
Alterations to secondary building units of metal–organic frameworks for the development of new functions
Junsu
Ha
 ,
Jae Hwa
Lee
* and
Hoi Ri
Moon
*
,
Jae Hwa
Lee
* and
Hoi Ri
Moon
*
Department of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 44919, Republic of Korea. E-mail: hoirimoon@unist.ac.kr
First published on 28th October 2019
Abstract
Secondary building units (SBUs) are the key components of metal–organic frameworks (MOFs) that help to build potentially porous periodic networks by linking multitopic organic ligands. Hence, metal SBUs are critical for determining the underlying topology of MOFs. Moreover, SBUs are the main MOF research topic nowadays, because of the simplicity of their synthesis, diverse directionality and their ability to easily harness open metal sites, compared to that of primary building units (comprising mononuclear metal centres) or tertiary building units (metal–organic polyhedra). Therefore, post-synthetic approaches for altering SBUs do not only include developing techniques for controlling the properties of MOFs but also involve a more in-depth understanding of their structure–function relationships from the materials science and engineering perspective. The SBU-related reviews published to date have successfully introduced and organised the chemistry of SBUs in MOFs. Because many recent studies have explored more diverse methods, such as metal exchange, oxidation-state transformation, defect generation, and incorporation of other species, in this review, we mainly focus on the recently developed methods for SBU alteration by classifying them into four groups and elaborate on how unique structures and properties can be achieved using those methods.
 Junsu Ha | Junsu Ha received his bachelor's degree in chemistry from Inha University, Korea, in 2018. He is currently pursuing a combined MS-PhD program at Ulsan National Institute of Science and Technology (UNIST) under the supervision of Prof. Hoi Ri Moon. His research interests are the development of MOFs for molecular separation and the design of MOF heterostructures. |
 Jae Hwa Lee | Jae Hwa Lee received his BS in 2013 from Interdisciplinary Green Energy, Ulsan National Institute of Science and Technology (UNIST), and PhD in 2019 from the Department of Chemistry, UNIST. Currently, he is working as a postdoctoral fellow at UNIST with Prof. Hoi Ri Moon, supported by National Research Foundation of Korea (NRF) and Samsung Research Funding & Incubation Centre of Samsung Electronics. His research focuses on the synthesis of metal–organic frameworks and their crystal engineering for gas storage, separation, molecular sensing, and catalytic reaction. |
 Hoi Ri Moon | Hoi Ri Moon received her BS in 2001 from Ewha Womans University and PhD in 2007 from Seoul National University. After working as a postdoctoral fellow at Molecular Foundry, Lawrence Berkeley National Laboratory, she joined Ulsan National Institute of Science and Technology (UNIST) in 2010 as an assistant professor and was promoted to an associate professor in 2014. Her group aims to understand the flexible behaviours of metal–organic frameworks (MOFs) and create unique MOFs for gas separation (especially hydrogen isotope separation), gas storage, molecular sensing and heterogeneous catalysis. |
1. Introduction
Metal–organic frameworks (MOFs) are the most prominent class of reticular compounds that link molecular building blocks (organic and inorganic) via coordinating bonds into periodic structures that contain potential voids.1–3 The properties and structures of MOFs have recently attracted the attention of scientists; moreover, they have been the subject of thousands of papers annually, owing to their modular topology, chemical variety, and controllable morphology, which are attributed to the combination of their organic and inorganic characteristics.4,5 In describing MOF structures, secondary building units (SBUs) designate clusters of metal (M) ions joined to more than one other metal via non-metal bonds (e.g., M–O–M and M–O–C–O–M; oxo and carboxylate bonds, respectively) that form 3D periodic networks.6 Therefore, the development of novel SBUs is essential for the advancement of the topological directionality of MOFs and the achievement of stable MOFs.In the late 1980s and 1990s, many researchers focused on assemblies of single metal ions and neutral donor linkers to construct coordination networks that could accommodate guest solvent molecules in their pores.7–11 However, most compounds irreversibly collapsed upon removing the guest molecules owing to their structural fragility which was attributed to the resultant coordination geometries based on single metals. Therefore, the discovery and use of polynuclear inorganic clusters (i.e., SBUs) has conferred thermodynamically and mechanically stable coordination environments, owing to the entanglement of SBUs via chelation and the bridging of multi-dentate ligands, such as carboxylate and phosphate. MOF-5, a representative MOF comprising octahedral SBUs of Zn4O tetrahedra, is regarded as a rigid and stable framework because the six extension point SBU restricts the movement of the linker and secures the position of the metal centres.12–14 Accordingly, flexible frameworks should be ascribed to SBUs that exhibit sufficiently strong coordination to preserve structural integrity yet allow hinge movements with certain degree of freedom.14,15 Based on the hard and soft acid and base (HSAB) theory, hard Lewis acids (Fe3+, Cr3+, Zr4+, and Ti4+) could react with hard Lewis bases (carboxyl-based ligands) to obtain hydrolytically and chemically ultra-stable MOFs.16–19
As previously described, metal ions/clusters and organic ligands are the two main parts of MOF structures, and are commonly considered to be the ‘joints’ and ‘struts’ of such structures. The direct assembly of the joints and struts dictates the structure and physicochemical properties of the MOF, and could limit the extent of their tunability. Thus, the concept of post-synthetic modification (PSM) emerged owing to the need of chemists and engineers to control the functions and expand the capability of MOFs. The initial studies on the PSM of MOFs were conducted in 1999 and became mainstream by 2007.12,20 However, because the introduction of versatile functional groups to organic linkers is more eidetic and straightforward compared to that of inorganic components, many previous reviews have focused more on the chemical alteration of ligands. The relevant PSM methods for the inorganic clusters in MOFs can be classified as: (1) substitution of cations, (2) variation of cation valence, (3) generation of vacancies, and (4) incorporation of new species (Scheme 1). These methods are also regarded as rational tools for modulating the properties of conventional inorganic solids, including minerals, nanocrystals, and organometallic complexes.21–29
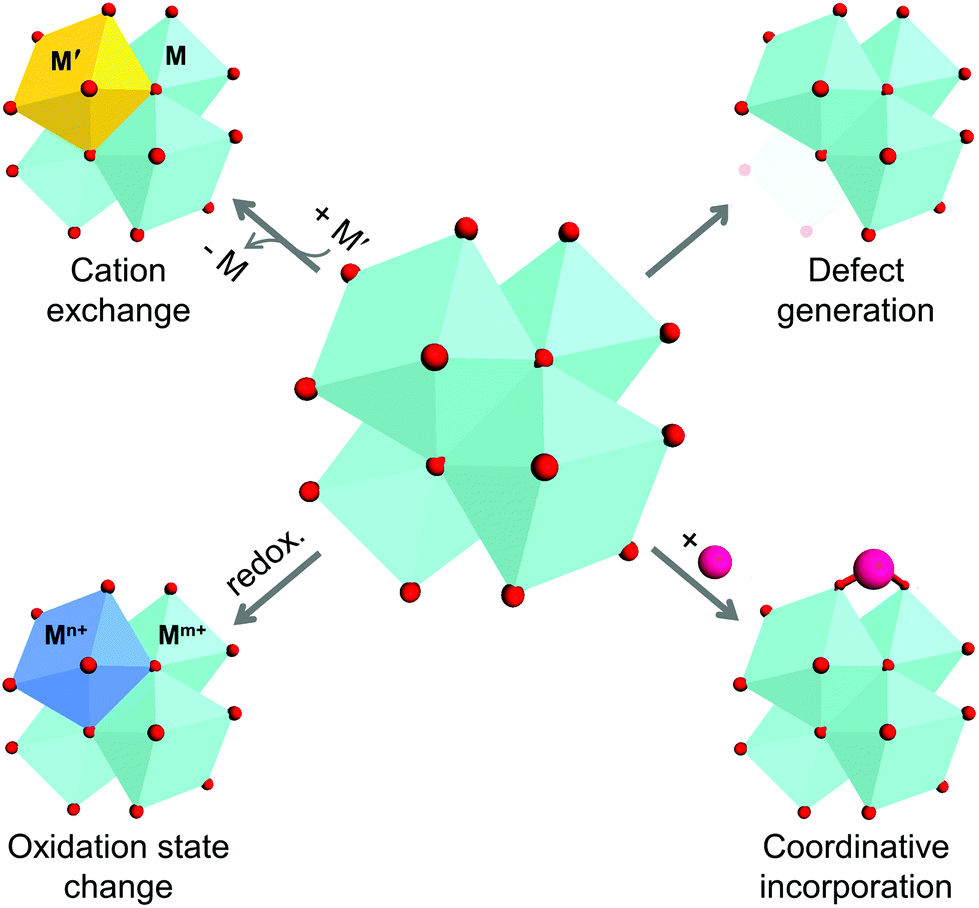 | ||
| Scheme 1 Schematic illustration of four methods for alteration of secondary building units of metal–organic frameworks. | ||
The SBU term should be differentiated from the ‘metal node’ one in certain cases; for example, single metal ions, such as those present in zeolitic imidazolate frameworks, could be designated as single-metal nodes or primary building units.11,30 In addition, neither the metal complexes located within the pores nor the metalloligands (ligands that contain metals) are considered to be SBUs. Hence, this review is limited to the chemical and/or mechanical alteration of SBUs. The PSM of SBUs has already generated many novel MOFs that could not have otherwise been obtained, and that present great potential for diverse applications. More specific reviews for each PSM method have been already published;31–35 however, broad terms comprehensive report has been scarcely published to date. For example, the previous reviews published by Wang et al.12 and Cohen et al.20 described the general concept of PSM for MOFs, but mainly focused on the covalent PSM of organic linkers, while the PSM of metal SBUs was only partially described. While several reviews addressed SBU alteration methods and results in detail, they only focused on one method each: the 2014 report published by Dincă et al.36 described the cation exchange alteration method and the 2015 and 2018 reports authored by Fischer et al.37,38 addressed defective engineering. Therefore, this review is intended to briefly introduce the general PSM strategies for SBUs and conceptualise them, to offer a toolbox that MOF researchers could use to design and engineer novel MOFs in a more systematic and versatile way.
2. Strategies for SBU alteration
2.1 Cation exchange at SBUs
Cation exchange of the SBUs is defined as a partial or complete substitution of a metal ion for another while retaining the framework34 and has been utilised by the MOF chemists to alter the inherent properties derived from metal clusters such as adsorption enthalpies and catalytic activities. The process usually involves mild and gradual reaction in a solution of the target cation. Dincă and Long, for the first time in 2007, performed a series of cation exchange experiments to vary the H2 adsorption energy by taking a tetrazolate-based Mn-MOF, Mn3[(Mn4Cl)3(BTT)8(MeOH)]2 (H3BTT = 1,3,5-tris(tetrazol-5-yl)benzene, MeOH = methanol) as the parent MOF.39 The compound includes coordinatively unsaturated extra- and intra-framework Mn2+ ions with the anionic skeleton (Fig. 1). Despite soaking in concentrated methanolic solutions of MCl2 (M = Fe2+, Co2+, Ni2+, Cu2+, Zn2+) at room temperature for a month, all the MOF crystals maintained its integrity yet exchanged a significant amount of the framework cations, which was proved by powder X-ray diffraction (PXRD) and inductively coupled plasma-atomic absorption (ICP-AA). Notably, the Fe2+, Co2+, and Ni2+ experiments showed the substitution of only extra-framework cations, while the Cu2+ and Zn2+ cases indicated that the intra-framework cation substitution occurred at the [Mn4Cl]7+ cluster as well. With monovalent cations, meanwhile, either very little (Li+) or negligible (Cu+) amounts of cation exchange was observed at the extra-framework sites. Likewise, mixed-metal derivatives could be readily prepared by using this strategy, which might be impossible to access via direct syntheses. From this initial work, several informative interpretations are obtainable, revealing certain cation exchange behaviours. The fact that the MOF crystal maintained its integrity during the exchange process excludes the scenario of ligand transfer to a new cation and the creation of new MOFs. One can notice that different metal sites have different preferences for cation exchange, while the inserted cations also showed different exchange behaviours even for the same substituting sites. This can be explained by their different electronegativities of the metals.53,54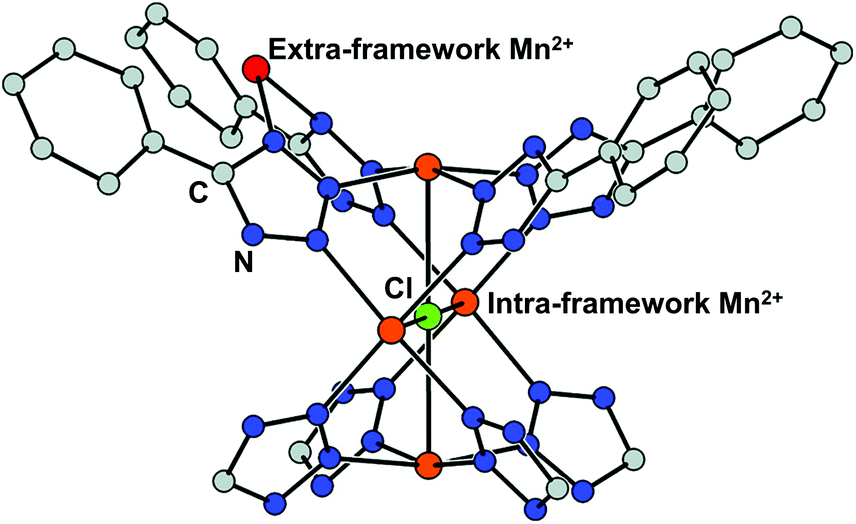 | ||
| Fig. 1 Coordinatively unsaturated extra- and intra-framework (red and orange, respectively) Mn2+ ions in the crystal structure of Mn and tetrazolate-based metal–organic framework, Mn3[(Mn4Cl)3(BTT)8(MeOH)]2. Reproduced with permission from ref. 40. Copyright 2007, American Chemical Society. | ||
Many reports on the cation exchange process in MOFs have been published in the decade that followed the publication of this paper. Dincă et al.36 surveyed those reports in 2014 and postulated four basic principles of cation exchange behaviours: (1) cation exchange of SBUs often occurred via open coordination sites, coordinating solvent molecules, or sites that could feature higher coordination numbers; (2) the weak field ligand environment of the SBUs enabled the coordinatively saturated metal sites to undergo cation exchange; (3) periodic trends existed depending on the types of cations (e.g., Cu2+ exhibited stronger replacement tendency than other second row transition metals); and (4) the distortion allowance in the structure could determine the potential extent of cation exchange. As emphasised in follow-up studies, these principles have helped us determine whether the compounds and experimental conditions for cation exchange would be ‘inert’ or ‘labile’.
