recorded on ground powder of Th-SINAP-1, Th-SINAP-2, Th-SINAP-3, Th-SINAP-4, and Th-SINAP-6 using a Thermo Nicolet 6700 FTIR spectrometer equipped with a diamond attenuated total reflectance (ATR) accessory.Thermal properties
Thermogravimetric analysis (TGA) of Th-SINAP-1, Th-SINAP-2, Th-SINAP-3, Th-SINAP-4, and Th-SINAP-6 was performed on a NETZSCH STA 449F3 instrument in the temperature range of 40–800 °C under a nitrogen flow and at a heating rate of 10 °C min−1.Element analysis
Scanning electron microscopy (SEM) and energy-dispersive spectroscopy (EDS) data were recorded on a Zeiss Merlin Compact LEO 1530 VP scanning electron microscope. The energy of the electron beam voltage was 10 keV for imaging and 15 keV for quantitative identification of elements. Samples were attached directly on carbon conductive tape. The SEM-EDS results are provided in Fig. S4 and S8.† Elemental analyses of C, H and N were performed with a Vario EL Cube elemental analyser.Iodine vapour adsorption measurements
An open vial (20 mL) containing 50 mg Th-SINAP-1 or Th-SINAP-4 was accurately weighted (m0) and introduced into a glass vessel (150 mL) containing 1 g iodine. The vessel was sealed and kept in an oven at 80 °C. After certain time intervals, the vial containing the sample was weighed (mt) until its mass did not change. The iodine adsorption capacity can be calculated as: wt% = (mt − m0)/mTh-SINAP-1/4.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (21876182, 21701184, and 21906163).Notes and references
- M. M. Waldrop, Nature, 2012, 492, 26 CrossRef CAS.
- C. Tamain, T. Dumas, C. Hennig and P. Guilbaud, Chem. – Eur. J., 2017, 23, 6864 CrossRef CAS.
- J. Lin, J. N. Cross, J. Diwu, N. A. Meredith and T. E. Albrecht-Schmitt, Inorg. Chem., 2013, 52, 4277 CrossRef CAS.
- J. Diwu, S. Wang and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 4088 CrossRef CAS PubMed.
- K. E. Knope and L. Soderholm, Inorg. Chem., 2013, 52, 6770 CrossRef CAS.
- K. E. Knope, R. E. Wilson, M. Vasiliu, D. A. Dixon and L. Soderholm, Inorg. Chem., 2011, 50, 9696 CrossRef CAS.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944 CrossRef CAS.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298 CrossRef CAS.
- R. E. Wilson, S. Skanthakumar and L. Soderholm, Angew. Chem., Int. Ed., 2011, 50, 11234 CrossRef CAS.
- R. E. Wilson, S. Skanthakumar, G. Sigmon, P. C. Burns and L. Soderholm, Inorg. Chem., 2007, 46, 2368 CrossRef CAS.
- C. Volkringer, I. Mihalcea, J.-F. Vigier, A. Beaurain, M. Visseaux and T. Loiseau, Inorg. Chem., 2011, 50, 11865 CrossRef CAS.
- R. D. Rogers, A. H. Bond and M. M. Witt, Inorg. Chim. Acta, 1991, 182, 9 CrossRef CAS.
- J. Lin, G. B. Jin and L. Soderholm, Inorg. Chem., 2016, 55, 10098 CrossRef CAS PubMed.
- J. Lin, M. Qie, L. Zhang, X. Wang, Y. Lin, W. Liu, H. Bao and J. Wang, Inorg. Chem., 2017, 56, 14198 CrossRef CAS PubMed.
- P. Woidy and F. Kraus, Z. Anorg. Allg. Chem., 2014, 640, 1547 CrossRef CAS.
- C. Falaise, C. Volkringer, J.-F. Vigier, N. Henry, A. Beaurain and T. Loiseau, Chem. – Eur. J., 2013, 19, 5324 CrossRef CAS.
- C. Falaise, C. Volkringer, C. Hennig and T. Loiseau, Chem. – Eur. J., 2015, 21, 16654 CrossRef CAS.
