
Substrate templated synthesis of single-phase and uniform Zr-porphyrin-based metal–organic frameworks†
Seong Won
Hong
 a,
Ju Won
Paik
ab,
Dongju
Seo
a,
Jae-Min
Oh
*c,
Young Kyu
Jeong
*b and
Jin Kuen
Park
*a
a,
Ju Won
Paik
ab,
Dongju
Seo
a,
Jae-Min
Oh
*c,
Young Kyu
Jeong
*b and
Jin Kuen
Park
*a
aDepartment of Chemistry, Hankuk University of Foreign Studies, Yongin, 17035, Gyeonggi-do, Republic of Korea. E-mail: jinkpark@hufs.ac.kr
bKorea Institute of Industrial Technology (KITECH), 137-41 Gwahakdanji-ro, Gangneung-si 25440, Republic of Korea. E-mail: immrc80@gmail.com
cDepartment of Energy and Materials Engineering, Dongguk University-Seoul, Seoul 04620, Republic of Korea. E-mail: jaemin.oh@dongguk.edu
First published on 24th October 2019
Abstract
In the synthesis of highly applicable metal–organic frameworks (MOFs), obtaining pure phases poses a great challenge since some MOFs have diverse phases with the same metal sources and ligands. Among such MOFs, PCN-223 and PCN-221 can easily be formed together in a one-pot reaction, making it hard to separate them in a single phase. It is known that some PCN series have a kinetic relationship in the early stages of their formation mechanisms. Thus, we have dedicated our efforts to finding the kinetic relationship between PCN-223 and PCN-221 by utilizing the chemical bath deposition (CBD) method to establish reaction parameters for their pure phases. We found that one out of the two phases can be transformed into the other phase by varying the Zr precursors, water amount, modulators, substrates, and reaction time. Furthermore, the pure phase of PCN-221 with a distinct open-pore channel of 100 nm, which has not been obtained previously, can be synthesized through the CBD method. In this work, we successfully demonstrate that the CBD method is a versatile method for synthesizing phase-pure and uniform MOFs by controlling their nucleation stage and pore structures.
Introduction
Metal–organic frameworks (MOFs) are low-density porous materials consisting of infinite networks formed by dative bonds between metals and organic ligands. They have stimulated interest for various applications such as gas storage,1,2 separation,3,4 purification5,6 and heterogeneous catalysts7–10 due to their unique and varied pore structures based on the choice of ligands and metals.11 However, due to the nature of dative bonds, some MOFs are vulnerable to hydrolysis, which limits their industrial applications. Therefore, many research groups have dedicated their efforts to developing water-stable MOFs, so numerous water-stable structures have been reported and some important criteria have been established for water-stable MOFs.12 Among the various water-stable MOFs, Zr4+-based MOFs such as UiO-66,13 the PCN family,14 NU-1000,15 and NU-1105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 have demonstrated exceptional hydro-stability even in highly acidic or basic conditions. These MOFs comprise Zr clusters as secondary building units (SBU) and carboxylate-functionalized ligands. Interestingly, one kind of ligand such as porphyrins can form diverse pore structures and framework topologies with Zr clusters, depending on the synthetic conditions because the phases and connectivity of Zr clusters vary with changing synthetic conditions.17 Due to the structural diversity of Zr cluster-based MOFs (Zr-MOFs) with porphyrin ligands, two or more crystalline phases are often produced as a mixture in one-pot reactions, which cannot be easily separated.18–20 Such phenomena cause serious problems in their applications because the properties of MOFs are highly related to their purity in terms of the crystalline phase and topologies.
16 have demonstrated exceptional hydro-stability even in highly acidic or basic conditions. These MOFs comprise Zr clusters as secondary building units (SBU) and carboxylate-functionalized ligands. Interestingly, one kind of ligand such as porphyrins can form diverse pore structures and framework topologies with Zr clusters, depending on the synthetic conditions because the phases and connectivity of Zr clusters vary with changing synthetic conditions.17 Due to the structural diversity of Zr cluster-based MOFs (Zr-MOFs) with porphyrin ligands, two or more crystalline phases are often produced as a mixture in one-pot reactions, which cannot be easily separated.18–20 Such phenomena cause serious problems in their applications because the properties of MOFs are highly related to their purity in terms of the crystalline phase and topologies.
Zhou and co-workers have reported pioneering works to address this issue, demonstrating that the phases and topologies of Zr-MOFs with porphyrin ligands can be kinetically controlled.21 Therefore, they revealed that the addition of nucleation seed crystals for desirable Zr-MOF phases with porphyrin ligands to solutions could successfully lead to pure phases of target MOFs because the seed could kinetically trigger the formation of corresponding MOFs by passing over their nucleation processes in the early stages of their growth mechanisms. Inspired by the work of Zhou et al.,22 in this study, we utilized the adsorption of chemical species on heterogeneous surfaces at the nucleation stage instead of adding seed MOFs. The controlled adsorption of the starting reactants, even on non-specific surfaces, would facilitate the formation of seed, resulting in the formation of the desired Zr-MOF with porphyrins.
Chemical bath deposition (CBD) has been widely utilized in the fabrication of various semiconductor and metal oxide thin films from aqueous solutions because it is an economical and simple method.23 With CBD, the size, morphology, and orientation of crystals grown on substrates can be strongly affected by specific experimental conditions and surface properties of substrates with various chemical speciation processes in the earlier stages of the growth mechanism on the surface.24 In general, such speciation can be modulated via condensation and/or hydrolysis reactions of the metal complexes and/or metal ions according to different interactions and concentrations of each active species on the surfaces of the substrates.21 Since the nucleation of MOFs can also be controlled by the same type of reactions between chemical species in solution, it implies that desirable MOF phases are obtainable and separable with the CBD method.
In this study, we disclose that PCN-221 and PCN-223 tend to grow in a mixed phase with irregular shapes and sizes when they are synthesized with ZrOCl2·8H2O in a conventional solvothermal reaction. Therefore, to synthesize uniform and single phases of PCN-221 and PCN-223, we adopted the CBD method with two different substrates (mica films and silicon wafers) without removing the native silicon oxide layers on the top of the wafers. We focused on optimizing the kinetic conditions upon changing metal sources such as ZrOCl2·8H2O and ZrCl4, water contents, the amount or kind of modulators and reaction time with the goal of demonstrating how such factors could affect the nucleation and growth of MOFs. In general, temperature is one of the key factors for the kinetic control of MOFs19 but in this contribution, we did not attempt to gauge the temperature effect since, to the best of our knowledge, PCN-221 and PCN-223 phases can be mixed even under the same temperature. Mica is known to have exposed aluminosilicate crystalline silica surfaces with honeycombed arrays of silicates and hydroxyl groups in the center of honeycombed arrays; thus, mica surfaces can absorb water molecules.25 The silicon wafers with native oxide layers are known to have amorphous silica surfaces with irregular arrangements of hydroxide groups that could simulate the surfaces of glassware for the conventional solvothermal reaction for MOF synthesis. Hence, we can also demonstrate the physicochemical relationship between the surface properties of substrates and the nucleation/growth of MOFs by utilizing two kinds of films for CBD.