The cation exchange method via simple soaking has been further modified to allow for its application for inert SBUs in MOFs. For example, UiO-66(Zr), a highly robust MOF that contains the Zr6O4(OH)4(CO2)12 SBU, yielded Hf4+-exchanged UiO-66(Zr/Hf) via soaking it in Hf4+ solution in dimethylformamide (DMF) while heating the mixture for 5 days at 85 °C.55 In addition, the unprecedented bimetallic UiO-66(Zr/Ti) MOF was generated by soaking UiO-66(Zr) in a Ti4+ solution in DMF under the same experimental conditions described above (Fig. 2a). The Ti4+ analogue of the robust UIO-66(Zr) MOF could not be directly synthesised because Ti4+ is not known to form the M6O4(OH)4(CO2)12 SBU structure of UIO-66(Zr). Similarly, Dincă et al.56 synthesised the unprecedented M-MOF-5 (M = Ti3+, V2+, V3+, Cr2+, Cr3+ Mn2+, and Fe2+), which also could not be obtained via direct synthesis (Fig. 2b). The authors indicated that the MOF-5 cluster acted as tripodal chelating ligand to construct the pseudo-tetrahedral geometry of MZn3O (M = V2+, Cr2+, Mn2+, and Fe2+) and the pseudo-trigonal bipyramidal geometry of ClMZn3O (M = Ti3+, V3+, and Cr3+) which featured a terminal Cl− moiety. These conventional solvothermal cation exchange methods typically require days or weeks and involve high energy consumption. To overcome these drawbacks, Zhou Long et al.57 reported a microwave-assisted approach for substituting the Zr4+ ions with Ti4+ ions in UiO-66. Within a few hours, approximately half the Zr4+ ions in the SBUs were exchanged, the crystallinity of the MOF was well maintained, and its photocatalytic activity significantly improved.
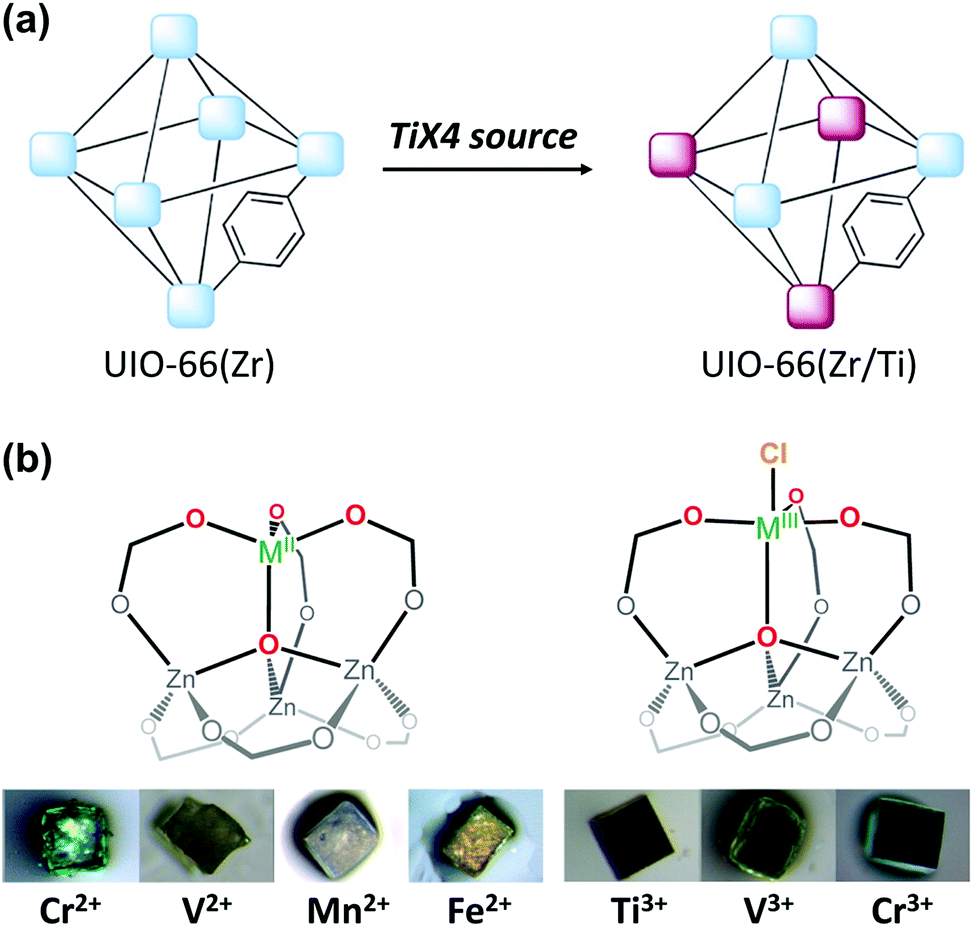 | ||
| Fig. 2 Illustrations of post-synthetic cation exchange of (a) UiO-66 and TiX4 (X = TiCp2Cl2, TiCl4, or TiBr4) and (b) MOF-5 after reacting with solutions of diverse metal salts. Reproduced with permission from ref. 55 and 56. Copyright 2012 and 2013, respectively, American Chemical Society. | ||
2.2 Change of cation valence in SBUs
In addition to the cation exchange approach, SBU metal centres can undergo redox reaction, which is a critical issue to enhance functionality in MOFs. However, altering the oxidation states of the metal centres, the frameworks are very susceptible of structural collapse because of a drastic electron reorganization on charge via post-redox reactions. Nevertheless, this method is advantageous to producing mixed-valence MOFs or isostructural analogues with different cation valances, inaccessible by direct syntheses. In addition, many works showed that the redox of SBUs endows unexpected properties to the MOFs (Table 1). In 2004, Suh group synthesised a MOF of pillared-bilayer framework by assembling bismacrocyclic Ni2+ complex [Ni2(C26H52N10)(Cl)4] and 1,3,5-benzenetricarboxylate (BTC).40 The authors also performed an oxidation reaction in a DMSO/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution of I2 (0.0136 M) for 10 h, reducing the I2 molecules to I3− anions in the channels, while two-thirds of the Ni2+ ions are oxidized to the low spin Ni3+ located in the framework. This yielded [Ni2(C26H52N10)]3[BTC]4(I3)4·nI2·17H2O via single-crystal-to-single-crystal transformation, which was amenable to X-ray diffraction structural analysis. Notably, this compound cannot be obtained directly by using a macrocyclic Ni3+ complex, which are stabilized in an octahedral geometry and thus have no coordination sites for ligands, and the Ni3+ species are easily reduced to Ni2+ in an aqueous media.
:1 v/v) solution of I2 (0.0136 M) for 10 h, reducing the I2 molecules to I3− anions in the channels, while two-thirds of the Ni2+ ions are oxidized to the low spin Ni3+ located in the framework. This yielded [Ni2(C26H52N10)]3[BTC]4(I3)4·nI2·17H2O via single-crystal-to-single-crystal transformation, which was amenable to X-ray diffraction structural analysis. Notably, this compound cannot be obtained directly by using a macrocyclic Ni3+ complex, which are stabilized in an octahedral geometry and thus have no coordination sites for ligands, and the Ni3+ species are easily reduced to Ni2+ in an aqueous media.
| Formula | Common name | Oxidation state variation | Extent | Agent | Conditions | Ref. |
|---|---|---|---|---|---|---|
| [Ni2(C26H52N10)3[BTC]4·6pyridine·36H2O | BOF-1 | Ni2+ to Ni3+ | ca. 66% | I2 | DMSO/H2O, 10 h | 40 |
| Fe3TMQPTC | PCN-426-Fe | Fe2+ to Fe3+ | ca. 97% | Bubbled O2 | DMF, 15 min | 41 |
| Cr3TMQPTC | PCN-426-Cr | Cr2+ to Cr3+ | ca. 91% | |||
| Cu3(BTC)2 | HKUST-1 | Cu2+ to Cu+ | ca. 66% | H2Q | MeCN, H2Q, | 42 |
| Mn2Cl2(bbta) | MAF-X25 | Mn2+ to Mn3+ | ca. 50% | H2O2 | H2O2 in H2O, MeCN, TEA, 2 days | 43 |
| Ni(C10H26N6)3(bpdc)3 | — | Ni2+ to Ni3+ | Unknown | Ag(I) | AgNO3 in MeOH, 10 min + 18 h | 44 |
| Cu3(BTC)2 | HKUST-1 | Cu2+ to Cu+ | ca. 40% | MeOH | MeOH vapor, 200 °C, 10 h | 45 |
| [{Cu12I(trz)8}·4Cl·8H2O]n | — | Cu+ to Cu2+ | Unknown | Air | Air, several weeks | 46 |
| [Cu(adp)(BIB)(H2O)]n | — | Cu2+ to Cu3+ | Unknown | NaOH | H2O2 in NaOH, | 47 |
| VIII(OH)(BDC) | MIL-47(VIII) | V3+ to V4+ | Complete | O2 | O2, 150 °C, 2 h, 200 °C 1 h | 48 |
| VIII2(OH)2C16H6O8 | MFM-300(VIII) | V3+ to V4+ | Complete | O2 | O2, 150 °C, 16 h | 49 |
| Ce(BTC) | Ce(III)-MOF | Ce3+ to Ce4+ | ca. 25% | NaOH, H2O2 | NaOH/H2O2, 2 min | 50 |
| Fe2(dobdc) | Fe-MOF-74 | Fe2+ to Fe3+ | Unknown | O2 | O2 | 51 |
| Fe(OH)(BDC) | MIL-100(Fe) | Fe3+ to Fe2+ | ca. 23% | Calcination | 250 °C, 12 h | 52 |
In contrast, Jeong et al.42 reported the coordinative reduction of the paddlewheel Cu2+ SBUs in HKUST-1. Hydroquinone (H2Q) was used as the reducing agent because it is an organic Lewis base that features lone-paired electrons. The reduction reaction was performed at 80 °C in acetonitrile (MeCN) solution of H2Q under anhydrous conditions. During the reaction, the coordinating H2Q molecule promptly dissociated into the HQ˙ radical by transferring a single electron and donating a proton (Fig. 3a). Approximately 30% Cu2+ ions were reduced to Cu+ ions, without being further reduced to Cu0. This was attributed to the single-electron transfer, as revealed by single-crystal X-ray diffraction, 1H nuclear magnetic resonance spectroscopy, and CO adsorption analyses. As illustrated in Fig. 3b, half of the reduced Cu+ ions still remained in the SBUs, and presented pseudo-square planar geometry, while the other half protruded and transformed into [Cu(MeCN)4]+ complexes that were trapped in the small cages of HKUST-1. Likewise, using the proper reductant is the key to achieving mild and gradual changes in cation valence while retaining the integrity of the MOF structure. Notably, the use of common reductants, such as hydrides, only resulted in the structural degradation of HKUST-1 and formation of metallic Cu0 nanoparticles, despite using exact stoichiometric amounts of reactants.