- N. P. Martin, J. Marz, H. Feuchter, S. Duval, P. Roussel, N. Henry, A. Ikeda-Ohno, T. Loiseau and C. Volkringer, Chem. Commun., 2018, 54, 6979 RSC.
- N. P. Martin, C. Volkringer, N. Henry, X. Trivelli, G. Stoclet, A. Ikeda-Ohno and T. Loiseau, Chem. Sci., 2018, 9, 5021 RSC.
- C. Falaise, C. Volkringer, J.-F. Vigier, A. Beaurain, P. Roussel, P. Rabu and T. Loiseau, J. Am. Chem. Soc., 2013, 135, 15678 CrossRef CAS.
- L. Chatelain, R. Faizova, F. Fadaei-Tirani, J. Pécaut and M. Mazzanti, Angew. Chem., Int. Ed., 2019, 58, 3021 CrossRef CAS.
- L. Chatelain, S. White, R. Scopelliti and M. Mazzanti, Angew. Chem., Int. Ed., 2016, 55, 14323 CrossRef.
- B. Biswas, V. Mougel, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed., 2011, 50, 5744 Search PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643 CrossRef CAS.
- J. I. Choi, H. Chun and M. S. Lah, J. Am. Chem. Soc., 2018, 140, 10915 CrossRef CAS.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068 CrossRef CAS.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Chem. Mater., 2016, 28, 3749 CrossRef CAS.
- J. J. Katz, L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Netherlands, 2011 Search PubMed.
- J. Veliscek-Carolan, J. Hazard. Mater., 2016, 318, 266 CrossRef CAS.
- K. P. Carter, J. A. Ridenour, M. Kalaj and C. L. Cahill, Chem. – Eur. J., 2019, 25, 7114 CrossRef CAS.
- P. Li, S. Goswami, K.-i. Otake, X. Wang, Z. Chen, S. L. Hanna and O. K. Farha, Inorg. Chem., 2019, 58, 3586 CrossRef CAS.
- P. Li, X. Wang, K.-i. Otake, J. Lyu, S. L. Hanna, T. Islamoglu and O. K. Farha, ACS Appl. Nano Mater., 2019, 2, 2260 CrossRef CAS.
- D. R. Fröhlich, A. Kremleva, A. Rossberg, A. Skerencak-Frech, C. Koke, S. Krüger, N. Rösch and P. J. Panak, Inorg. Chem., 2017, 56, 6820 CrossRef.
- C. Hennig, A. Ikeda-Ohno, W. Kraus, S. Weiss, P. Pattison, H. Emerich, P. M. Abdala and A. C. Scheinost, Inorg. Chem., 2013, 52, 11734 CrossRef CAS.
- W. Liu, L. Liu, Y. Wang, L. Chen, J. A. McLeod, L. Yang, J. Zhao, Z. Liu, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt, G. Liu and S. Wang, Chem. – Eur. J., 2016, 22, 11170 CrossRef CAS.
- S. Takao, K. Takao, W. Kraus, F. Emmerling, A. C. Scheinost, G. Bernhard and C. Hennig, Eur. J. Inorg. Chem., 2009, 2009, 4771 CrossRef.
- J. Ling, H. Lu, Y. Wang, K. Johnson and S. Wang, RSC Adv., 2018, 8, 34947 RSC.
- C. Lucks, A. Rossberg, S. Tsushima, H. Foerstendorf, K. Fahmy and G. Bernhard, Dalton Trans., 2013, 42, 13584 RSC.
- C. Falaise, C. Volkringer and T. Loiseau, Cryst. Growth Des., 2013, 13, 3225 CrossRef CAS.
- N. P. Martin, C. Volkringer, C. Falaise, N. Henry and T. Loiseau, Cryst. Growth Des., 2016, 16, 1667 CrossRef CAS.
- C. Falaise, A. Assen, I. Mihalcea, C. Volkringer, A. Mesbah, N. Dacheux and T. Loiseau, Dalton Trans., 2015, 44, 2639 RSC.
- C. Falaise, J.-S. Charles, C. Volkringer and T. Loiseau, Inorg. Chem., 2015, 54, 2235 CrossRef CAS PubMed.
- J. Ye, R. F. Bogale, Y. Shi, Y. Chen, X. Liu, S. Zhang, Y. Yang, J. Zhao and G. Ning, Chem. – Eur. J., 2017, 23, 7657 CrossRef CAS.