Results and discussion
PCN-221 and PCN-223 were synthesized with ZrOCl2·8H2O and porphyrin ligands in a conventional solvothermal reaction by varying the amount and kind of modulators such as acetic acid and benzoic acid and adopting specific reaction conditions for each MOF with ZrCl4. PCN-223 was synthesized with benzoic acid as the modulator at 100 °C for 12 hours based on the published method,26 except that ZrOCl2·8H2O was used instead of ZrCl4. The powder X-ray diffraction (P-XRD) pattern revealed that the resulting product had mixed phases of PCN-221 and 223 as shown in Fig. 1a (red-line). Conversely, P-XRD confirmed that the PCN-223 phase was obtained with ZrOCl2·8H2O by following the published method,27 which used ZrCl4 (acetic acid at 120 °C for 12 hours) as shown in the blue-line of Fig. 1a. Therefore, it was thought that the dynamic equilibrium, e.g., the association and dissociation reactions during nucleation processes, for each MOF could be varied depending on the modulators and/or the amount of water molecules in the precursors for this homogeneous nucleation system with ZrOCl2·8H2O. To obtain additional evidence for the phase of each MOF, the morphologies of the products shown in Fig. 1a (blue and red lines) were monitored by using scanning electron microscopy (SEM). Mixed morphologies such as rods and globular features are clearly shown in Fig. 1b while only globular shapes were observed in Fig. 1c, consistent with the results of P-XRD analysis. Since PCN-221 and PCN-223 have different numbers of Zr atoms in their clusters (Zr8 and Zr6, respectively), with 12 connectivity for both as shown in Fig. 1d and e, PCN-221 and PCN-223 should have different three-dimensional networks of porphyrin ligands and pore structures as displayed in Fig. 1f and g.26,27 In general, the acidity of the modulator depends on the water content. For example, acetic acid (pKa = 4.8) is slightly less acidic than benzoic acid (pKa = 4.2) in water. However, the pKa of acetic acid (pKa = 12.6) is much greater than that of benzoic acid (pKa = 11.0) under dimethylsulfoxide conditions.28,29 By comparing the literature results using ZrCl4 for the synthesis of PCN-221 and PCN-223, pKa values of the modulators would be different when using ZrOCl2·8H2O due to the different water contents. Therefore, the condensation and hydrolysis reactions of metal complexes could be thermodynamically and or kinetically affected by the water content in the metal source. Although we could not precisely calculate the pKa values of the modulators or carboxylates in porphyrins in the reaction system with ZrOCl2·8H2O, the difference in pKa between modulators and carboxyl acid groups in porphyrin ligands upon changing the water content seems to be an important factor in controlling the dynamic equilibrium between PCN-221 and PCN-223.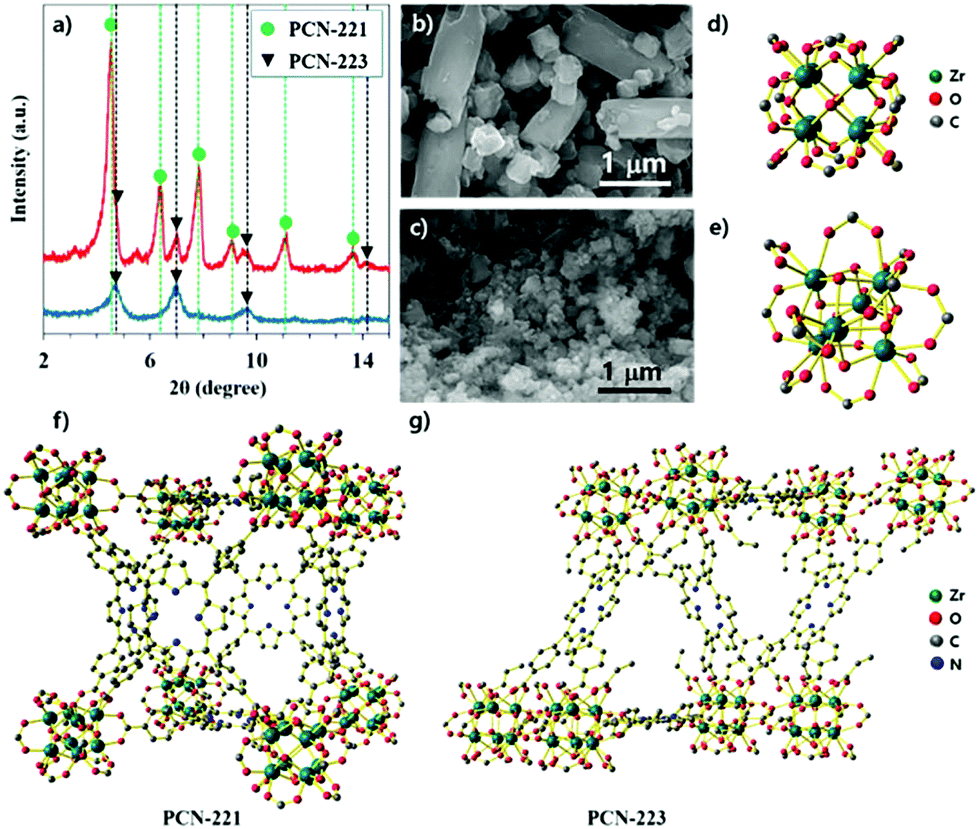 | ||
| Fig. 1 (a) Powder X-ray diffraction patterns of as-synthesized MOFs with ZrOCl2·8H2O by adopting procedures with ZrCl4, (b) images of scanning electron microscope for the mixture of PCN-221 and PCN-223 corresponding to the red-line pattern in (a), (c) images of scanning electron microscope for the mixture of PCN-223 corresponding the blue-line pattern in (a), (d) chemical structure of Zr8 cluster for PCN-221, (d) chemical structure of Zr6 cluster for PCN-223, (f) schematic illustration of networking fashion for PCN-221 with a combination of Zr8 clusters and porphyrin ligands, (g) schematic illustration of networking fashion for PCN-223 with a combination of Zr6 clusters and porphyrin ligands. | ||
To demonstrate the effect of water on the growth of the PCN series atop the silica surface, the morphologies of the resulting particles from the precursor solution of ZrCl4 with porphyrins and benzoic acid (250 mg) upon purposely adding water were investigated via SEM as shown in Fig. 2. For these series of experiments, all chemicals and organic solvents were as anhydrous as possible. They were controlled in a glove box charged with N2 gas. First, the silicon wafer surface resulting from the precursor solution without the addition of water did not show any particularly known morphologies of MOFs that could be attributed to PCN families (Fig. 2a). However, it was clearly noticed that the shapes of particles gradually transformed from fibrous rods to ellipsoidal rods as the amount of water for ZrCl4 increased from 2 equivalents (eq.) of water to 4 eq. The ellipsoidal rod shape was known as the PCN-223 morphology26 (from Fig. 2b–e). In particular, the surface driven with 2 eq. of water seemed to be a middle stage showing grain boundaries of ellipsoidal rod shapes represented by the red dotted lines in Fig. 2b and the particles on the surface resulting from 3 eq. of water displayed a fairly uniform size and shape. Furthermore, particles on the surface resulting from 4 eq. of water (Fig. 2e) were crowded and seemed to be well developed into PCN-223. Besides, when the added water amount was 6 eq. (Fig. 2f), the morphology seemed to show an amorphous feature like a disordered agglomerate of organic materials. Presumably, the state driven by the addition of 6 eq. of water could be considered as another middle state for the next phase transition. When the amount of water was further increased from 8 eq. to 10 eq., ellipsoidal rod shapes disappeared while globular polyhedral shapes appeared; this could be the correct morphology of PCN-22127 (Fig. 2g and f). Therefore, it was inferred that the water amount should be precisely controlled to form the desired MOF phases; otherwise, several phases of MOFs could be mixed as shown in Fig. 1a.