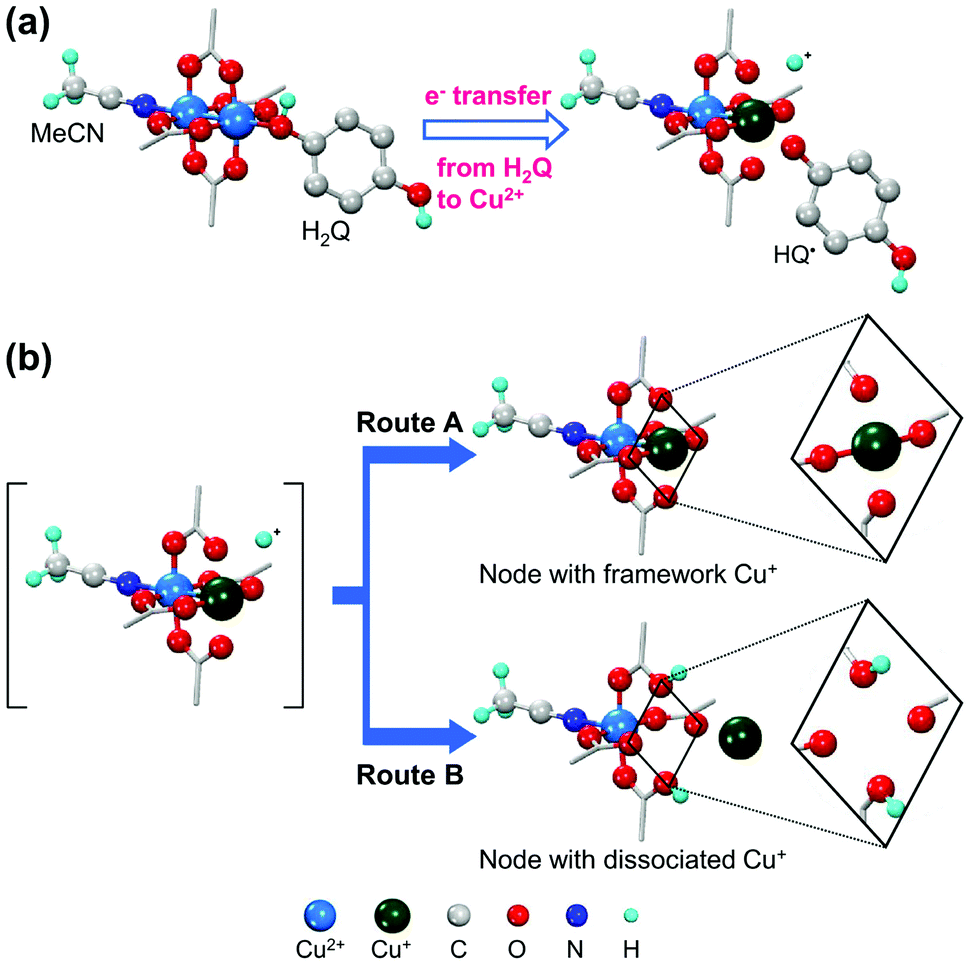 | ||
| Fig. 3 Illustrations of (a) coordinative reduction of Cu2+ in paddlewheel node of HKUST-1 and (b) two possible routes for association of Cu+ ions with paddlewheel node or dissociation of Cu+ ions from framework. Here, H2Q is hydroquinone. Reproduced with permission from ref. 42. Copyright 2019, American Chemical Society. | ||
To obtain MOFs that feature oxidation states that could not be achieved using conventional synthesis methods, cation exchange using metal species that could readily change their oxidation states could be conducted first. This strategy was reported in 2014 by Zhou et al.41 who synthesised PCN-426-Mg, that comprised [Mg3(μ3-O)] clusters of three Mg2+ octahedrons. Because the Mg–O bonds are relatively labile according to the HSAB theory, the synthesised MOF easily underwent cation exchange with common transition metal ions, and acted as template for the next step: changing the oxidation states. During this step, divalent cations (Fe2+ and Cr2+) were first inserted to exchange the cations and preserve the structure of the MOF, then, they were oxidised to higher oxidation states (Fe3+ and Cr3+) using a stream of air for 15 min while suspended in DMF. Conversely, when high-valence metals were used for direct synthesis, the MOFs scarcely crystallise because of the inertness of the pre-formed strong metal–ligand bonds during the synthesis reaction.18,58–62
2.3 Generation of defects of SBUs
Crystal defects indicate the presence of characteristic imperfections (or irregularities) of the host lattice, such as vacancies, compositional impurities, disorders, and dislocations.37,38 For real-life systems, the crystalline materials mostly contain inherent defect sites, and therefore, so do MOFs. The defects decrease the crystallinity of MOFs and are considered to be detrimental for some applications, such as optoelectronics.63 However, defects could also be advantageous for generating novel functions, such as higher porosity, higher sorption, and enhanced catalytic performance, which cannot be achieved using the original structures.In broad terms, defective MOFs are frameworks that feature heterogeneous composition; in this sense, the afore-mentioned processes of cation exchange and oxidation state change can also be understood within defect chemistry and engineering if they occur incomplete and disordered. Nevertheless, among MOF scholars, the definition is typically considered to be the absence of linkers and/or metal nodes (i.e., Schottky-type point defects). Thus, for differentiation, we will denote those types of defects as ‘vacancy defects’. Moreover, this report strictly refers to the PSM methods that generate defects (mostly vacancies) in SBUs while preserving the characteristics of MOFs, as illustrated in the Venn diagram in Fig. 4. The report Li et al.64 published in 2014 could help clarify the boundaries of definition. They used Zn4O(PyC)3 (PyC = 4-pyrazolecarboxylate) and eliminated the metals and linkers in the framework (Fig. 5). Upon the immersion of the MOF crystals in water, their structure underwent SCSC transformation, and their architectural stability was fully retained. This facilitated the crystallographic characterisation of the missing sites, i.e., vacancies, via the removal of 25% of the metal ions and 50% of the linkers (structure 2 in Fig. 5). This metal and linker elimination reaction even varied the type of SBUs from octahedral to triangular ones. However, the resultant compounds would not be considered to be defective MOFs owing to the crystallographic transformation with ordered vacancies. Subsequently, those vacant sites were filled with other metal ions by immersing the MOF crystals in solutions of different metal ions, and the frameworks of heterometallic octahedral SBUs could be obtained.
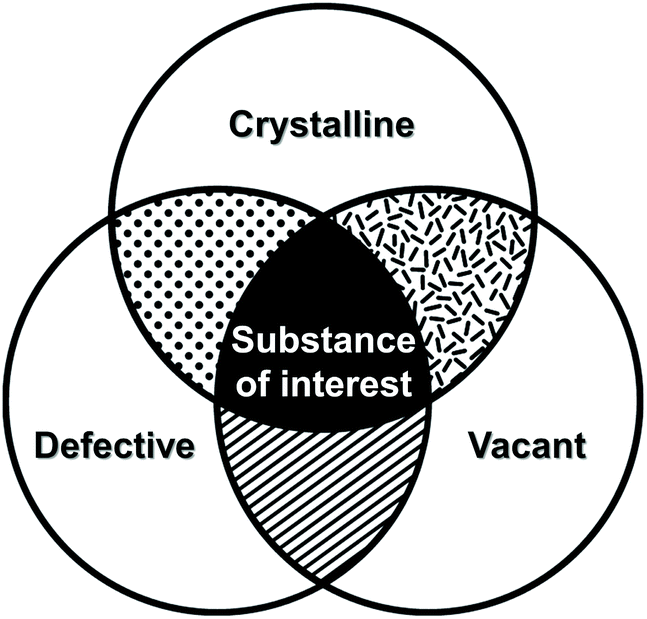 | ||
| Fig. 4 Venn diagram of three properties of metal–organic frameworks after post-synthetic modification of secondary building units. | ||
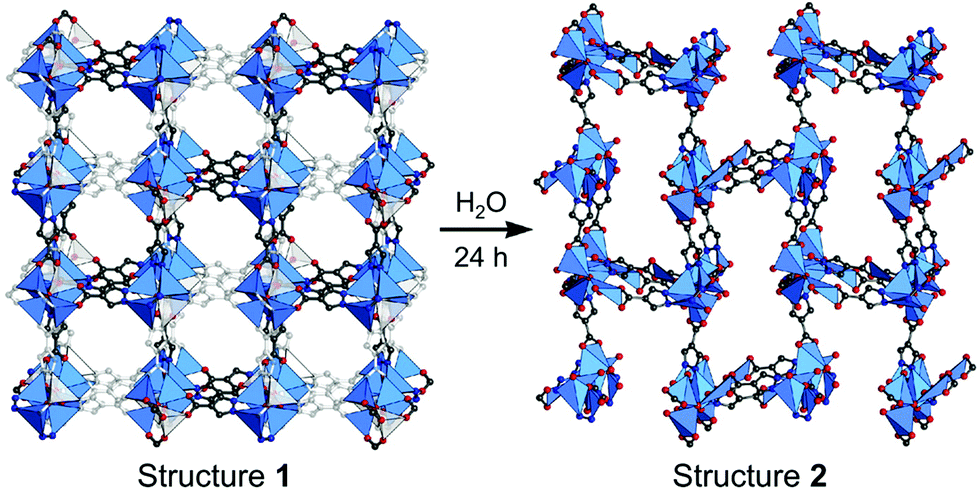 | ||
| Fig. 5 Single-crystal structures of Zn4O(PyC)3 before and after the creation of ordered vacancies. Here, PyC is 4-pyrazolecarboxylate. Reproduced with permission from ref. 65. Copyright 2014, American Chemical Society. | ||
According to Fischer et al.,38 two methods are available for creating defect sites: a de novo synthesis (e.g., modulation, mixed-linker approach, and fast crystal growth) and the PSM of pre-synthesised MOF crystals, including mechanical treatment, harsh activation, and acid/base treatment. For the PSM procedures, it is important that the MOF structure be tolerant of certain levels of missing constituents to prevent complete structure degradation. Therefore, the key for generating vacancy defects in SBUs would lie in finding highly defect-tolerant MOFs and/or partially decomposing the structure. In 2014, Guo et al.65 reported the use of thermal decarboxylation to generate vacancy defects. They broke the Zn-carboxylate bonds of MOF-5, which were the weakest points of the structure that would be lost first upon heating. Structural characterisation revealed that MOF-5 annealed at 380 °C possessed local vacancy defects owing to the partial decomposition of carboxylates while maintaining the overall framework. In 2017, De Vos et al.66 further developed this method using the mixed-linker UiO-66 that featured trans-1,4-cyclohexane-dicarboxylate (CDC) and 1,4-benzenedicarboxylate (BDC) as ligands. The relatively thermo-labile CDC linker was decomposed at 325 °C, whereas the BDC linker remained unchanged at this temperature. Advantageously, this process eliminated the CDC linker moieties completely and remained no by-products, unlike MOF-5, where benzoate moieties were detected in the framework after annealing.
In 2014, Lillerud et al.67 observed that the concentration of missing linker defects of UiO-66 increased as the number of washings increased, which was attributed to its hydrolysis. Similarly, external solvents or chemicals often cause the cleavage of the framework bonds and the generation of defects. Therefore, the metal–carboxylate bonds of MOFs could be broken via deprotonation in acidic environment. For example, De Vos et al.68 reported the use of acid treatment for the Fe3(μ3-OH) SBU of MIL-100(Fe). As presented in Fig. 6a, soaking the MOF in common acids, including CF3COOH and HClO4, which are stronger acids than the BTC linker of MIL-100(Fe), generated defective Brønsted acid sites. Conversely, Kim et al.69 recently proposed a Ag-catalysed decarboxylation, which involved HKUST-1 crystals, AgNO3, and K2S2O8. During the reaction, Ag+ transferred two electrons to S2O82− and was oxidised to Ag3+ while generating SO4˙−. The generated SO4˙− radical reacted with the square planar Cu2+ centre, and formed six-coordinated octahedral Cu-sulphate complexes; this was accompanied by decarboxylation, which generated CO2. This Ag-catalysed decarboxylation reaction removed a C–C bond and directly cleaved the M–O bond, unlike the conventional decarboxylation methods.