- C. Y. Sun, X. L. Wang, C. Qin, J. L. Jin, Z. M. Su, P. Huang and K. Z. Shao, Chem. – Eur. J., 2013, 19, 3639 CrossRef CAS.
- J. Andreo, E. Priola, G. Alberto, P. Benzi, D. Marabello, D. M. Proserpio, C. Lamberti and E. Diana, J. Am. Chem. Soc., 2018, 140, 14144 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Theory and Applications in Inorganic Chemistry, Wiley, 1997 Search PubMed.
- O. A. Ejegbavwo, C. R. Martin, O. A. Olorunfemi, G. A. Leith, R. T. Ly, A. M. Rice, E. A. Dolgopolova, M. D. Smith, S. G. Karakalos, N. Birkner, B. A. Powell, S. Pandey, R. J. Koch, S. T. Misture, H.-C. z. Loye, S. R. Phillpot, K. S. Brinkman and N. B. Shustova, J. Am. Chem. Soc., 2019, 141, 11628 CrossRef CAS PubMed.
- E. A. Dolgopolova, O. A. Ejegbavwo, C. R. Martin, M. D. Smith, W. Setyawan, S. G. Karakalos, C. H. Henager, H.-C. zur Loye and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 16852 CrossRef CAS.
- Y. Wang, W. Liu, Z. Bai, T. Zheng, M. A. Silver, Y. Li, Y. Wang, X. Wang, J. Diwu, Z. Chai and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 5783 CrossRef CAS.
- H. Xu, C.-S. Cao, H.-S. Hu, S.-B. Wang, J.-C. Liu, P. Cheng, N. Kaltsoyannis, J. Li and B. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6022 CrossRef CAS.
- W. Xie, D. Cui, S.-R. Zhang, Y.-H. Xu and D.-L. Jiang, Mater. Horiz., 2019, 6, 1571 RSC.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther III, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598 CrossRef.
- X. Zhang, I. da Silva, H. G. W. Godfrey, S. K. Callear, S. A. Sapchenko, Y. Cheng, I. Vitórica-Yrezábal, M. D. Frogley, G. Cinque, C. C. Tang, C. Giacobbe, C. Dejoie, S. Rudić, A. J. Ramirez-Cuesta, M. A. Denecke, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 16289 CrossRef CAS.
- M. H. Rand, F. J. Mompean, J. Perrone and M. Illemassène, Chemical Thermodynamics of Thorium, OECD Pub., 2008 Search PubMed.
- S. Wang, E. V. Alekseev, D. W. Juan, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057 CrossRef CAS PubMed.
- C. Falaise, K. Kozma and M. Nyman, Chem. – Eur. J., 2018, 24, 14226 CrossRef CAS.
- K. M. Ok and D. O'Hare, Dalton Trans., 2008, 5560 RSC.
- J.-Y. Kim, A. J. Norquist and D. O'Hare, J. Am. Chem. Soc., 2003, 125, 12688 CrossRef CAS.
- K. M. Ok, J. Sung, G. Hu, R. M. J. Jacobs and D. O'Hare, J. Am. Chem. Soc., 2008, 130, 3762 CrossRef CAS.
- Y. Li, Z. Yang, Y. Wang, Z. Bai, T. Zheng, X. Dai, S. Liu, D. Gui, W. Liu, M. Chen, L. Chen, J. Diwu, L. Zhu, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 1354 CrossRef.
- Y. Li, Z. Weng, Y. Wang, L. Chen, D. Sheng, Y. Liu, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Dalton Trans., 2015, 44, 20867 RSC.
- X.-Y. Qian, T.-H. Zhou and J.-G. Mao, Dalton Trans., 2015, 44, 13573 RSC.
- G. M. Sheldrick, SADABS, program for empirical absorption correction of area detector data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The polyhedral representation of the Th-SINAP-1, solid circular dichroism spectra, TGA, PXRD, crystal images, SEM-EDS results, crystallographic data, selected bond distances, and synthesis details. CCDC 1934855–1934860. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01263j |
| This journal is © the Partner Organisations 2020 |