 | ||
| Fig. 2 Scanning electron microscopy images showing silicon wafers treated with ZrCl4, porphyrin ligands and benzoic acid (250 mg) in DMF at 100 °C for 12 hours upon varying the water content. (a) Without adding water (0 equimolar amount of water for ZrCl4); (b) adding 1 equimolar amount of water for ZrCl4; (c) adding 2 equimolar amounts of water for ZrCl4; (d) adding 3 equimolar amounts of water for ZrCl4; (e) adding 4 equimolar amounts of water for ZrCl4; (f) adding 6 equimolar amounts of water for ZrCl4; (g) adding 8 equimolar amounts of water for ZrCl4; (h) adding 10 equimolar amounts of water for ZrCl4. | ||
To study the effects of modulators such as acetic acid and benzoic acid on the growth of the PCN series atop the silica surface, the morphologies of the resulting particles from the precursor solution of ZrOCl2·8H2O with porphyrins upon purposely varying each modulator amount on were investigated with SEM as shown in Fig. 3. Fig. 3a–e represent the transformation of morphologies after adding different amounts of acetic acid and Fig. 3f–h reveal the transformation of morphologies with various amounts of benzoic acid. Fig. 3a shows small globular rod shapes driven from 0.3 mL of acetic acid which could be those of PCN-223. Then, with 0.7 mL of acetic acid, the clear cubic morphology could be observed as shown in Fig. 3b. This morphology might belong to MOF-525. It has been previously reported30 that MOF-525 particles have a cubic morphology with the same topology as PCN-221, except that the Zr cluster of MOF-525 is Zr6 with 12 connectivity like PCN-223. Besides, with 0.9 mL of acetic acid, as shown in Fig. 3c, the points of the cubic morphology became blunt, which may have been due to the transitioning process to a polyhedron shape to form PCN-221. Currently, we do not exactly know the thermodynamic or kinetic relationship among the mechanisms of PCN-221, PCN-223, and MOF-525 formation. However, it presumably implies that thermodynamically or kinetically favorable Zr clusters could exist under the reaction conditions at earlier stages of nucleation for each MOF because one can clearly see mixed morphologies such as polyhedra, ellipsoidal rods, and simple cubic shapes, respectively, with 1.1 mL of acetic acid (Fig. 3d). Ellipsoidal rods and polyhedra disappeared upon the addition of 1.9 mL of acetic acid on transitioning to the cubic shape (Fig. 3e). Another modulator, benzoic acid, also showed a similar trend to acetic acid. With 107 mg of benzoic acid, an irregular ellipsoidal rod shape (PCN-223) was formed on the surface of the silicon wafer as depicted in Fig. 3f. Upon adding 250 mg of benzoic acid, crowded nanoparticles with polyhedron shapes implying PCN-221 were obtained on the surface of silicon wafer, which corresponded well with Fig. 2g. On increasing the amount of benzoic acid up to 392 mg, irregular sized cubic particles were clearly observed, where small cubic particles seemed to be grown on the surface of large cubic particles. Notably, since ZrOCl2·8H2O contains 8 eq. water for Zr in the crystal form, it was found that the results shown in Fig. 3g were almost identical to the results depicted in Fig. 2g obtained with ZrCl4, 250 mg of benzoic acid, and 8 eq. of water. Therefore, the absorbed amount of water and modulator on the surface of the silicon wafer could be among the important factors for the desired MOF phases influencing the condensation and hydrolysis reaction rates of metal complexes.
 | ||
| Fig. 3 Scanning electron microscopy images showing silicon wafers treated with ZrOCl2·8H2O and porphyrin ligands in DMF at 100 °C for 12 hours upon varying the amount of modulator, i.e., acetic acid and benzoic acid: (a) 0.3 mL of acetic acid; (b) 0.7 mL of acetic acid; (c) 0.9 mL of acetic acid; (d) 1.1 mL of acetic acid; (e) 1.9 mL of acetic acid; (f) 107 mg of benzoic acid; (g) 250 mg of benzoic acid; (h) 392 mg of benzoic acid. | ||
We demonstrated the possibility of a kinetic relationship between PCN-223 and PCN-221 upon varying Zr sources, water contents, and modulators. The transformation between the two phases could also possibly occur on the surfaces of substrates as time elapses even under the same conditions. It could also be presumed that according to the phase changes on the surface, the phases of those MOFs in suspension above the substrates could be influenced. To gauge the phase change on silicon wafers with the native oxide layer under optimized PCN-223 conditions using ZrCl4, porphyrins and benzoic acid with 3 eq. of water, SEM analysis was conducted as shown in Fig. 4a–d. The morphological changes in the particles on the substrate seemed to be very dynamic as time elapsed as shown in Fig. 4a–d. After 12 hours, the uniform distribution of ellipsoidal rod-shaped particles on the silicon wafer could clearly be seen (Fig. 4a). After five more hours elapsed, the size of PCN-223 became greater and some polyhedron-shaped particles such as PCN-221 (inset image in Fig. 4b) were obviously formed as depicted in Fig. 4b. However, as the reaction time increased to 18 hours (Fig. 4c) and 19 hours (Fig. 4d), the resulting products on the surfaces could not be easily identified based on their morphologies.
 | ||
| Fig. 4 Scanning electron microscopy images of the resulting products on the surface of silicon wafers (a) to (d) and images of powder in supernatants simultaneously produced with silicon wafers (e) to (h) using ZrCl4, 3 eq. of water, and porphyrin ligands in DMF at 100 °C upon varying reaction time: (a) and (e) for 12 hours; (b) and (f) for 17 hours; (c) and (g) for 18 hours; (d) and (h) for 19 hours. | ||
To compare the particle morphologies in suspension above the amorphous SiO2 surface of the silicon wafer with those on the substrate during the same time period, we also conducted SEM analysis under the same conditions for PCN-223. In contrast to the morphological changes in the particles on the substrate, the morphological changes in the particles in corresponding suspensions seemed to be less dynamic. The ellipsoidal shape was well maintained for a long time, although the shape or size was not very uniform (Fig. 4e–h). Interestingly, the particle morphology after 12 hours was almost identical to each other, representing some freestanding particles (see high-magnified inset image in Fig. 4e), which might be small polyhedra in a relatively low magnified image as shown in Fig. 4e. Nonetheless, as the reaction time increased (Fig. 4f–h), thicker ellipsoidal particles and many chunks were observed (Fig. 4f). Particularly, surfaces of ellipsoidal particles in suspensions were not smooth or clean. Unknown shapes of phases shown on silicon substrates in the same time period seemed to adhere to the surface of the ellipsoidal particles in suspensions (Fig. 4g and h). Therefore, it is worth noting that the desired phase was favorably nucleated on the substrates in a relatively short time period rather than both on the surface and in the bulk solution and grown particles could be diffused into the suspension phase following the convection flow. Furthermore, as the reaction time elapsed, particles could grow further. Sometimes other impurities that formed on the surface of substrates could be combined with particles of the desired phase in suspensions. Although at this point we lack insight on why impurity phases are formed on the surface of substrates, it could be speculated that they would be formed since the stoichiometry between chemical species for a pure phase could be temporarily disproportionate on the surface of substrates as building blocks such as Zr ions and porphyrins are consumed.