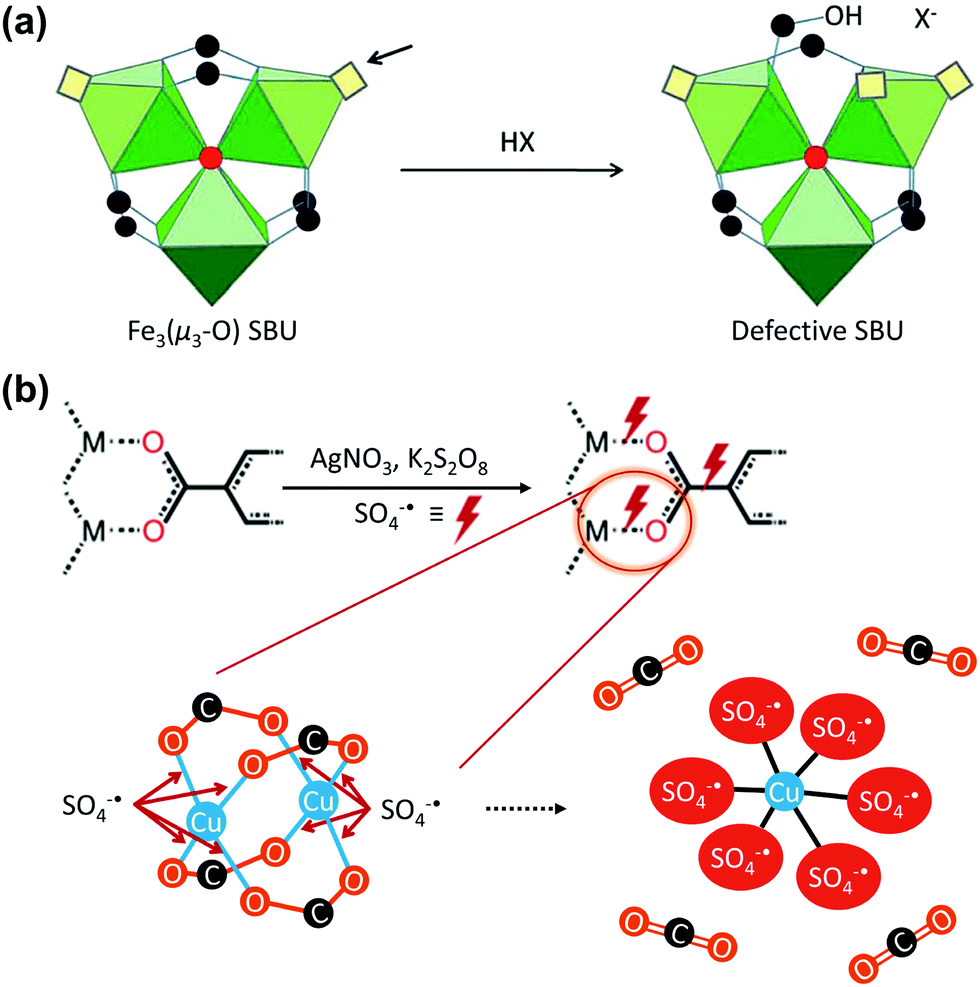 | ||
| Fig. 6 Proposed vacancy-generation routes of (a) acid activation of the Fe3(μ3-O) secondary building unit (SBU) using a protonic acid and (b) chemical etching via Ag-catalysed decarboxylation under mild conditions. Reproduced with permission from ref. 68 and 69. Copyright 2012, Royal Society of Chemistry, and Copyright 2018, Springer Nature, respectively. | ||
2.4 Incorporation of new species into SBUs
Open metal sites (OMSs) are a noteworthy feature of MOFs, and could be used as catalytic or specific adsorption sites. In addition, OMSs can be functionalised via coordination bonds between them and functional organic molecules (Table 2). For example, Long et al.70 synthesised and functionalized Mg2(dobpdc) (H4dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) by coordinating a diamine ligand at its five-coordinated OMS (Fig. 7a). Aqueous amine solutions are widely utilised as CO2 absorbents for amine scrubbing systems owing to their high affinity for CO2. Therefore, combining the amine functional group with Mg2(dobpdc) achieved a synergistic effect for capturing CO2. Recently, Chen et al.71 decorated the Fe3(μ3-OH) SBUs of flexible MIL-88B with coordinating molecules: 4-cyanopyridine, 4-ethynylpyridine and 4-vinylpyridine, which contained unsaturated nitrile, ethynyl, and vinyl groups, respectively, for [2 + 2 + 2] cyclotrimerisation. Owing to the confined porous environment with suitable orientation, the incorporated monomers could undergo cyclotrimerisation and generated trimeric products. This consecutive incorporation of monomers followed by PSM via chemical reaction transformed the flexible MIL-88B into a rigid framework of unprecedentedly large void volume. Farha et al.72 decorated the Zr6 SBUs of NU-1000 with perfluoroalkane to form defect-free NU-1000 that comprised [Zr6(μ3-OH)8(–OH)8]8+ clusters with carboxylate linkers (Fig. 7b). Similar to the OMS-containing MOFs, these Zr6 SBUs allowed the addition of functional species to it. The functional ligand could be coordinated to the Zr6 SBUs by replacing the terminal OH–/H2O groups via the acid–base reaction known as solvent-assisted ligand incorporation. Thus, the generated perfluoroalkane-functionalised NU-1000 exhibited high CO2 affinity owing to the presence of the C–F dipoles of the introduced ligand which interacted with the CO2 quadrupole. | ||
| Fig. 7 (a) Synthetic route for the alkylamine-appended mmen-Mg2(dobpdc) (mmen = N,N′-dimethylethylenediamine and H4dobpdc = 4,4′-dihydroxy-(1,1′-biphenyl)-3,3′-dicarboxylic acid) and (b) solvent assisted ligand incorporation approach for functionalisation of perfluoroakane. Here, DMF is dimethylformamide. Reproduced with permission from ref. 70 and 72. Copyright 2012 and 2013, respectively, American Chemical Society. | ||
| MOF | Incorporated molecule | Incorporated position in SBU | Applications | Ref. |
|---|---|---|---|---|
| Mg2(dobpdc) | Dimethylethylenediamine | OMS | Capture | 70 and 78 |
| Mg2(dobpdc) | 2,2-Dimethyl-1,3-diaminopropane | OMS | Capture | 100 |
| Mg2(dobpdc) | 2-(Aminomethyl)piperidine | OMS | Capture | 79 |
| Ni-MOF-74 | Imidazole | OMS | Separation | 87 and 101 |
| Magnetic | ||||
| HKUST-1 | 4-(Methylamino)-pyridine | OMS | Capture | 102 |
| HKUST-1 | Tetracyanoquinodimethane | OMS | Conductivity | 94 |
| HKUST-1 | H2O | OMS | Proton conductor | 96 |
| MIL-101(Cr) | L-Proline | OMS | Catalysis | 103 |
| MIL-101(Cr) | Ethylenediamine | OMS | Catalysis | 104 |
| UIO-66 | LitBuO | μ3-OH | Detoxification | 90 |
| UIO-66 | SO4 | Terminal –OH/H2O | Catalysis | 105 |
| NH2-UIO-66 | Phenylsilane | Termianl –OH | Hydrophobicity | 106 |
| NU-1000 | Perfluoroalkane | Terminal –OH/H2O | Capture | 72 |
| NU-1000 | Boron-dipyrromethene | Terminal –OH/H2O | Detoxification | 107 |
| NU-1000 | Non-polar organic carboxylate | Terminal –OH/H2O | Water stability | 108 |
| MOF-808 | SO4 | Terminal –OH/H2O | Catalysis | 109 and 110 |
| MOF-808 | Trimethylsilyl triflate | Terminal –OH/H2O | Catalysis | 111 |
| MOF-808 | Ethylenediaminetetraacetic acid | Terminal –OH/H2O | Capture | 97 and 99 |
| Proton conductor |
Using a similar ligand installation procedure on a Zr-based MOF, Zhou et al.73 synthesised a mixed-linker Zr-MOF which could not be obtained via one-pot synthesis owing to the thermodynamic unfavorability of the process (Fig. 8). PCN-700, a Zr-based MOF, consists of eight-connected Zr6O4(OH)8(H2O)4 with two vacant pockets (A and B, 16.4 and 7.0 Å in length, respectively) between neighbouring SBUs. By inserting linkers of the suitable size into the pockets, mixed-linker structures were successfully obtained via a kinetically controlled synthetic pathway. Because the BDC linker (6.9 Å) was fitted to pocket B, the PCN-701 structure was constructed via sequential installation by replacing the terminal OH–/H2O groups of the Zr SBU, accompanied by the slight elongation of pocket A to 16.5 Å (pocket A′). Because the biphenyl rings of BPDC (BPDC = biphenyl-4,4′-dicarboxylate) allow for changes in the dihedral angle from 78.1° to 89.5°, the framework could self-adjust its structure. Similarly, pocket A was linked with a Me2-TPDC (TPDC = p-terphenyl-4,4′′-dicarboxylate) linker to construct PCN-702 and pocket B was lengthened to 8.2 Å (pocket B′). Because only the slight elongation of pocket A was induced via the installation of the BDC linker, Me2-TPDC was installed into pocket A′ to construct PCN-703, which generated the elongated pocket A′′ (17.4 Å). However, pocket B′ was too large to bridge the BDC linker, and therefore PCN-703 could not be constructed using PCN-702. As such, multiple functional groups could be installed to the coordinatively unsaturated Zr6 clusters in a crystallographically controlled manner. Following this study, Zhou et al.74 more extensively controlled the arrangement of functionalities in PCN-700 using linkers of different lengths and/or substituents.
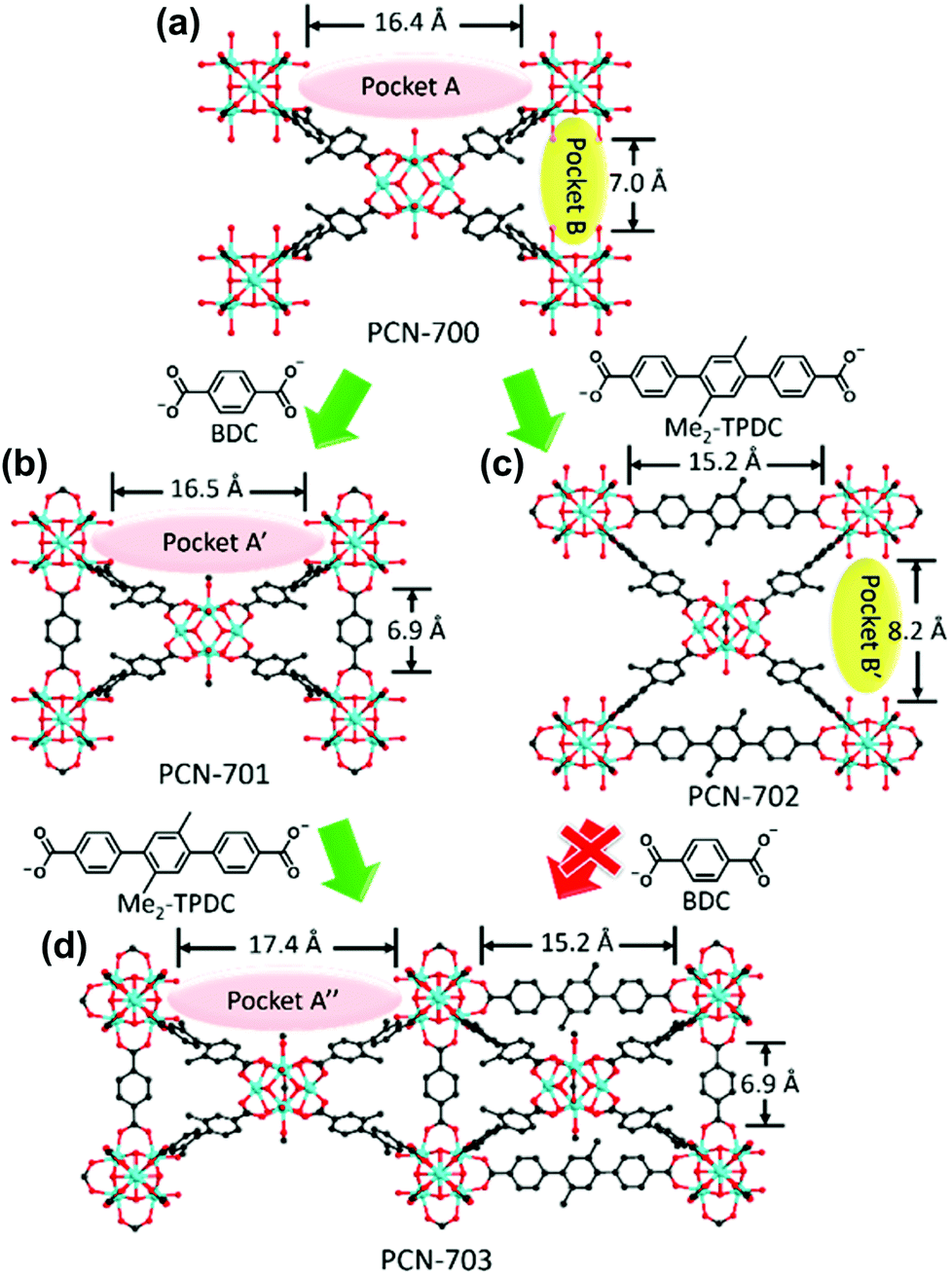 | ||
| Fig. 8 Structures of (a) PCN-700, (b) PCN-701, (c) PCN-702 and (d) PCN-703. Here, BDC and TPDC are 1,4-benzenedicarboxylate and p-terphenyl-4,4′′-dicarboxylate, respectively. Reproduced with permission from ref. 73. Copyright 2015, American Chemical Society. | ||
In addition to decoration with organic ligands, incorporation of inorganic complexes to impart functionalities that cannot be observed in organic species has being extensively studied (Table 3). Fischer et al.75 first combined the inorganic complex, 1,1′-ferrocenediyl-dimethylsilane with MIL-53(Al) that consisted of an Al3+ cation node and bridging OH group (Fig. 9). Because the bridging OH group was available for functionalisation, the Si atoms of the ferrocene complex could bind to the OH site followed by the migration of protons to the η5-C5H5 rings, which resulted in the slight improvement in the catalytic performance of the MOF. Gates et al.76 introduced Ir complexes (Ir(CO)2 and Ir(C2H4)2) to UiO-66 and NU-1000 by reacting Ir precursors, Ir(CO)2(acac) (acac− = acetylacetonate) and Ir(C2H4)2(acac), with the OH–/H2O sites. The Zr6 clusters of UIO-66 and NU-1000 could be decorated with inorganic complexes owing to their defective OMSs in the SBUs, which were occupied by OH–/H2O functional groups. Zaworotko et al.77 reported a PSM approach that represents the transformation from dinuclear Cd2+ SBUs to novel tetranuclear Cu2+ SBUs, via stepwise metal ion exchange and metal salt incorporation. During the transformation, the Cu2+ paddlewheels derived from Cd2+ ones bound two more Cu2+ cations to form a Cu4 intermediate, and subsequently underwent the molecular rearrangement to generate the tetrametallic [Cu4X2(COO)6(H2O)2] SBU (X = CH3O−, OH−).
