
Alterations to secondary building units of metal–organic frameworks for the development of new functions

Abstract
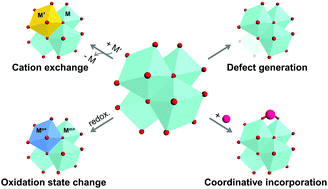
Secondary building units (SBUs) are the key components of metal–organic frameworks (MOFs) that help to build potentially porous periodic networks by linking multitopic organic ligands. Hence, metal SBUs are critical for determining the underlying topology of MOFs. Moreover, SBUs are the main MOF research topic nowadays, because of the simplicity of their synthesis, diverse directionality and their ability to easily harness open metal sites, compared to that of primary building units (comprising mononuclear metal centres) or tertiary building units (metal–organic polyhedra). Therefore, post-synthetic approaches for altering SBUs do not only include developing techniques for controlling the properties of MOFs but also involve a more in-depth understanding of their structure–function relationships from the materials science and engineering perspective. The SBU-related reviews published to date have successfully introduced and organised the chemistry of SBUs in MOFs. Because many recent studies have explored more diverse methods, such as metal exchange, oxidation-state transformation, defect generation, and incorporation of other species, in this review, we mainly focus on the recently developed methods for SBU alteration by classifying them into four groups and elaborate on how unique structures and properties can be achieved using those methods.
 Please wait while we load your content...
Please wait while we load your content...