Subsequent SEM analysis was performed to determine the influence of the substrate's surface properties on the morphological formation of PCN-223 (Fig. 5). Instead of the silicon wafer, mica films with crystalline silica surfaces were immersed in the reaction media under the conditions for PCN-223. The mica has two unique properties that can influence the crystal growth of MOFs as illustrated in Fig. 5a. First, the top surface of mica can display very crystalline SiO2 layers and a regular array of hydroxyl groups that can interact with water molecules, metal ions, and other hydrophilic molecules. Besides, mobile cations are intercalated in the lattice of the mica structure, which could be exchanged with Zr4+ ions, Zr4+ complexes with modulators or porphyrins, and/or Zr clusters.25 Morphological changes in the particles on mica substrates did not seem to be dynamic as the time elapsed as shown in Fig. 4b–e when they were compared with those of particles on silicon wafers. After 12 hours, one could clearly see the uniform distribution of ellipsoidal rod-shaped particles on the mica surface, whose chemical nature was identical to SiO2 (Fig. 5b). After 17 hours, the resulting products on surfaces could not be easily classified, with morphologies showing small globular shapes as presented in Fig. 5c. However, after around 19 hours (Fig. 5d), it was found that PCN-223 phases were re-established, although the sizes of particles were bigger than those of particles formed after 12 hours. The PCN-223 phase that was almost identical to the phase formed after 12 hours was surprisingly reconstructed after one more hour passed, as displayed in Fig. 5e.
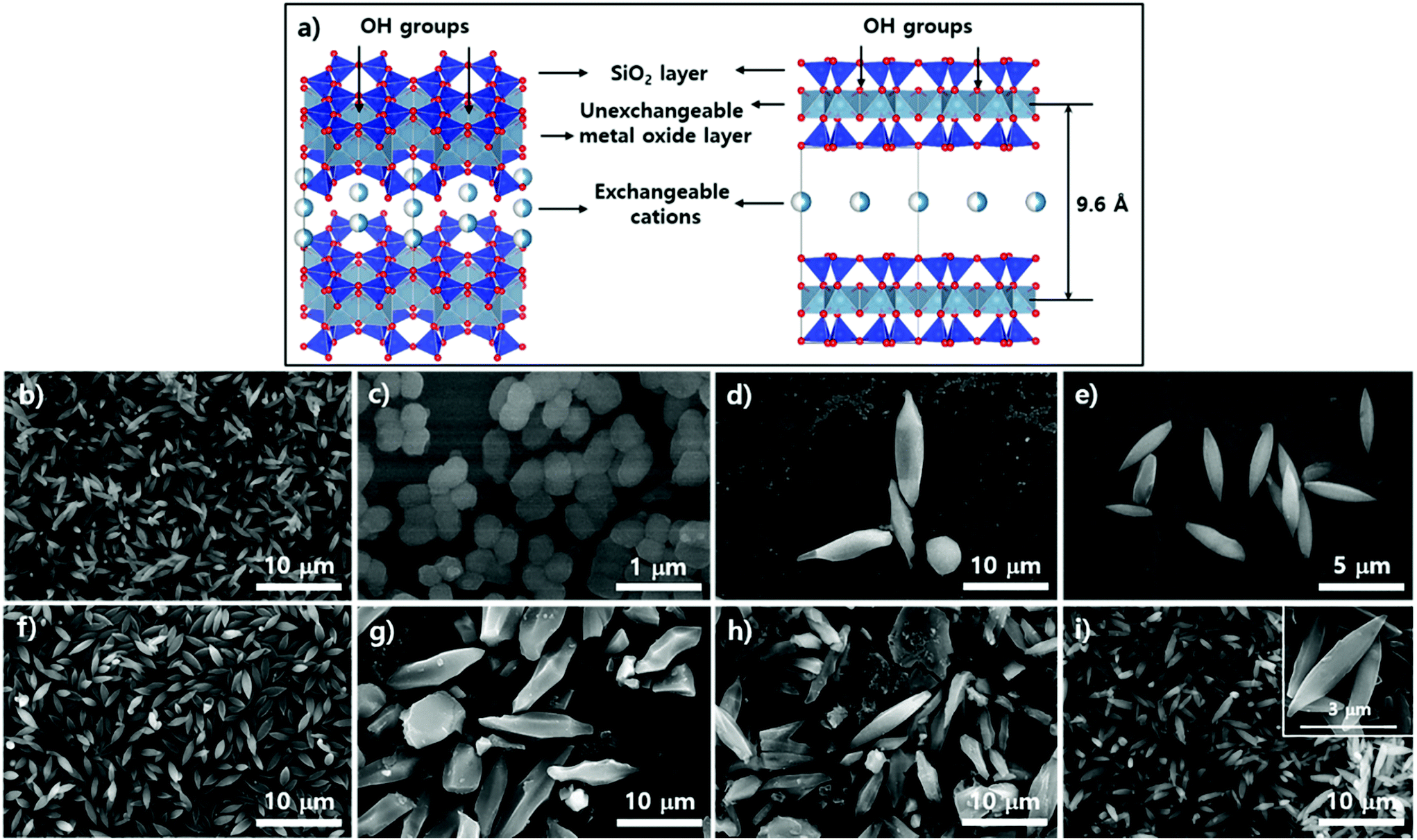 | ||
| Fig. 5 Schematic illustration of the mica lattice structure and surface topology (a) and scanning electron microscopy images of resulting products on the surface of mica films from (b) to (e), and images of samples in suspension simultaneously produced with mica films from (f) to (i) using ZrCl4, 3 eq. of water, and porphyrin ligands in DMF at 100 °C upon varying reaction time: (b) and (f) for 12 hours; (c) and (g) for 17 hours; (d) and (h) for 19 hours; (e) and (i) for 20 hours. | ||
The morphologies of particles in suspensions above the film surfaces at the same time point were also demonstrated with SEM. Like silicon wafer cases, the overall morphologies of particles from the suspension were quite well maintained for a long time as shown in Fig. 5f–i, while optimal uniformity and size identical to those on mica surfaces were obtained from 12 hours of reaction as clearly shown in Fig. 5f. As the reaction time increased (Fig. 5 from g to h), larger ellipsoidal particles and some chunks were observed, similar to those from silicon wafers. Interestingly, unlike the silicon wafer cases, rough PCN-223 particles were transformed back to smaller and quite uniform smooth ellipsoidal particles after 20 hours, resembling particles simultaneously formed on the mica surface. Hence, it is worth suggesting that the particles were also possibly nucleated on the substrate rather than on both the substrate and in bulk solution. After the particles grew to a certain size, they could diffuse into the solution phase by following the convection flow as mentioned for the silicon wafer. Therefore, it could be considered that the degree of crystallinity of the surface would not affect the growth mechanism of PCN-223. However, another interesting finding was that as the reaction was pursued on mica films, the morphologies and phases of particles were well maintained as compared to those on amorphous SiO2 surfaces. This unique observation is presumably due to the fact that some Zr sources and/or Zr-complexes would be exchanged with the mobile cations on/in mica layers and then, as they would be slowly released from the lattices, the molar ratio between chemical species for one phase could be re-established on the mica surface.