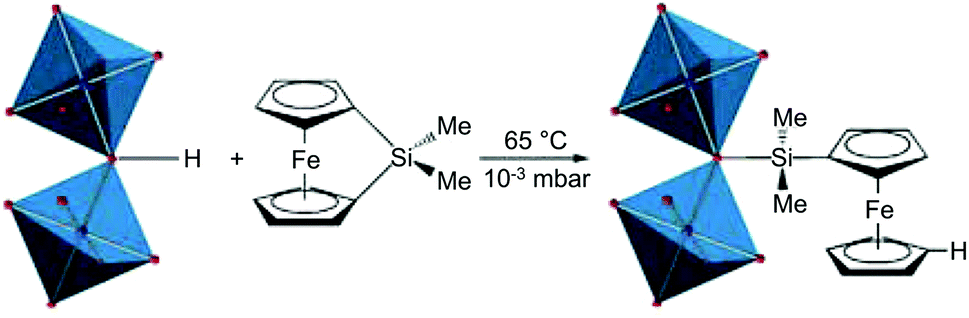 | ||
| Fig. 9 Ring-opening reaction of the bridging HO-group in the secondary building units of MIL-53(Al) and 1,1′-ferrocenediyl-dimethylsilane. Reproduced with permission from ref. 75. Copyright 2009, American Chemical Society. | ||
| MOF | Incorporated metal | Incorporated position in SBU | Applications | Ref. |
|---|---|---|---|---|
| NU-1000 | Zn, Al | Terminal –OH | Catalysis | 112 |
| NU-1000 | Ir(III) | Terminal –OH/H2O | Catalysis | 113 |
| NU-1000 | Cu(II) | Terminal –OH | Catalysis | 89 |
| NU-1000 | In-Me3 | Terminal –OH/H2O, μ3-OH | Catalysis | 114 |
| NU-1200 | Vanadium oxide | Terminal –OH/H2O | Catalysis | 115 |
| UIO-66 | Cu | Terminal –OH/H2O | Catalysis | 88 |
| NH2-UIO-66 | Ti(IV) | Terminal –OH/H2O | Catalysis | 116 |
| UIO-68(Zr) | Co–Cl | μ3-OH | Catalysis | 117 |
| Zr12-bpdc MOF | Cu(I) | μ2-OH, μ3-OH | Catalysis | 118 |
| Zr-MTBC | Co | μ2-OH, μ3-OH | Catalysis | 119 |
| Ti8-BDC MOF | Co | (μ2-O), (μ2-O−) | Catalysis | 120 |
| TPHN-MOF | Mg–Me | μ3-OH | Catalysis | 121 |
| Zr12-TPDC | Co–H | μ2-OH, μ3-OH | Catalysis | 122 |
| PCN-700 | Ni | Terminal –OH/H2O, μ3-OH | Catalysis | 123 |
| MOF-808 | Pd | Terminal –OH/H2O | Catalysis | 124 |
| Hf-MOF-808 | Vanadium oxide | Terminal –OH/H2O | Catalysis | 125 |
| MIL-53(Al) | Ferrocene | –OH group | Electrochemical system | 75 |
| MIL-125 | NiH | μ2-OH | Catalysis | 126 |
3. Property modulation and applications via SBU alteration
Four approaches for the alteration of SBUs were summarised and discussed in the previous section. Relevant studies over the last two decades have indicated that even minor alterations of SBUs could significantly influence the physicochemical properties of MOFs, and could engender novel functions. Therefore, in this section, several representative results are discussed where SBU alterations successfully modified the MOF properties, and rendered them useful for applications such as sorption, separation, and catalysis.3.1 Sorption properties
To overcome the microporosity of MOFs and enhance the ability to capture larger molecules, hierarchically porous MOFs have been studied. Recently, Sun et al.45 achieved hierarchical porosity in HKUST-1 by reducing Cu2+ to Cu+ using CH3OH vapor at 200 °C as a reducing agent. The resultant compound was used to capture thiophene, a large aromatic sulphide molecule, which was too large and could not be captured using pristine HKUST-1. Similar to the case of H2Q-treated HKUST-1, Cu2+ changed its coordination geometry to two-coordinated complex by releasing two of the four carboxylates on the paddlewheel SBU. The interaction between the aromatic sulphide molecule and Cu+via robust phi-complexation rendered this hierarchically porous MOF a good thiophene adsorbent. The amounts of Cu+ and resulting mesopores were controlled by tuning the reaction time. The optimum MOF for thiophene capture was obtained after 10 h, and comprised 30 nm mesopores and 44% Cu+ ions.As described above, Mg2(dobpdc) decorated with N,N′-dimethylethylenediamine (mmen) presented large CO2 capture capacity and high efficiency owing to the phase-changing properties that contributed to unusual step-shaped adsorption isotherm.73,78 As illustrated in Fig. 10, mechanistically, the capturing process followed several steps: first, the coordinated amine was deprotonated by the neighbouring amine, which could act as strong base and led to the formation of ammonium groups. CO2 was simultaneously added to the coordinated amine and formed a carbamate species. This ammonium carbamate species destabilised the coordinated neighbouring amine and facilitated the cooperative insertion of CO2 molecules between the metal and amine groups via a chain reaction. Consequently, this mmen-decoration approach on Mg2(dobpdc) led to the enhanced CO2 capture performance of the MOF under flue gas conditions (15% CO2 in N2 at 40 °C), and the low energy consumption for the process. Very recently, 2-(aminomethyl)piperidine was incorporated into the same MOF, and a significant improvement of CO2 uptake was achieved even under high humidity conditions, which has been considered to be a significant drawback for the practical use of this MOF in flue gas environment.79
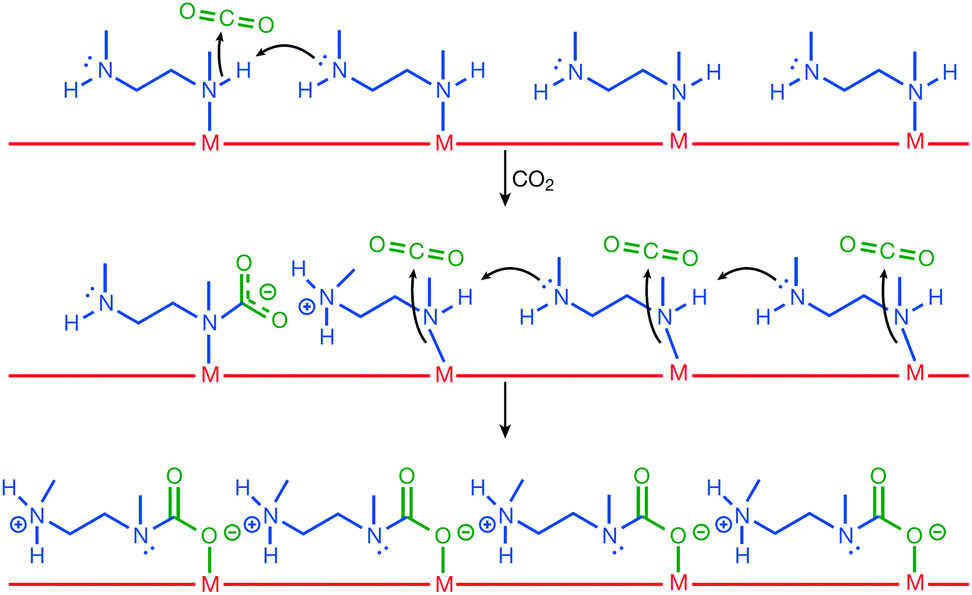 | ||
| Fig. 10 Mechanism for CO2 adsorption at four neighbouring M-mmen sites within an infinite one-dimensional chain of such sites running along the channel of a mmen-M2(dobpdc) compound. Here, mmen is N,N′-dimethylethylenediamine. Reproduced with permission from ref. 78. Copyright 2015, Springer Nature. | ||
3.2 Gas separation
Unlike conventional gas mixture separation that could be conducted via simple molecular sieving,80 the isotope mixture separation procedure requires special techniques owing to the almost identical properties of the isotopes. Therefore, researchers have recently used nanoporous materials to separate hydrogen isotopes using two types of the so-called ‘quantum sieving effect’. Heavier isotopes with shorter de Broglie wavelength possess higher mobility than lighter isotopes in confined spaces, where the size difference between the pores and molecules becomes comparable to the de Broglie wavelength, which results in the enrichment of the product with heavier isotope materials. This phenomenon is known as kinetic quantum sieving (KQS), and thus, it is important to design a porous material that contains optimal pore size (3.0–3.4 Å).81,82 Another quantum sieving mechanism, chemical affinity quantum sieving (CAQS), has been proposed to separate hydrogen isotopes via the strong interaction sites with hydrogen molecules, such as OMSs, where the heavier molecular mass of deuterium (D2) led to the higher adsorption enthalpy and resulted in its preferential adsorption.83,84To achieve such quantum sieving effects in MOFs, SBU alteration methods can be utilised. Heine et al.85 used Cu(I)-MFU-4l, which exhibited high adsorption enthalpies for hydrogen molecules (32 kJ mol−1) for this purpose. Cu(I)-MFU-4l was synthesised by replacing half of the terminal Zn sites with Cu2+ followed by exchanging the Cl− ions with formate and subsequent heating under vacuum for 1 h.86 At high temperature (above 90 K), the difference in the adsorption enthalpies of D2 and H2 was large (2.5 kJ mol−1), which was attributed to the strongly binding Cu(I) sites that exhibited the high selectivity of 11. Recently, we described the imidazole-decorated Ni-MOF-74 (left side of Fig. 11).87 Ni-MOF-74 is known to be one of the most effective H2/D2 separation materials owing to its high isosteric heat of adsorption for hydrogen via OMSs, and also its optimal pore size achieved via SBU alteration. The partially occupied imidazole molecules in the OMSs of Ni-MOF-74 could act as diffusion barrier and enhance the KQS effect by modifying the pore structures; moreover, the remaining OMSs provided selectively strong binding sites which exhibited CAQS effect. Based on control experiments on the degree of occupancy, 10% of imidazole decoration on the OMSs of Ni-MOF-74 (MOF-74-IM-10, right side of Fig. 11) presented the highest D2/H2 selectivity (26 at 77 K).
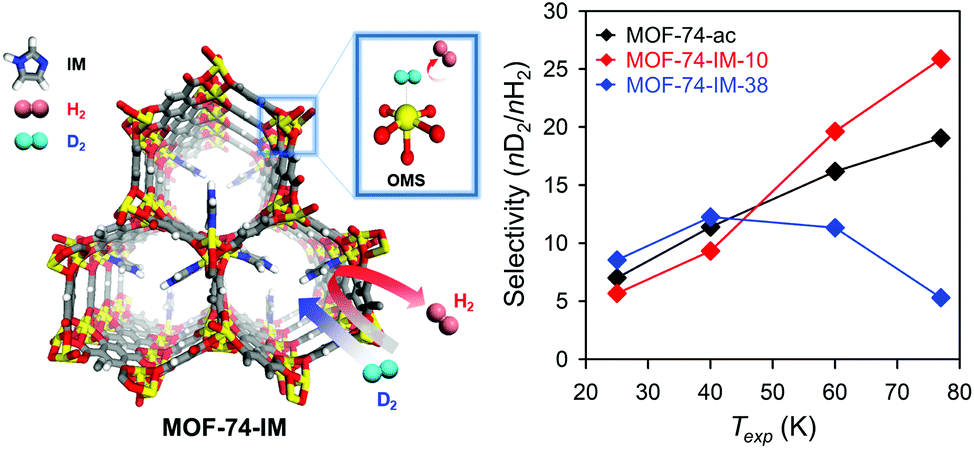 | ||
| Fig. 11 Selectivity of MOF-74-IMs as function of exposure temperature (Texp) after exposure to 1:1 H2/D2 mixture. Here, OMS is open metal site. Reproduced with permission from ref. 87. Copyright 2017, American Chemical Society. | ||
3.3 Catalysis
The catalytic activity of MOFs is typically attributed to the metal centres of SBUs, which could act as Lewis acidic sites. The report published by Long et al.57 in 2017 is an interesting example, and described inducing photocatalysis via the microwave-induced cation exchange of Zr6O4(OH)4 SBU in UiO-66 with Ti4+ ions. The energy band of pristine UiO-66(Zr) was calculated to be 4.00 eV using UV–vis spectroscopy and Mott–Schottky measurements. This indicated that pristine UiO-66(Zr) presented n-type semiconductor characteristics and its photocatalytic activity towards the reduction of Se6+ was almost negligible. Conversely, the cation exchange product of UiO-66(Zr/Ti) (50% cations exchanged) presented the energy band value of 3.75 eV because the inserted Ti4+ cations increased the lower level of the conduction band. As illustrated in Fig. 12, because the probability of the electron transfer from the excited BDC linker to Ti4+ was higher than that from the excited BDC linker to Zr4+, Ti4+ and Zr4+ acted as electron acceptor and trap, respectively, and thus, Ti4+ in the (Ti4+/Zr4+)6O4(OH)4 SBU could donate electrons to Zr4+ more easily. This improved the photocatalytic performance for the reduction of Se6+ by increasing the interfacial charge transfer from the linker to the metal SBU.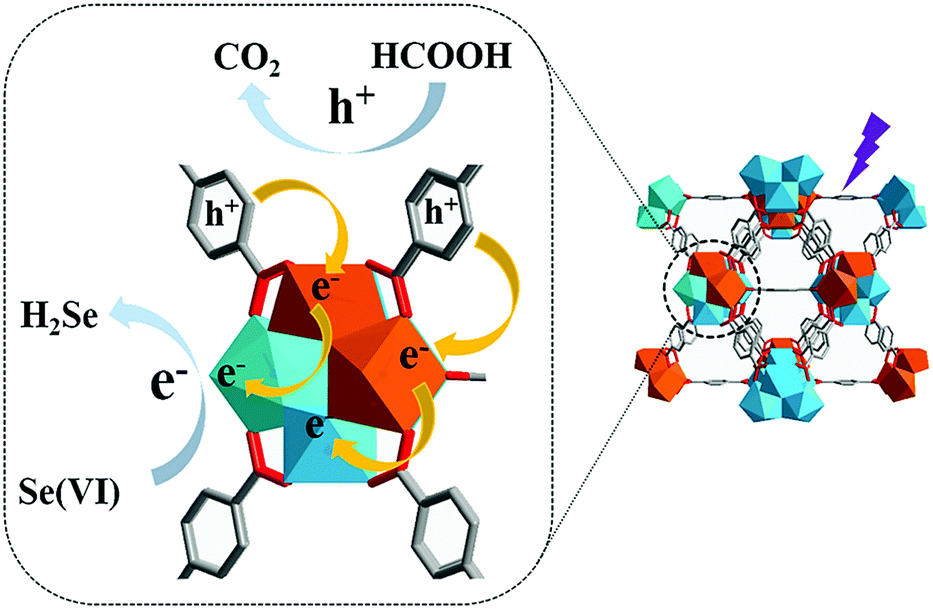 | ||
| Fig. 12 Proposed mechanism for enhanced photocatalytic performance of UiO-66(Zr/Ti). Here, the Zr4+ and Ti4+ ions are blue and orange, respectively. Reproduced with permission from ref. 57. Copyright 2017, Royal Society of Chemistry. | ||
Incorporating new metal species into SBUs is another facile method for imparting additional catalytic activities to MOFs. Behm et al.88 recently synthesised a Cu/UiO-66 catalyst for CO oxidation, where single Cu metal was deposited into SBUs. After soaking the UiO-66 crystals in a Cu2+ solution in DMF at 85 °C, it was observed that Cu2+ was coordinatively bridged to the OH–/H2O terminal groups of the defective Zr6 SBU in UiO-66 along with coordinated Cl−. The attached Cu sites were again reduced to active Cu+ species, and the Cl− ions were removed via subsequent activation with H2 (Fig. 13). The CO oxidation activity of this single-Cu catalyst was at least three times higher than that of conventional catalysts, such as Cu/CeO2 and Cu/ZrO2, even under O2-rich atmosphere. Similarly, Lercher et al.89 synthesised Cu-oxo dimer-supported NU-1000, by depositing Cu2+ ions on the Zr6 SBU followed by oxidation at 200 °C in O2 atmosphere. The resultant catalyst was able to oxidise methane to methanol with high efficiency under the methane pressure of 1 bar at 150 °C.
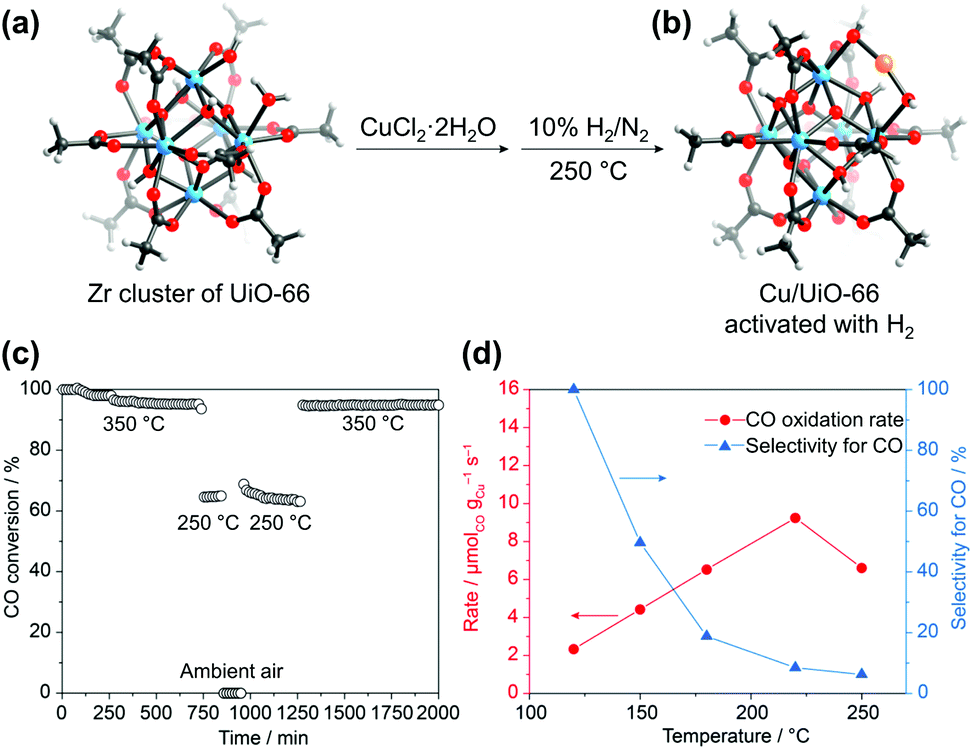 | ||
| Fig. 13 Secondary building unit structures of (a) UiO-66 and (b) Cu/UiO-66. (c) CO conversion at different temperatures (cooling and shutdown of the reaction, and restart by heating in a reaction gas mixture), and (d) reaction rates of CO oxidation upon cycles. Here the Zr4+ and Cu+ ions are blue and orange, respectively. Reproduced with permission from ref. 88. Copyright 2019, American Chemical Society. | ||
Detoxification of chemical warfare agents (CWAs) is one of the most important research fields where MOFs are used as catalysts. UiO-66 presumably contains different types of inherent or intentionally created defects. In 2015, Navarro et al.90 reported the reaction of activated pristine UiO-66 with LitBuO and introduced additional basic catalytic sites to UiO-66 by replacing the μ3-OH sites in its SBUs (Fig. 14). In addition, UiO-66 was also reacted with acetic acid and KHSO4, stepwise, which generated additional acidic sites (UiO-66@SO4H) that could control the degradation activity of various CWAs. UiO-66@LiOtBu presented good performance for the degradation of CWA analogues, particularly diisopropylfluorophosphate (DIFP) (turnover frequency (TOF) = 0.13 min−1), while both UiO-66@AcOH and UiO-66@SO4H exhibited relatively low CWA degradation kinetics and conversion ratios.
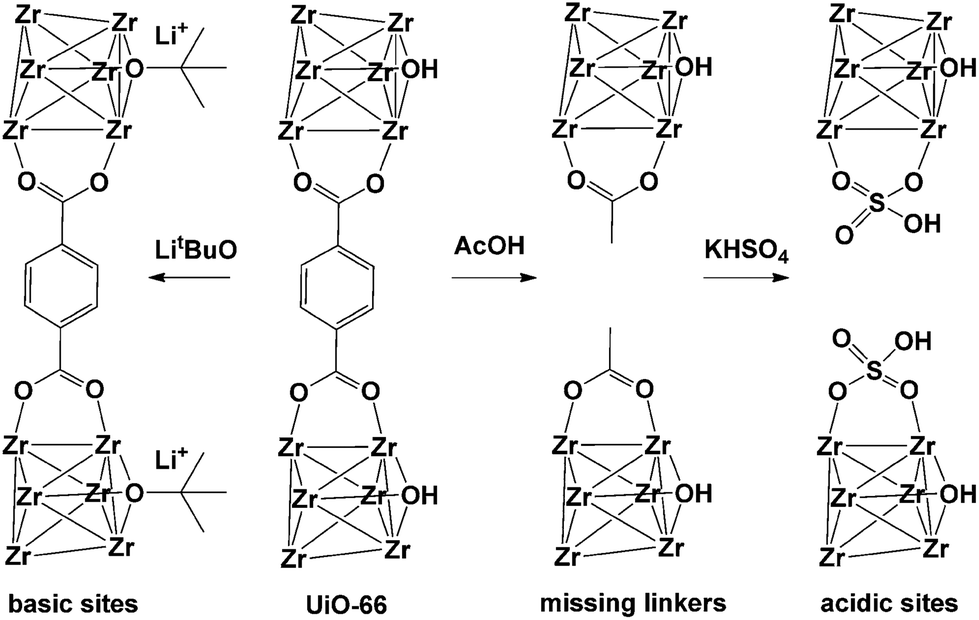 | ||
| Fig. 14 Improvement of catalytic activity of [Zr6O4(OH)4(bdc)6] (UiO-66) by introducing missing-linker defects and/or acidic and basic sites in its structure. Reproduced with permission from ref. 90. Copyright 2015, Wiley VCH. | ||
Despite the significantly enhanced degradation ability caused by the incorporated lithium alkoxide moieties, the material was still air-sensitive and could not be utilised for real-life systems. Recently, Navarro et al.91 investigated the substitution of the Zr6O4(OH)4 SBUs of NU-1000, MOF-808, and UiO-66 with MgZr5O4(OH)4, which involved a basic magnesium alkoxide and exhibited good catalytic activity and excellent sustainability. For this process, [Mg(OMe)2(MeOH)2]4 was used as Mg precursor and replaced one Zr site for each SBU of NU-1000 and MOF-808 with Mg(OMe)2 (Fig. 15). UiO-66 did not undergo cation exchange because its pores could not accommodate the bulky Mg complex. MOF-808@Mg(OMe)2 exhibited significantly high degradation performance for DIFP (TOF = 0.26 min−1) compared with pristine MOF-808 (TOF = 0.01 min−1). In addition, this compound could retain its activity for 20 days, while the activity of UiO-66@LiOtBu was significantly decreased within a month. That was attributed to the incorporation of Mg(OMe)2 in the centre of the SBUs, whereas LiOtBu was located at the periphery of SBUs. NU-1000@Mg(OMe)2 also exhibited good catalytic activity, but lower than that of MOF-808, because the nerve agents could access the Mg-decorated mesopores and non-decorated micropores of NU-1000, which led to the lower exposure of the agents to catalytic Mg2+ sites, as explained using the Monte Carlo simulation.
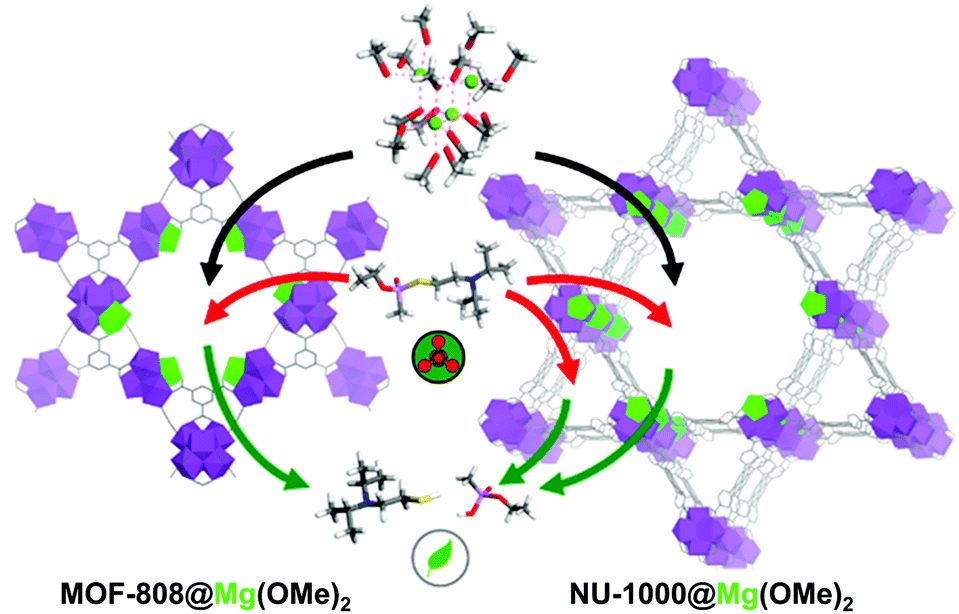 | ||
| Fig. 15 Schematic representation of post-synthetic modification of metal–organic framework mesopores with [Mg(OMe)2(MeOH)2]4 to yield MgZr5O2(OH)6 clusters (Mg is highlighted in green) and ulterior detoxification of nerve agents that took place in the pore structure of MOF-808@Mg(OMe)2 and NU-1000@Mg(OMe)2. Reproduced with permission from ref. 91. Copyright 2019, American Chemical Society. | ||
3.4 Electron conductivity
Low electron conductivity is one of the drawbacks of MOFs, and thus it is a hindrance for the use of MOFs for many practical applications. The intrinsic design and synthesis of conductive MOFs are very challenging because MOFs present low atomic density and are usually constructed via the coordination between hard metal ions and redox-inactive organic ligands. This brings about significant differences in orbital energy between bonding atoms and reduces orbital overlap, which leads to the charge carriers being energetically trapped on lattice sites. Consequently, typical MOFs present little or no band dispersion and behave as insulators. To construct electrically conductive MOFs by closing the electronic band gap, two basic strategies can be used: the use of redox-active components in de novo synthesis processes and doping or introducing defects that could increase the electron conductivity of the MOFs. Using the second strategy, one can move the Fermi level of the structures to increase the charge carrier concentration in the band gap. Nonetheless, the goal is to facilitate charge transport via the lattice sites of MOFs and to lower their band gap so they can conduct electricity.92,93In 2014, Allendorf et al.94 reported engendering and controlling the conductivity of HKUST-1 by introducing redox-active 7,7,8,8-tetracyanoquinodimethane (TCNQ) molecules to its OMSs. As depicted in Fig. 16, the TCNQ molecules bridged two Cu paddlewheel SBUs; moreover, ab initio calculations revealed that TCNQ could strongly bind to the MOF (binding energy of 83.9 kJ mol−1) and insert unoccupied molecular orbitals into the band gap of the MOF, thus creating new charge transfer paths and enabling electronic coupling between the HKUST-1 framework and TCNQ. The importance of guest–host interactions was further demonstrated by replacing TCNQ with its fully hydrogenated form, (cyclohexane-1,4-diylidene)dimalononitrile (H4-TCNQ), which lacked a conjugated π electron network. Consequently, the electron conductivity of the obtained MOF was poor. Conversely, when 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ), which presented similar band gap with, but higher electron affinity then TCNQ, was introduced to the OMSs of HKUST-1, the obtained MOF presented lower conductivity than HKUST-1, because the high electron affinity of F4-TCNQ inhibited electron mobility. Volkmer et al.92 used the cation exchange method and reported the modulation of the band gap and electron conductivity of MFU-4. Exchanging the octahedral Zn sites of the SBUs with Co2+ cations (i.e., Co-MFU-4), the highest occupied molecular orbital energy state of Co-MFU-4 was no different than that of MFU-4 (−6.14 eV) as it originated from the ligand, while the insertion of the partially occupied d-orbitals of CO2+ generated bands below the lowest unoccupied molecular orbital energy state of the ligand, and subsequently decreased the band gap.