The phase transition of particles in suspensions formed with substrates such as silicon wafers and mica films was also demonstrated with P-XRD profiles as illustrated in Fig. 6. The intensity of the characteristic peak26 of PCN-223 formed with silicon wafer in suspension was gradually reduced and the peak width became broader as time elapsed, suggesting that the phase in suspension lost crystallinity (Fig. 6a). Therefore, the PCN-223 phase formed on the surface of the silicon wafer was gradually decomposed in the suspension with increasing reaction time. This trend was consistent with the corresponding SEM analysis. However, different characteristics of the transformation process for products in suspensions formed with mica films were detected in P-XRD profiles as shown in Fig. 6b. After 16 hours, the PCN-221 phase, in the presence of the PCN-223 phase, started to be formed in suspensions when the reactions were modulated with mica films. The PCN-221 phase was dominant under the same conditions as the reaction time was increased to 18 hours. However, characteristic peaks of the PCN-223 phase appeared again in the presence of the PCN-221 phase. When the reaction time was increased to more than 19 hours, the PCN-223 phase became more pronounced with decreased characteristic peaks of the PCN-221 phase, corroborating the findings of the SEM analysis. It was also found that the large chunks observed in the SEM image could be ascribed to the PCN-221 phase.
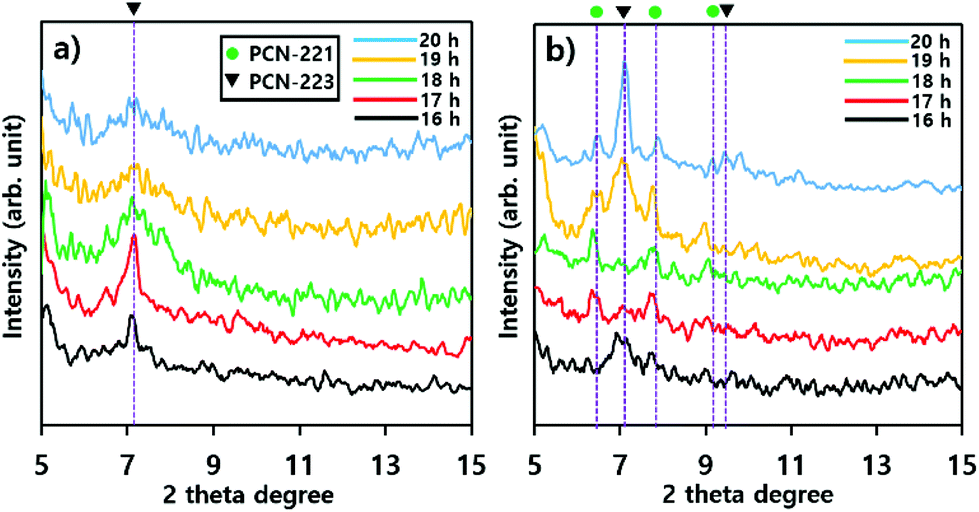 | ||
| Fig. 6 Powder X-ray diffraction profiles of products in suspensions formed with substrates such as (a) silicon wafers and (b) mica films upon varying the reaction time. In each profile, green filled circles represent the PCN-221 phase and black triangles represent the PCN-223 phase. | ||
To examine the role of water in the phase construction, we attempted to produce the PCN-221 phase on the Si/SiO2 layer using ZrOCl2·8H2O, porphyrins, and benzoic acid without adding additional water. SEM analysis was then conducted; results are shown in Fig. 7. First, morphological changes in the particles on substrates seemed to be less dynamic with respect to the reaction time as compared to the PCN-223 case as demonstrated in Fig. 7a–e. After about 13 hours, one could clearly see polyhedron-shaped particles on the silicon wafer with size distribution of approximately 300–400 nm (Fig. 7a), which was close to the seeding state of PCN-221. After three more hours elapsed, PCN-221 particles with sizes bigger than before with open pores of approximately 100 nm in size were formed, possibly due to the ordered aggregation of polyhedron-shaped particles (Fig. 7b with inset image). Such aggregation and seeding processes were continuously observed for up to 18 hours since both open-pored particles and small polyhedron particles were simultaneously observed. However, when the reaction time was increased to 19 hours (Fig. 7d), the seeding state seemed to be dominant again. At around 20 hours (Fig. 7e), the aggregation of seeds was repeated; therefore, it could be that the PCN-221 formation (polyhedrons) was slightly retarded as compared to the formation of PCN-223 (ellipsoids), while the formation and re-dissolution processes of PCN-221 repeatedly occurred on the silicon oxide layer.
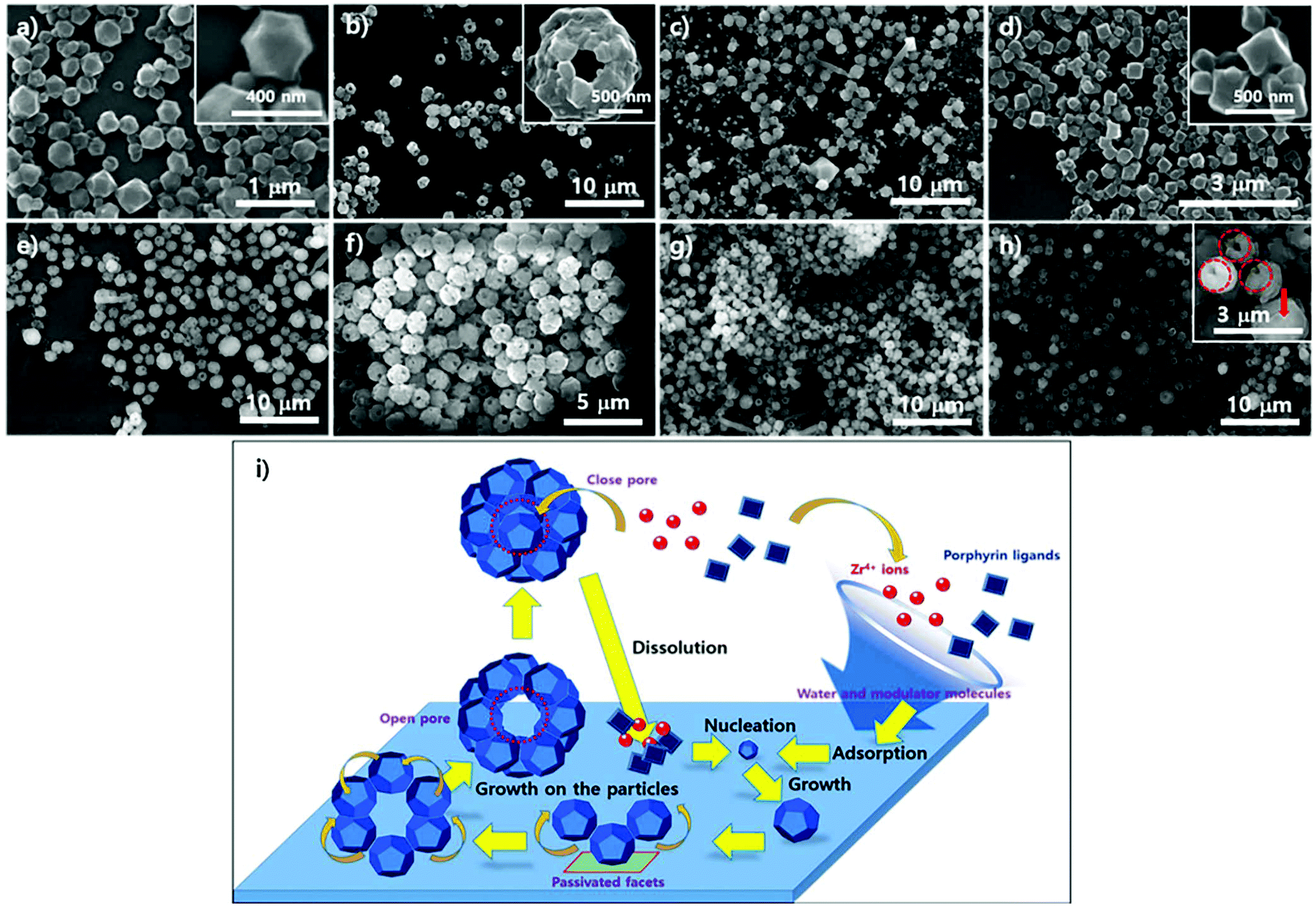 | ||
| Fig. 7 Scanning electron microscopy images of the resulting products on the surface of silicon wafers: images of powders in supernatants simultaneously produced (a) to (e) with silicon wafers, (f) to (h) using ZrOCl2·8H2O and porphyrin ligands in DMF at 100 °C upon varying the reaction time. (i) Schematic illustration of the growth mechanism for the resulting particles formed on the surface and in suspension upon varying reaction time: (a)13 hours 30 minutes; (b) and (f) 16 hours; (c) and (g) 18 hours; (d) and (h) 19 hours; (e) 20 hours. Red dotted-line circles in (h) represent open pores and the red filled arrow represents a closed pore. | ||
The particle morphology in suspension above the amorphous SiO2 surface of the silicon wafer was also analyzed with SEM under the same conditions as PCN-221. Unlike the morphological changes in particles on the substrate, significant morphological changes in particles in corresponding suspensions were not detectable upon varying the reaction time, as shown in Fig. 7f–h, displaying quite uniformly sized globular particles with hollow open channels. Although angled rod shapes were scarcely obtained after 18 hours (Fig. 7g), which were of unknown phases, angled rod shapes disappeared after 19 hours (Fig. 7h). Therefore, it is also evident that there were dynamics of formation and dissolution processes in the case of the PCN-221 formation mechanism. Interestingly, there was considerable formation of open channels of particles after 19 hours as depicted by red-colored circles in the inset image of Fig. 7h. Since particles with closed pores (red-colored arrow in the inset image of Fig. 7h) were more often observed in the suspension than those on the surface of substrates, it could be that the PCN-221 phase was nucleated on the substrate rather than in bulk solution, while grown particles diffused into the solution phase following the convection flow of solution. Once detached from the substrate, pores formed by ordered aggregates could be closed in the suspension. Hence, the detailed mechanism for the PCN-221 formation can be summarized as illustrated in Fig. 7i. One possible rationale for open channel formation was that the one stationary facet of polyhedrons on the surface could be passivated for further chemical reaction as particles grew on substrates.
Fig. 8 shows the particle morphology on the crystalline SiO2 surface of mica films (from 8a to 8d) and in the suspensions (from 8e to 8h) as reaction time varied under the same conditions as PCN-221. In contrast to the morphological changes in the particles on the silicon wafers, there were no significant morphological changes in the particles on mica films or in the suspension as reaction time elapsed, which displayed regular globular shapes with macropores (∼100 nm). However, few angled rod shapes both on mica films and in the suspension were often observed more than those on silicon wafers. In addition, the rod shapes gradually disappeared for up to 20 hours (Fig. 8h), while most angled shapes developed with silicon wafers disappeared after 18 hours. Furthermore, one could see uneven surfaces, as shown in Fig. 8c, emphasized by red-dotted circles, which might be concerned with a state forming macropores of PCN-221 particles from the aggregation of small seeds as illustrated in Fig. 7i. Therefore, the phase could be nucleated on the surface. This implies that the rate of formation of the PCN-221 phase on mica surfaces was more retarded than that on silicon wafers due to the capturing effect of the mica surface toward Zr4+ and/or Zr-complexes as expected for PCN-223 formation on mica films.
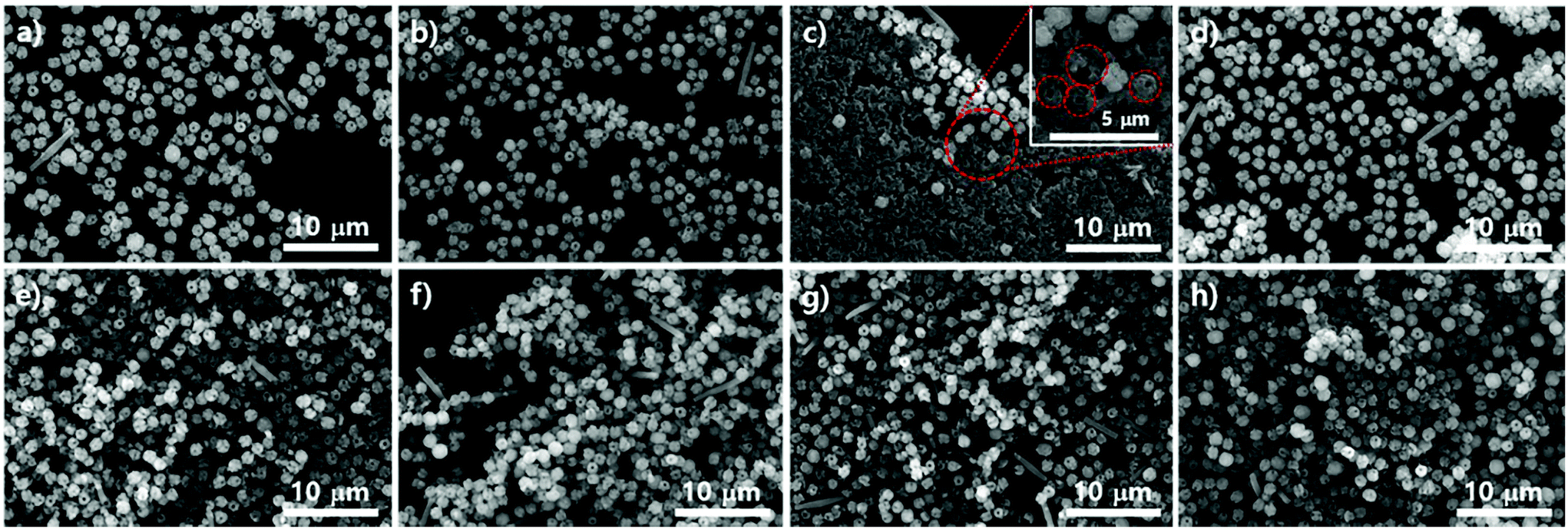 | ||
| Fig. 8 (a)–(d) Scanning electron microscopy images of the resulting products on the surfaces of mica films and (f)–(h) images of powders in supernatants simultaneously produced with mica films using ZrOCl2·8H2O and porphyrin ligands modulated with benzoic acid in DMF at 100 °C upon varying the reaction time: 16 hours for (a) and (e); 18 hours for (b) and (f); 19 hours for (c) and (g); 20 hours for (d) and (h). | ||
Phase transitions of PCN-221 in suspensions as studied above (Fig. 8) were also evident in P-XRD profiles shown in Fig. 9a and b. When resulting particles were formed with silicon wafers, characteristic peaks of PCN-221 clearly appeared as the reaction time increased and the peak width became narrower as time elapsed, in contrast to PCN-223 cases. This could mean a lower rate of PCN-221 formation than that of PCN-223 (Fig. 9a). However, P-XRD patterns for the resulting powders in suspensions formed with mica films showed broad and unclear characteristic peaks for PCN-221 (Fig. 9b). This could mean a lower crystallinity than PCN-221 formed with silicon wafers. Besides, PCN-221 could be slowly formed with mica films relative to silicon wafers. Furthermore, the morphology of PCN-221 formed with substrates such as silicon wafers and mica films was observed using transmission electron microscopy (TEM). As shown in Fig. 9c, TEM results clearly demonstrated that the open pore was formed by the aggregation of small particles and the pore width was about 100 nm. Since the inset image provided in Fig. 9c as a side view of resulting particles obviously showed the channel, TEM analysis also supported the possibility of the PCN-221 formation mechanism as illustrated in Fig. 7i.
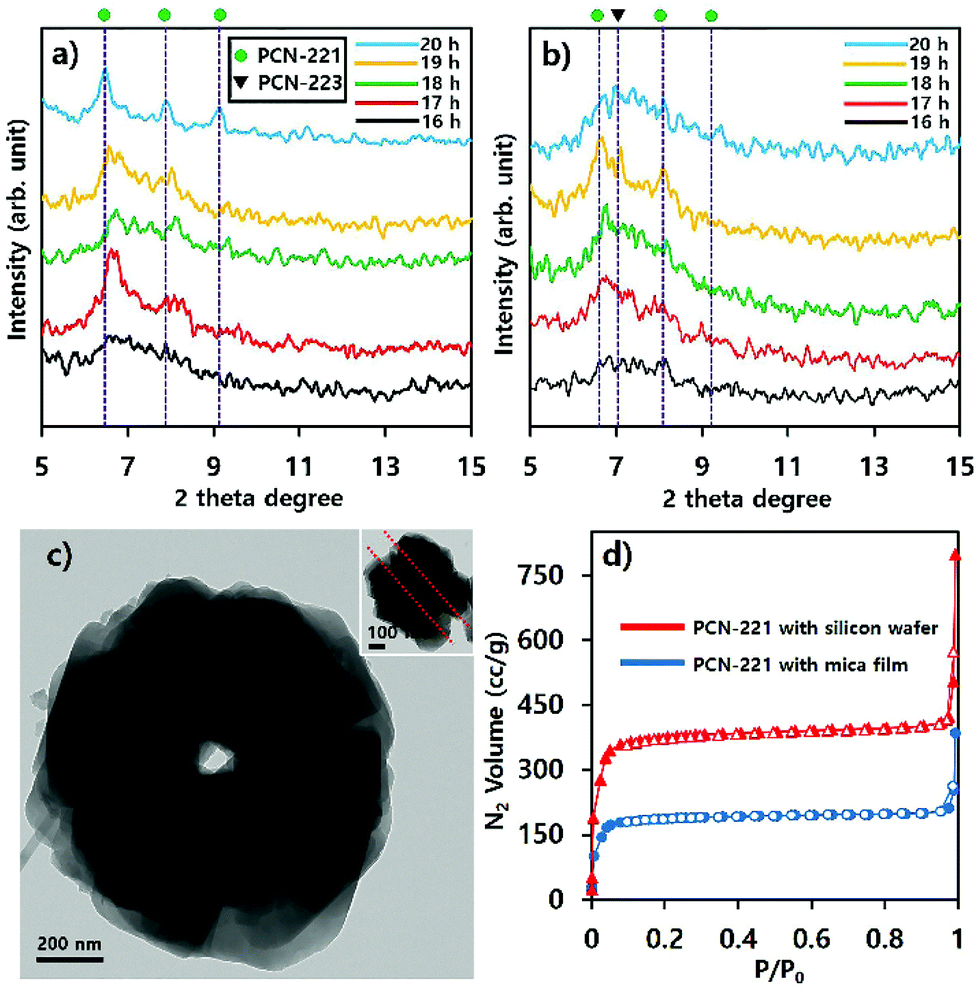 | ||
| Fig. 9 Powder X-ray diffraction profiles of products in suspensions formed with substrates such as (a) silicon wafers, (b) mica films upon varying reaction time. (c) Transmission electron microscopy Image formed with substrates; (d) N2 adsorption–desorption isotherm curves for PCN-221 induced by substrates after 20 hours. Empty red triangles and blue circle shapes with corresponding lines represent N2 desorption curves. Color-filled triangles and circle with corresponding lines represent N2 adsorption curves. | ||
N2 adsorption/desorption isotherm profiles at 77 K for PCN-221 with substrates after 20 hours indicated that the resulting PCNs had predominantly microporous characteristics (type I) with calculated Brunauer–Emmet–Teller (BET) surface areas of 1246.2 m2 g−1 for PCN-221 from silicon wafer and 652.15 m2 g−1 for PCN-221 from mica films. Particularly, both cases showed sharp gas uptake amounts in two regimes: the P/P0 < 0.1 regime and the P/P0 ≅ 1 regime. The first regime was due to micropores as well-known features of PCN-221. The sharp uptake of N2 gas in the second regime can be considered as the capillarity condensation of filled gas in nano-sized channels.31,32 However, the rationale for the smaller BET surface area of PCN-221 formed with silicon wafer relative to the known value in some literature27 could be ascribed to reduced micropore volume due to the formation of open channels. In addition, the reason for the smaller BET surface area of PCN-221 produced by mica films as compared to that from silicon wafers could be the low crystallinity of the products from mica films as compared to PCN-221 from silicon wafers in the same time period. Therefore, gas adsorption/desorption isotherm profiles also demonstrated that resulting particles produced by substrates simultaneously possessed both micropores and macropores, which corresponded well to SEM and TEM analyses. Furthermore, to estimate their detailed pore structure in their bulk state, their pore size distribution was obtained by using the Barret–Joyner–Halenda (BJH) method based on their N2 adsorption branches as presented in Fig. 1S.† These BJH profiles were also consistent with the microscopic analysis and N2 isotherm adsorption/desorption profiles showing quite significant pore volumes for the 100 nm open-pore channels. Finally, it is worth noting here that the CBD method would be useful for obtaining MOFs with uniform size and shape and unique pore structure. This method was preliminarily applied to other MOFs such as UiO-66 and MOF-525 systems showing the uniform size and shape of particles as illustrated in Fig. S2.†
Conclusions
We obtained mixed phases of two MOFs using ZrOCl2·8H2O and porphyrins, and the goal of this study was to establish a kinetic relationship between these two MOFs (PCN-223 and PCN-221) and find some factors controlling their kinetics. We postulated that the early stages of both MOFs could be formed on the surface of a glassy reaction bath for a conventional solvothermal reaction system without agitating solutions. To demonstrate this phenomenon, silicon wafers having amorphous SiO2 layers were immersed into reaction systems to simulate the surface of the glassy bath for the CBD technique and a substantial series of reactions was examined by varying Zr sources, modulators, water content, and reaction times. Initially, we found that the PCN-223 phase was well optimized with ZrCl4 and benzoic acid upon varying the amount of water. However, on increasing the amount of water, the PCN-221 phase was formed under the same conditions. The PCN-221 phase was well formulated with a relatively larger amount of water and benzoic acid modulator as compared to the formation of PCN-223 on silicon wafers. This implies that the relevant condensation and/or hydrolysis reactions of metal complexes should occur quickly for the uniformed PCN-223 phase. However, since various MOFs could be formed with acetic acid as a modulator under the same conditions, the appropriate acidity of modulators should be sought for desirable MOFs. Upon varying the reaction time, a uniform PCN-223 phase was obtained both on the silicon wafer and from the suspension above the wafer surface for a fast period time relative to the PCN-221 phase. With increasing reaction time, the PCN-223 phase was gradually decomposed on the surface of the substrate and in its suspension. However, the decomposition of the PCN-221 phase was prevented upon replacing a silicon wafer with a mica film under the same conditions because mica films can reserve and slowly release some of the building blocks from their lattices, while the better crystallinity of the SiO2 surfaces on mica films as compared to the silicon wafers was not related to the phase formation. Unlike PCN-223, the serious deterioration of PCN-221 was not detected on silicon wafers or mica films by varying the time, although the required reaction time for PCN-221 was slightly longer than that for PCN-223. Nonetheless, it took even more time to form PCN-221 with mica films, which could be because the unique lattice property may have affected PCN-223 formation. Another remarkable feature of PCN-221 formation was that the resulting particles had open channels with windows of about 100 nm in size. Those were gradually closed in its suspension phase as time elapsed. Therefore, it is worth suggesting that the PCN series of MOFs was nucleated on solid surfaces rather than in bulk solutions. Depending on the surface properties, the formation process of MOFs can be modulated. Studies to understand the physicochemical attributes of organic components for MOF crystal formation can help us to develop new types of MOFs and control their properties by seeking a combination of various building blocks and appropriate substrates.Experimental
Materials
Zirconyl chloride octahydrate (ZrOCl2·8H2O, 98%), zirconium tetrachloride (ZrCl4, 99.9%), and N,N-dimethylformamide (DMF, anhydrous, 99.8%) were purchased from Sigma-Aldrich. Tetrakis(4-carboxyphenyl)porphyrin (TCPP, 97%) and benzoic acid (99%) were purchased from Tokyo Chemical Industry Co., Ltd. Acetic acid (glacial, 99.7%) was purchased from Samchun Chemicals. Mica disc was purchased from Probes Inc. All reagents were used without any further purification.Measurements
Powder X-ray diffraction (XRD) patterns were measured using an Empyrean XRD diffractometer (PANalytical B.V., Netherlands) with Cu-Kα radiation (λ = 1.5406 Å) having a 5 mm fixed incident beam mask and 1/2° fixed anti-scatter slit. The measurement angle ranged from 5° to 15° with time step increments of 0.0262° and 60 s per step. Scanning electron microscopy (SEM) images were obtained using a Quanta 250 FEG (FEI Company, Hillsboro, OR, USA). Sample surfaces were coated with Au/Pd plasma for 60 s and sample images were obtained using a 15 kV electron beam. N2 adsorption–desorption isotherm curves were measured with a Belsorp-mini II instrument (Bel Japan Inc.) using N2 gas at 77 K. Transmission electron microscopy (TEM) images were taken using a JEM-2100F (JEOL) with an accelerating voltage of 200 kV.Synthesis of Zr-porphyrinic MOFs
Substrate templated synthesis of Zr-porphyrinic MOFs via the CBD method
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants (NRF-2018R1D1A1B07050671 and NRF-2017R1A2B4006352) of the National Research Foundation of Korea (NRF) funded by the Korea Government (Ministry of Science and ICT) and by Industrial Core Technology Development Program (no. 20005342, ‘Development of VOC oxidation system’) funded by the Ministry of Trade, Industry & Energy (MI, Korea). This work was also supported by the Hankuk University of Foreign Studies Research Fund of 2019. This work has been conducted with the financial support of the Korea Institute of Industrial Technology (Project No. EE-19-0022).Notes and references
- D. Alezi, Y. Belmabkhout, M. Suyetin, P. M. Bhatt, L. J. Weselinski, V. Solovyeva, K. Adil, I. Spanopoulos, P. N. Trikalitis, A.-H. Emwas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 13308–13318 CrossRef CAS.
- Q. Al-Naddaf, M. Al-Mansour, H. Thakkar and F. Rezaei, Ind. Eng. Chem. Res., 2018, 57, 17470–17479 CrossRef CAS.
- A. Ibrahim and Y. S. Lin, Ind. Eng. Chem. Res., 2016, 55, 8652–8658 CrossRef CAS.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeulen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 2292–2301 CrossRef CAS.
- A. Gokay, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 33693–33706 CrossRef.
- Q. Al-Naddaf, H. Thakkar and F. Rezaei, ACS Appl. Mater. Interfaces, 2018, 10, 29656–29666 CrossRef CAS.
- J. L. Harding and M. M. Reynolds, J. Am. Chem. Soc., 2012, 134, 3330–3333 CrossRef CAS.
- E. D. Metzger, R. J. Comito, Z. Wu, G. Zhang, R. C. Dubey, W. Xu, J. T. Miller and M. Dincă, ACS Sustainable Chem. Eng., 2019, 7, 6654–6661 CrossRef CAS.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS.
- L. Jiao and H.-L. Jiang, Chem., 2019, 5, 786–804 CAS.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS.
- C. Wang, X. Liu, N. K. Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- D. Ma, G. Han, S. B. Peh and S. B. Chen, Ind. Eng. Chem. Res., 2017, 56, 12773–12782 CrossRef CAS.
- G.-B. Hu, C.-Y. Xiong, W.-B. Liang, X.-S. Zeng, H.-L. Xu, Y. Yang, L.-Y. Yao, R. Yuan and D.-R. Xiao, ACS Appl. Mater. Interfaces, 2018, 10, 15913–15919 CrossRef CAS.
- A. Pankajakshan, M. Sinha, A. A. Ojha and S. Mandal, ACS Omega, 2018, 3, 7832–7839 CrossRef CAS.
- P. Deria, D. A. Gómez-Gualdrón, W. Bury, H. T. Schaef, T. C. Wang, P. K. Thallapally, A. A. Sarjeant, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 13183–13190 CrossRef CAS.
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef CAS.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS.
- X. Gong, H. Noh, N. C. Gianneschi and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 6146–6151 CrossRef CAS.
- S. M. Shaikh, P. M. Usov, J. Zhu, M. Cai, J. Alatis and A. J. Morris, Inorg. Chem., 2019, 58, 5145–5153 CrossRef CAS PubMed.
- H.-Q. Xu, K. Wang, M. Ding, D. Feng, H.-L. Jiang and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 5316–5320 CrossRef CAS.
- L. Zhang, S. Yuan, W. Fan, J. Pang, F. Li, B. Guo, P. Zhang, D. Sun and H.-C. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 22390–22397 CrossRef CAS.
- M. Kokotov and G. Hodes, J. Mater. Chem., 2009, 19, 3847–3854 RSC.
- X. Liu, M. Afzaal, K. Ramasamy, P. O'Brien and J. Akhtar, J. Am. Chem. Soc., 2009, 131, 15106–15107 CrossRef CAS.
- H. Heinz, H. J. Castelijns and U. W. Suter, J. Am. Chem. Soc., 2003, 125, 9500–9510 CrossRef CAS.
- D. Feng, Z.-Y. Gu, Y.-P. Chen, J. Park, W. Zhangwen, Y. Sun, M. Bosch, S. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 17714–17717 CrossRef CAS.
- D. Feng, H.-L. Jiang, Y.-P. Chen, Z.-Y. Gu, Z. Wei and H.-C. Zhou, Inorg. Chem., 2013, 52, 12661–12667 CrossRef CAS.
- F. G. Bordwell and D. Algrim, J. Org. Chem., 1976, 41, 2507–2508 CrossRef CAS.
- W. N. Olmstead and F. G. Bordwell, J. Org. Chem., 1980, 45, 3299–3305 CrossRef CAS.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS.
- M.-S. Kim, C. S. Phang, Y. K. Jeong and J. K. Park, Polym. Chem., 2017, 8, 5655–5659 RSC.
- J. K. Park, Bull. Korean Chem. Soc., 2017, 38, 153–154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9QI01045A |
| This journal is © the Partner Organisations 2020 |