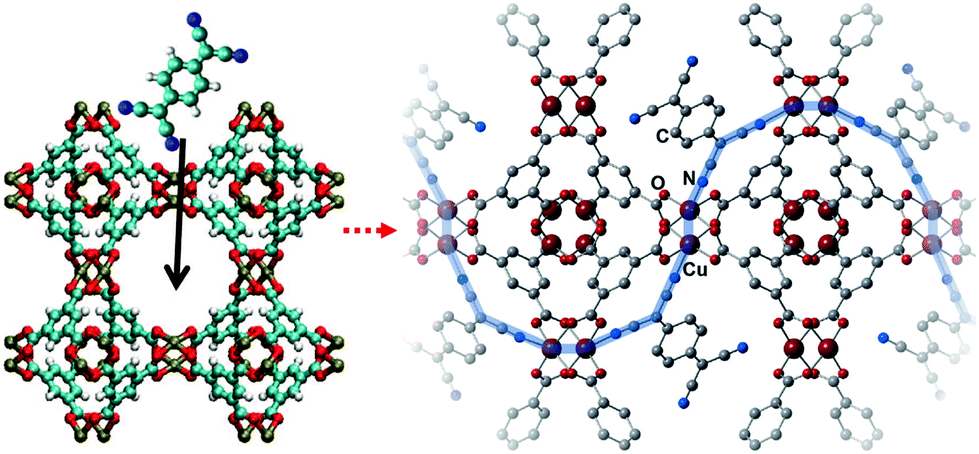 | ||
| Fig. 16 Addition of 7,7,8,8-tetracyanoquinododimethane molecules to HKUST-1 and suggested electron pathway through the framework. Reproduced with permission from ref. 94 and 93. Copyright 2014, American Association for the Advancement of Science, and Copyright 2016, Wiley VCH. | ||
3.5 Proton conductivity
Many studies on proton conducting MOFs have mostly focused on the introduction of guest molecules/ions and the incorporation of functional groups. In particular, the guests accommodated in pores, such as water and imidazole, could act as proton source, and provide proton transfer pathways via hydrogen bonding.95 The proton conductivity of MOFs could be significantly increased by incorporating guest molecules and also by decorating SBUs with proper functional molecules. Hupp et al.96 investigated the relationship between the proton conductivity and coordination environment at the SBUs of HKUST-1. The OMSs of HKUST-1 were initially coordinated with synthetic solvent molecules, such as H2O and ethanol (EtOH), and could be substituted with other coordinating solvent molecules, such as methanol (MeOH) and MeCN. As depicted in Fig. 17a. The MOF that contained H2O-coordinated Cu2+ and MeOH molecules in the pores exhibited much larger proton conductivity (15 μS cm−1) than the MOF that contained only pristine MeOH in the pores (0.17 μS cm−1). In contrast, EtOH- or MeCN-coordinated Cu2+ moieties did not present any increase in proton conductivity (0.2 μS cm−1). This result was attributed to MeOH molecules being protonated by coordinating H2O molecules, and the increased concentration of protonated MeOH increased the proton conductivity of the MOF. Conversely, the MeCN-coordinated compound lacked dissociable protons for MeOH, and therefore, it could not improve the proton conductivity of the target. Because the acidity of EtOH is slightly lower than that of MeOH (pKa of 15.9 and 15.5, respectively), the increase in conductivity of the EtOH-coordinated HKUST-1 should also be limited. Such functional groups modified the pore environment and generated an abundance of carboxyl groups by substituting the detachable formate anions grafted to the SBUs.97 The resultant MOF-808-OX exhibited higher proton conductivity (1.94 × 10−4 S cm−1) than pristine MOF-808 (1.25 × 10−6 S cm−1).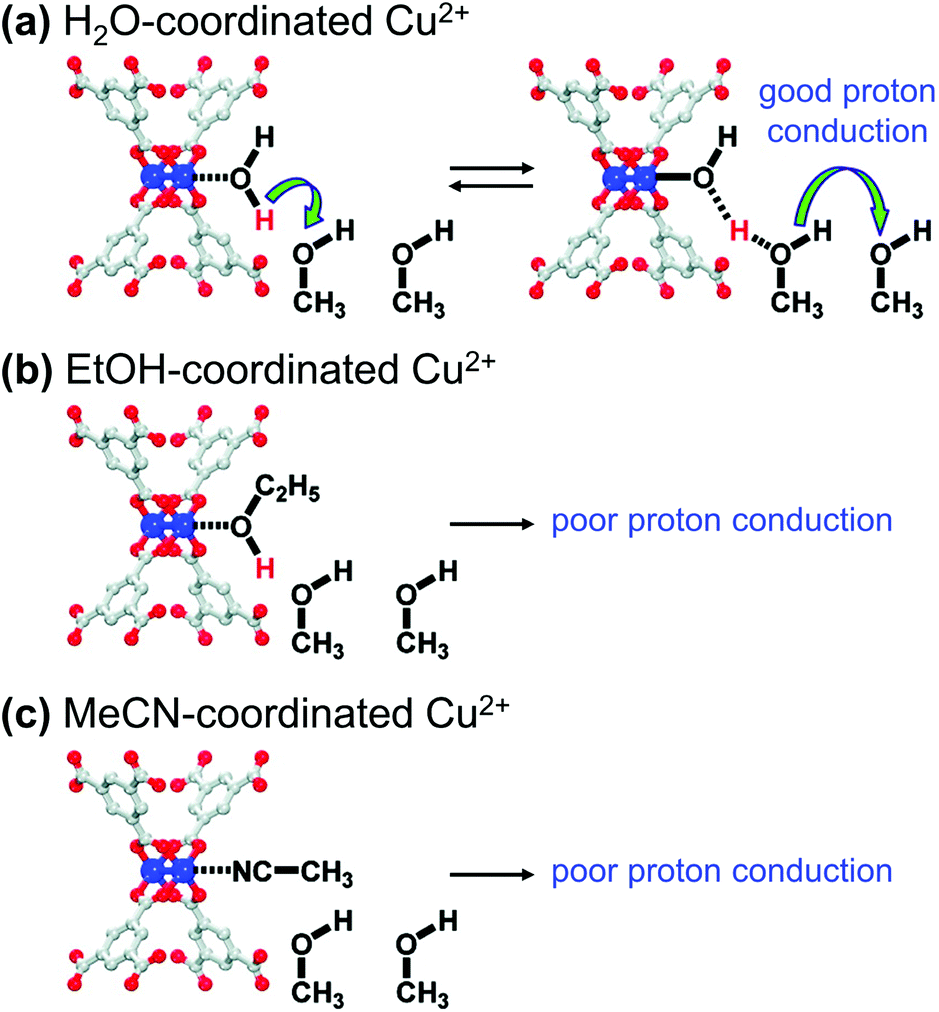 | ||
| Fig. 17 Qualitative representations of proton transfer from Cu2+ sites coordinated with (a) H2O, (b) ethanol (EtOH), and (c) acetonitrile (MeCN). Reproduced with permission from ref. 96. Copyright 2012, American Chemical Society. | ||
3.6 Sensing
Chemical sensing of metal ions in MOFs usually occurs via coordinative ligand or is induced by guest molecules. If MOF cation exchange could occur in fast and easily detectable fashion, it can be exploited for the sensing of metal cations.31 Yan et al.98 utilised MIL-53(Al) to “turn-off” the detection of Fe3+, as this MOF quenched the fluorescence emission that resulted from the ligand-to-metal charge transfer from the BDC moieties to the Al3+ cations. Of various aqueous cation solutions, including K+, Na+, Ca2+, Mg2+, Cu2+, Zn2+, Co2+, Mn2+, Ni2+, Hg2+, Cd2+, Pb2+, Fe2+, Fe3+, Cr3+, and Al3+, only Fe3+ presented quenching effect for MIL-53(Al) within the 3–200 μM concentration range, and thus, MIL-53(Al) could be utilised for selective Fe3+ sensing. In 2018, Zhong et al.99 adopted ethylenediaminetetraacetic acid (EDTA) as decorating functional group with many coordination sites (four hard carboxyl groups and two softer tertiary amine groups) and MOF-808, which consisted of Zr6O4(OH)4 SBUs and BTC linkers and presented large cavities and high chemical stability, as support material.92 The detachable formate anions on the SBUs of the MOF were substituted with the carboxylate groups of the EDTA molecules to produce MOF-808-EDTA. Given the five remaining coordination sites of EDTA, MOF-808-EDTA presented excellent performance (>99%) for capturing a wide range of heavy metal ions, such as La3+, Hg2+, and Pb2+ from polluted water sources.4. Conclusions
Owing to the modular properties of MOFs attributed to the diverse combination of inorganic species and organic linkers, many researches have focused on synthesising new structures for new functions. To overcome the limits of conventional direct synthesis, the PSM approach led to facile methods for producing desired materials. In this review, we provided recent representative examples of using the PSM approach on the SBUs of MOFs and divided the synthesis strategies into four categories: substitution of cations, redox reactions of metal centres, generation of defects, and incorporation of new species in the SBUs of MOFs. In addition, we discussed the modulation of the MOF functions owing to these simple alteration strategies as well as some interesting applications induced using the PSM approach on SBUs, such as sorption, separation, catalysis including detoxification, electron conductivity, and sensing. Unprecedented MOFs, which could not be obtained using conventional synthesis methods could be readily constructed via the PSM alteration of SBUs; moreover, small alterations of SBUs significantly affected the properties of the entire MOF. These simple but powerful tools present great potential for constructing MOFs for applications that have not been widely investigated yet. Moreover, it is believed that more breakthrough applications for MOFs will be uncovered in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Science and ICP (No. NRF-2016R1A5A1009405, NRF-2017R1A2B4008757, and NRF-2019R1A6A3A01096867).References
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, CrystEngComm, 2012, 14, 3001–3004 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- K. E. Cordova and O. M. Yaghi, Mater. Chem. Front., 2017, 1, 1304–1309 RSC.
- F.-L. Li, Q. Shao, X. Huang and J.-P. Lang, Angew. Chem., Int. Ed., 2018, 57, 1888–1892 CrossRef CAS.
- F.-L. Li, P. Wang, X. Huang, D. J. Young, H.-F. Wang, P. Braunstein and J.-P. Lang, Angew. Chem., 2019, 131, 7125–7130 CrossRef.
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1995, 34, 2127–2129 CrossRef CAS.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS.
- Z. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315–1329 RSC.
- L. Sarkisov, R. L. Martin, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2014, 136, 2228–2231 CrossRef CAS.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- L. Wang, C. Wang, Y. Sun, K. Shi, S. Deng and H. Lu, Mater. Chem. Phys., 2016, 175, 138–145 CrossRef CAS.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822, 10.1039/B704325B.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS.
- J. Gao, J. Miao, P.-Z. Li, W. Y. Teng, L. Yang, Y. Zhao, B. Liu and Q. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC.
- S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS.
- V. M. Goldschmidt, J. Chem. Soc., 1937, 655–673 RSC.
- K. S. Hagen, D. W. Stephan and R. H. Holm, Inorg. Chem., 1982, 21, 3928–3936 CrossRef CAS.
- H. Li and J. D. Otvos, Biochemistry, 1996, 35, 13929–13936 CrossRef CAS.
- A. Putnis, Mineral. Mag., 2002, 66, 689–708 CrossRef CAS.
- V. Autissier and R. A. Henderson, Inorg. Chem., 2008, 47, 6393–6403 CrossRef CAS.
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361 CrossRef CAS.
- K. C. Misra, Introduction to geochemistry : principles and applications, Wiley-Blackwell, Chichester, West Sussex, Hoboken, NJ, 2012 Search PubMed.
- J. B. Rivest and P. K. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- H. Wang, W. Meng, J. Wu, J. Ding, H. Hou and Y. Fan, Coord. Chem. Rev., 2016, 307, 130–146 CrossRef CAS.
- R. E. Morris and L. Brammer, Chem. Soc. Rev., 2017, 46, 5444–5462 RSC.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS.
- S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501 CrossRef.
- M. Dincă and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef CAS.
- T.-F. Liu, L. Zou, D. Feng, Y.-P. Chen, S. Fordham, X. Wang, Y. Liu and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 7813–7816 CrossRef CAS PubMed.
- D. Song, J. Bae, H. Ji, M.-B. Kim, Y.-S. Bae, K. S. Park, D. Moon and N. C. Jeong, J. Am. Chem. Soc., 2019, 141, 7853–7864 CrossRef CAS.
- P.-Q. Liao, X.-Y. Li, J. Bai, C.-T. He, D.-D. Zhou, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Chem. – Eur. J., 2014, 20, 11303–11307 CrossRef CAS.
- H. R. Moon, J. H. Kim and M. P. Suh, Angew. Chem., Int. Ed., 2005, 44, 1261–1265 CrossRef CAS.
- S.-C. Qi, X.-Y. Qian, Q.-X. He, K.-J. Miao, Y. Jiang, P. Tan, X.-Q. Liu and L.-B. Sun, Angew. Chem., 2019, 58, 10104–10109 CrossRef CAS.
- Y.-G. Huang, B. Mu, P. M. Schoenecker, C. G. Carson, J. R. Karra, Y. Cai and K. S. Walton, Angew. Chem., Int. Ed., 2011, 50, 436–440 CrossRef CAS.
- C. Zhang, M. Wang, L. Liu, X. Yang and X. Xu, Electrochem. Commun., 2013, 33, 131–134 CrossRef CAS.
- H. Leclerc, T. Devic, S. Devautour-Vinot, P. Bazin, N. Audebrand, G. Férey, M. Daturi, A. Vimont and G. Clet, J. Phys. Chem. C, 2011, 115, 19828–19840 CrossRef CAS.
- Z. Lu, H. G. W. Godfrey, I. da Silva, Y. Cheng, M. Savage, F. Tuna, E. J. L. McInnes, S. J. Teat, K. J. Gagnon, M. D. Frogley, P. Manuel, S. Rudić, A. J. Ramirez-Cuesta, T. L. Easun, S. Yang and M. Schröder, Nat. Commun., 2017, 8, 14212 CrossRef CAS.
- Y. Xiong, S. Chen, F. Ye, L. Su, C. Zhang, S. Shen and S. Zhao, Chem. Commun., 2015, 51, 4635–4638 RSC.
- M. Märcz, R. E. Johnsen, P. D. C. Dietzel and H. Fjellvåg, Microporous Mesoporous Mater., 2012, 157, 62–74 CrossRef.
- J. W. Yoon, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, H. Leclerc, S. Wuttke, P. Bazin, A. Vimont, M. Daturi, E. Bloch, P. L. Llewellyn, C. Serre, P. Horcajada, J.-M. Grenèche, A. E. Rodrigues and G. Férey, Angew. Chem., Int. Ed., 2010, 49, 5949–5952 CrossRef CAS.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- K. Li and D. Xue, J. Phys. Chem. A, 2006, 110, 11332–11337 CrossRef CAS.
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS.
- J. Tu, X. Zeng, F. Xu, X. Wu, Y. Tian, X. Hou and Z. Long, Chem. Commun., 2017, 53, 3361–3364 RSC.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS.
- H.-L. Jiang, D. Feng, T.-F. Liu, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 14690–14693 CrossRef CAS.
- P. Capper, Bulk Crystal Growth of Electronic, Optical and Optoelectronic Materials, Wiley, New York, 2005 Search PubMed.
- B. Tu, Q. Pang, D. Wu, Y. Song, L. Weng and Q. Li, J. Am. Chem. Soc., 2014, 136, 14465–14471 CrossRef CAS.
- S. Gadipelli and Z. Guo, Chem. Mater., 2014, 26, 6333–6338 CrossRef CAS.
- B. Bueken, N. Van Velthoven, A. Krajnc, S. Smolders, F. Taulelle, C. Mellot-Draznieks, G. Mali, T. D. Bennett and D. De Vos, Chem. Mater., 2017, 29, 10478–10486 CrossRef CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- F. Vermoortele, R. Ameloot, L. Alaerts, R. Matthessen, B. Carlier, E. V. R. Fernandez, J. Gascon, F. Kapteijn and D. E. De Vos, J. Mater. Chem., 2012, 22, 10313–10321 RSC.
- G.-Y. Jeong, A. K. Singh, M.-G. Kim, K.-W. Gyak, U. Ryu, K. M. Choi and D.-P. Kim, Nat. Commun., 2018, 9, 3968 CrossRef.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS.
- Y.-S. Wei, M. Zhang, P.-Q. Liao, R.-B. Lin, T.-Y. Li, G. Shao, J.-P. Zhang and X.-M. Chen, Nat. Commun., 2015, 6, 8348 CrossRef CAS.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135, 16801–16804 CrossRef CAS.
- S. Yuan, W. Lu, Y.-P. Chen, Q. Zhang, T.-F. Liu, D. Feng, X. Wang, J. Qin and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 3177–3180 CrossRef CAS.
- S. Yuan, Y.-P. Chen, J.-S. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 8912–8919 CrossRef CAS.
- M. Meilikhov, K. Yusenko and R. A. Fischer, J. Am. Chem. Soc., 2009, 131, 9644–9645 CrossRef CAS PubMed.
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS.
- Z. Zhang, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 5982–5985 CrossRef CAS PubMed.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303 CrossRef CAS.
- R. L. Siegelman, P. J. Milner, A. C. Forse, J.-H. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS.
- R.-B. Lin, L. Li, H.-L. Zhou, H. Wu, C. He, S. Li, R. Krishna, J. Li, W. Zhou and B. Chen, Nat. Mater., 2018, 17, 1128–1133 CrossRef CAS.
- J. J. M. Beenakker, V. D. Borman and S. Y. Krylov, Chem. Phys. Lett., 1995, 232, 379–382 CrossRef CAS.
- H. Oh, K. S. Park, S. B. Kalidindi, R. A. Fischer and M. Hirscher, J. Mater. Chem. A, 2013, 1, 3244–3248 RSC.
- S. A. FitzGerald, C. J. Pierce, J. L. C. Rowsell, E. D. Bloch and J. A. Mason, J. Am. Chem. Soc., 2013, 135, 9458–9464 CrossRef CAS PubMed.
- J. Y. Kim, H. Oh and H. R. Moon, Adv. Mater., 2019, 31, 1970147 CrossRef.
- I. Weinrauch, I. Savchenko, D. Denysenko, S. M. Souliou, H. H. Kim, M. Le Tacon, L. L. Daemen, Y. Cheng, A. Mavrandonakis, A. J. Ramirez-Cuesta, D. Volkmer, G. Schütz, M. Hirscher and T. Heine, Nat. Commun., 2017, 8, 14496 CrossRef CAS.
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., Int. Ed., 2014, 53, 5832–5836 CrossRef CAS.
- J. Y. Kim, R. Balderas-Xicohténcatl, L. Zhang, S. G. Kang, M. Hirscher, H. Oh and H. R. Moon, J. Am. Chem. Soc., 2017, 139, 15135–15141 CrossRef CAS.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS.
- J. Zheng, J. Ye, M. A. Ortuño, J. L. Fulton, O. Y. Gutiérrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef PubMed.
- E. López-Maya, C. Montoro, L. M. Rodríguez-Albelo, S. D. Aznar Cervantes, A. A. Lozano-Pérez, J. L. Cenís, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2015, 54, 6790–6794 CrossRef PubMed.
- R. Gil-San-Millan, E. López-Maya, A. E. Platero-Prats, V. Torres-Pérez, P. Delgado, A. W. Augustyniak, M. K. Kim, H. W. Lee, S. G. Ryu and J. A. R. Navarro, J. Am. Chem. Soc., 2019, 141, 11801–11805 CrossRef CAS PubMed.
- P. Sippel, D. Denysenko, A. Loidl, P. Lunkenheimer, G. Sastre and D. Volkmer, Adv. Funct. Mater., 2014, 24, 3885–3896 CrossRef CAS.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS PubMed.
- A.-L. Li, Q. Gao, J. Xu and X.-H. Bu, Coord. Chem. Rev., 2017, 344, 54–82 CrossRef CAS.
- N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CrossRef CAS PubMed.
- X. Meng, H.-N. Wang, L.-S. Wang, Y.-H. Zou and Z.-Y. Zhou, CrystEngComm, 2019, 21, 3146–3150 RSC.
- C.-X. Yang, H.-B. Ren and X.-P. Yan, Anal. Chem., 2013, 85, 7441–7446 CrossRef CAS PubMed.
- Y. Peng, H. Huang, Y. Zhang, C. Kang, S. Chen, L. Song, D. Liu and C. Zhong, Nat. Commun., 2018, 9, 187 CrossRef PubMed.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed.
- K. Son, J. Y. Kim, G. Schütz, S. G. Kang, H. R. Moon and H. Oh, Inorg. Chem., 2019, 58, 8895–8899 CrossRef CAS PubMed.
- M. J. Ingleson, R. Heck, J. A. Gould and M. J. Rosseinsky, Inorg. Chem., 2009, 48, 9986–9988 CrossRef CAS.
- M. Banerjee, S. Das, M. Yoon, H. J. Choi, M. H. Hyun, S. M. Park, G. Seo and K. Kim, J. Am. Chem. Soc., 2009, 131, 7524–7525 CrossRef CAS.
- Y. K. Hwang, D.-Y. Hong, J.-S. Chang, S. H. Jhung, Y.-K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS.
- J. M. Fernández-Morales, L. A. Lozano, E. Castillejos-López, I. Rodríguez-Ramos, A. Guerrero-Ruiz and J. M. Zamaro, Microporous Mesoporous Mater., 2019, 290, 109686 CrossRef.
- D. Sun, P. R. Adiyala, S.-J. Yim and D.-P. Kim, Angew. Chem., 2019, 131, 7483–7487 CrossRef.
- A. Atilgan, T. Islamoglu, A. J. Howarth, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 24555–24560 CrossRef CAS.
- P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC.
- P. Liu, E. Redekop, X. Gao, W.-C. Liu, U. Olsbye and G. A. Somorjai, J. Am. Chem. Soc., 2019, 141, 11557–11564 CrossRef CAS PubMed.
- C. A. Trickett, T. M. O. Popp, J. Su, C. Yan, J. Weisberg, A. Huq, P. Urban, J. Jiang, M. J. Kalmutzki, Q. Liu, J. Baek, M. P. Head-Gordon, G. A. Somorjai, J. A. Reimer and O. M. Yaghi, Nat. Chem., 2019, 11, 170–176 CrossRef CAS PubMed.
- P. Ji, X. Feng, P. Oliveres, Z. Li, A. Murakami, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 14878–14888 CrossRef CAS PubMed.
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- M. Rimoldi, A. Nakamura, N. A. Vermeulen, J. J. Henkelis, A. K. Blackburn, J. T. Hupp, J. F. Stoddart and O. K. Farha, Chem. Sci., 2016, 7, 4980–4984 RSC.
- I. S. Kim, J. Borycz, A. E. Platero-Prats, S. Tussupbayev, T. C. Wang, O. K. Farha, J. T. Hupp, L. Gagliardi, K. W. Chapman, C. J. Cramer and A. B. F. Martinson, Chem. Mater., 2015, 27, 4772–4778 CrossRef CAS.
- X. Wang, X. Zhang, P. Li, K.-i. Otake, Y. Cui, J. Lyu, M. D. Krzyaniak, Y. Zhang, Z. Li, J. Liu, C. T. Buru, T. Islamoglu, M. R. Wasielewski, Z. Li and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 8306–8314 CAS.
- J. G. Santaclara, A. I. Olivos-Suarez, A. Gonzalez-Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS.
- K. Manna, P. Ji, Z. Lin, F. X. Greene, A. Urban, N. C. Thacker and W. Lin, Nat. Commun., 2016, 7, 12610 CrossRef CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- P. Ji, K. Manna, Z. Lin, A. Urban, F. X. Greene, G. Lan and W. Lin, J. Am. Chem. Soc., 2016, 138, 12234–12242 CrossRef CAS.
- P. Ji, Y. Song, T. Drake, S. S. Veroneau, Z. Lin, X. Pan and W. Lin, J. Am. Chem. Soc., 2018, 140, 433–440 CrossRef CAS.
- K. Manna, P. Ji, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2016, 138, 7488–7491 CrossRef CAS.
- P. Ji, K. Manna, Z. Lin, X. Feng, A. Urban, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 7004–7011 CrossRef CAS.
- S. Yuan, Y.-P. Chen, J. Qin, W. Lu, X. Wang, Q. Zhang, M. Bosch, T.-F. Liu, X. Lian and H.-C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 14696–14700 CrossRef CAS.
- N. Van Velthoven, S. Waitschat, S. M. Chavan, P. Liu, S. Smolders, J. Vercammen, B. Bueken, S. Bals, K. P. Lillerud, N. Stock and D. E. De Vos, Chem. Sci., 2019, 10, 3616–3622 RSC.
- K.-i. Otake, Y. Cui, C. T. Buru, Z. Li, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 8652–8656 CrossRef CAS.
- Y. Song, Z. Li, P. Ji, M. Kaufmann, X. Feng, J. S. Chen, C. Wang and W. Lin, ACS Catal., 2019, 9, 1578–1583 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |