
A reverse slipping strategy for bulk-reduced TiO2−x preparation from Magnéli phase Ti4O7†
Jiantao
Huang
 ab,
Xiangli
Che
a,
Jijian
Xu
a,
Wei
Zhao
a,
Fangfang
Xu
ac and
Fuqiang
Huang
*ab
ab,
Xiangli
Che
a,
Jijian
Xu
a,
Wei
Zhao
a,
Fangfang
Xu
ac and
Fuqiang
Huang
*ab
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P.R. China. E-mail: huangfq@mail.sic.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
cInorganic Materials Analysis and Testing Center, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, P.R. China
First published on 22nd October 2019
Abstract
Black TiO2−x is an attractive material due to its adjustable band structure, useful in different applications, and has been heavily investigated and developed. The current reduction of TiO2 involves a process from surface to center: thus, the issue of either insufficient or excessive reduction seems inevitable. To the best of our knowledge, it is rarely reported that a homogeneous defect distribution can be facilely achieved inside black TiO2−x. In this study, a bulk-reduced rutile TiO2−x was obtained via a circuitous two-step approach, with an intermediate Magnéli phase (TinO2n−1). With the decomposition of a solid atmosphere creator (e.g. KClO4 grains) at a high temperature, the missing oxygen ions in Ti4O7 could be replenished quantitatively. The TEM and SAED results reveal that the oxidation process is not just a surface reaction, but it involves reverse displacement or structural rearrangement inside the crystal. In particular, the periodic variation of crystallographic sheer planes was the direct evidence of the above bulk reaction, i.e. unified long period for Ti4O7 and varied long period for critical ratio sample. The as-prepared samples showed different band gaps and colors, based on their oxygen content. KClO4, with a critical mixing ratio of around 17.5 mol%, could almost oxidize Ti4O7 to black rutile TiO2−x. Furthermore, the sample formed at the critical ratio showed a considerable photo-thermal response speed of 1.7 °C s−1 and an equilibrium temperature of up to 70 °C under irradiation with light above 400 nm, which can be an evidence for the enhancement of non-intrinsic absorption. In short, this study provides a new route for the controllable preparation of black TiO2−x, and a possibility for further development of other solid atmosphere creators.
Introduction
Titanium dioxide (TiO2) has been widely investigated since its first significant application in the photo-electrochemistry field in 1972 (Fujishima and Honda1). In order to improve the light utilization of intrinsic TiO2, phase-level composition2–5 and atomic-level doping6–11 are two main effective methods. The former can effectively separate photo-generated carriers through matching energy band structures,5 while the latter can introduce new energy levels into the band gap to obtain additional light absorption.7,8 The upsurge in research on black titania (TiO2−x) began in 2011, as reported by Chen et al.12 Black TiO2−x exhibits higher utility in the visible and IR regions via a hydrogen reduction process. Up to now, the reduction of TiO2 is still widely adopted to prepare black TiO2−x.13 The atmosphere (e.g. H2,12,14–19 Ar,14,20 NH3,21,22 C/CO,23,24 and vacuum17,25) treatment and active metal (e.g. Li,26 Mg,27–30 and Al31,32) reduction are two main reduction methods reported in the literature, while it is uncommon to adopt other solutions or solids (e.g. imidazole,33 NaBH4,34 GaH2,35–37 and TiH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38). The thermodynamic analysis reveals that the reduction of well-crystallized TiO2 indeed requires much harsher reaction conditions.12,13,25 The reducing degree of these white precursors results in the different colors of the products, as reported in the literature, which varies with the mixing ratio, treatment temperature and reaction time.15,38 Alternatively, the subscript x in black TiO2−x and the color intensity are not easy to control. Moreover, the above reduction methods usually only affect the elemental composition of the surface layer, where a disordered TiO2−x layer12,25 is introduced. Furthermore, the reported atmosphere treatment usually employs a CVD method with different gas flows,21 which is always accompanied with an insufficient utilization of gases. For active metal reduced products, the reaction uniformity and removal of oxide impurities should be taken into consideration. It seems to be a huge challenge to obtain a bulk-reduced black TiO2−x, and further investigation is needed. Herein, a more controllable and economical method for preparing bulk-reduced TiO2−x with great solar absorption has been presented.
38). The thermodynamic analysis reveals that the reduction of well-crystallized TiO2 indeed requires much harsher reaction conditions.12,13,25 The reducing degree of these white precursors results in the different colors of the products, as reported in the literature, which varies with the mixing ratio, treatment temperature and reaction time.15,38 Alternatively, the subscript x in black TiO2−x and the color intensity are not easy to control. Moreover, the above reduction methods usually only affect the elemental composition of the surface layer, where a disordered TiO2−x layer12,25 is introduced. Furthermore, the reported atmosphere treatment usually employs a CVD method with different gas flows,21 which is always accompanied with an insufficient utilization of gases. For active metal reduced products, the reaction uniformity and removal of oxide impurities should be taken into consideration. It seems to be a huge challenge to obtain a bulk-reduced black TiO2−x, and further investigation is needed. Herein, a more controllable and economical method for preparing bulk-reduced TiO2−x with great solar absorption has been presented.
The excessive reduction of TiO2, which means a change in its phase here, has been heavily studied in the literature. When the ratio of O/Ti changes in the range between 1.75 and 2, a series of nonstoichiometric titanium oxides are obtained, denoted as TinO2n−1 (n ≥ 4, integers).39 These new structures are known as line or Magnéli phase, generated by a sliding in the crystallographic shear (CS) planes.40–42 From the perspective of symmetry, a tetragonal rutile structure is transformed into a triclinic Magnéli structure after sliding.41 Structurally, rutile (1 2 1) planes can be seen as two layers of oxygen ions sandwiched with one layer of Ti ions (denoted as a O–Ti–O unit).43 If a layer of oxygen ions is removed from each of the n O–Ti–O units, and a relative displacement is produced on the rutile (1 2 1) plane, then a Magnéli phase TinO2n−1 is obtained.43 On the flip side, if oxygen element is introduced into TinO2n−1, the reverse slipping in the (1 2 1) plane should happen, which is the basis of structural design here.
In this study, a circuitous two-step approach was adopted to prepare black TiO2−x with high oxygen defects. First, as a representative of TinO2n−1, Ti4O7 was obtained via the reduction of TiO2 with specific proportions of the Ti metal.44 The excessive reduction degree was controlled by adjusting the Ti ratio.45 Second, the appropriate oxidation of Ti4O7 to black TiO2−x was controlled using a solid gas creator (a compound, such as KClO4, which can produce an oxygen atmosphere by decomposition at high temperatures), realizing the reverse slipping strategy by replenishing the missing oxygen ions in Ti4O7. As all the reactions happened in a sealed vacuum tube, the amount of KClO4 grains determined the degree of oxidation, which meant that the subscript “x” in black TiO2−x could be manually adjusted. Moreover, TinO2n−1 is black, with its Fermi level located in the conduction band,46 showing full-spectrum absorption.44 Therefore, this circuitous two-step approach ensures the efficient light utilization of the products. Lastly, the properties of the as-prepared products were evaluated via a photo-thermal response study, showing an excellent response speed and equilibrium temperature when illuminated under visible and IR radiations.
Experimental section
Raw materials
Magnéli Ti4O7 powder (pure, 300–500 nm) was synthesized at 1000 °C in 1000 min using previous methods.44 Briefly, the well-mixed powders of Ti (200 mesh) and TiO2 (rutile, 25 nm) were heated in a closed vacuum silica tube. The mixing amount was determined by the following reaction: Ti + 7TiO2 → 2Ti4O7.45 Potassium perchlorate (KClO4, AR, 99%) was the oxygen source in this study, which was purchased from Aladdin Industrial Corporation.Preparation of TiO2−x
To control the oxidation process, different molar ratios of KClO4, varying from 6.25 to 25 mol%, were mixed with Ti4O7 in a sealed vacuum tube (Fig. 1b). When heated at 600 °C for 2 h, the decomposition of KClO4 provided the reaction atmosphere. As a result, it has little relationship with mixing uniformity. The amount of solid KClO4 determined the O2 content in the closed vacuum tube, which was easy to control. Through this adjustment, black TiO2−x samples were obtained with an appropriate Ti/O ratios, with high oxygen defects. The KCl impurity in the product mixture can be washed with large amounts of deionized water; but this washing process can also be avoided by pre-isolating KClO4 and Ti4O7, as illustrated in Fig. 1b.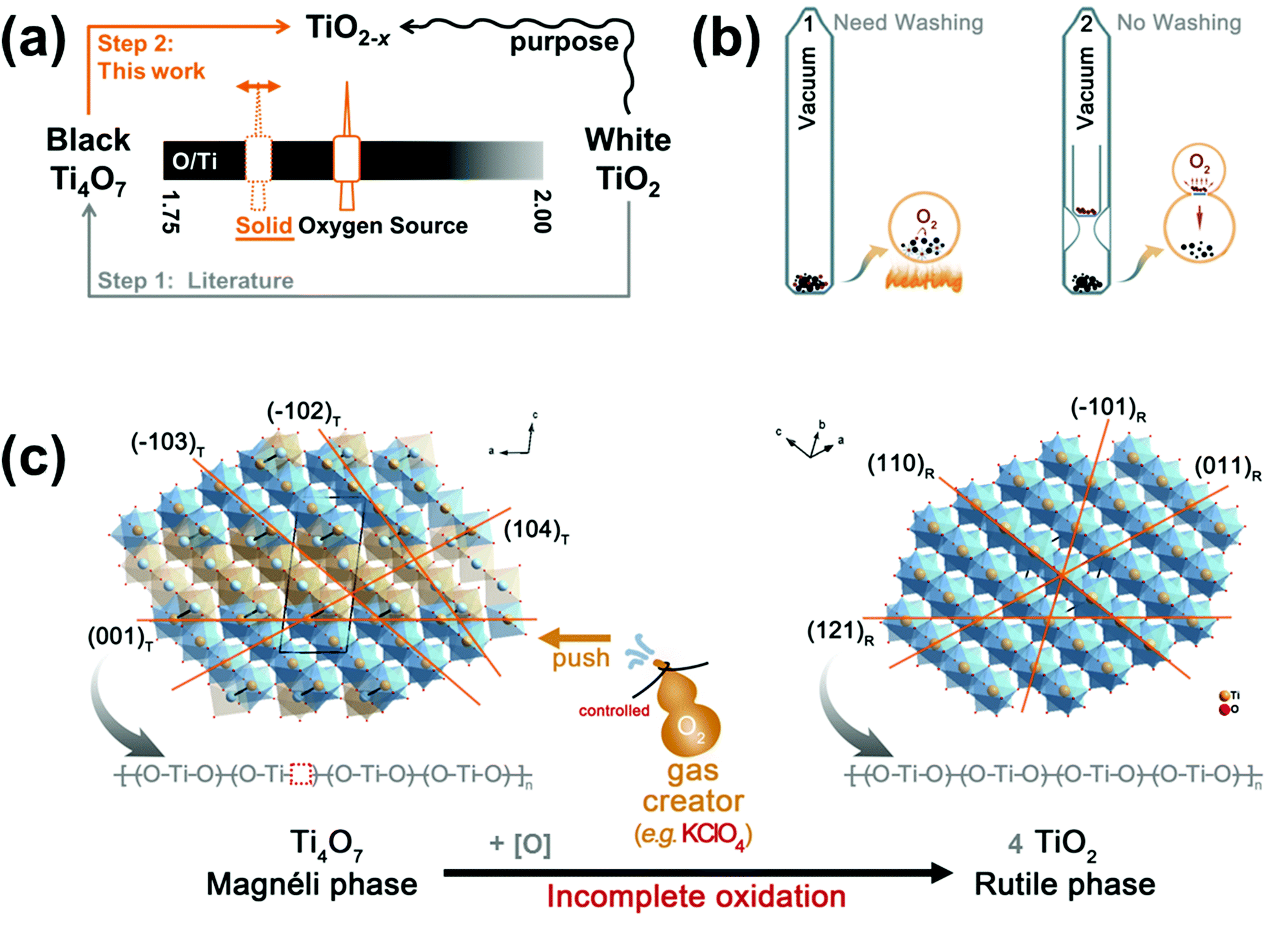 | ||
Fig. 1 (a) Scheme of the circuitous two-step approach in this study. (b) Illustration of the vacuum vessel devices. Left one needs a washing step, while the right one does not. (c) Schematic of black TiO2−x preparation by incompletely oxidized Ti4O7. The crystal structure of the Magnéli phase Ti4O7 [0 1 0]T is similar to that of rutile phase TiO2 [1 ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1]R, as they can be converted after slipping in the crystallographic shear plane (1 2 1)R. The rearrangement of the structure caused by oxygen deficiency can be reversed with the increase in the O/Ti ratio. In this study, solid KClO4 was used as the O2 gas creator, which oxidized Ti4O7 to TiO2−x. Alternatively, it was KClO4 that pushed the structure rearrangement along the CS plane. 1]R, as they can be converted after slipping in the crystallographic shear plane (1 2 1)R. The rearrangement of the structure caused by oxygen deficiency can be reversed with the increase in the O/Ti ratio. In this study, solid KClO4 was used as the O2 gas creator, which oxidized Ti4O7 to TiO2−x. Alternatively, it was KClO4 that pushed the structure rearrangement along the CS plane. | ||
Measurement
X-ray diffraction (XRD) results of as-prepared products were recorded on a Bruker D8 Focus using a Cu Kα radiation (wavelength λ = 1.5418 Å, current I = 40 mA, voltage U = 40 kV). The range of 2-theta was from 10° to 80° at a scanning rate of 12° min−1. Based on the peak areas of rutile TiO2 (1 1 0) and Ti4O7 (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0), the amounts of residual Ti4O7 in the product were calculated by the standard curves.44 More structural information on the products was obtained via characterization with high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED), taking the raw materials and critical sample as references. For the purpose of (a) observing whether there was a clear phase interface and (b) characterizing the orientation relationships, the product synthesized in the presence of 15 mol% KClO4 was chosen. To compare the structures, information from the zone axis [1 0 0]T was carefully extracted. The content of oxygen in the products was measured via thermo-gravimetric analysis (TGA) at a rate of 10° min−1 from room temperature to 950 °C. UV-visible diffuse reflection spectroscopy (DRS) was used to compare the different light absorption capacities and band gaps as the amount of KClO4 increased. The scanning speed was 600 nm min−1, and the analysis range went from 1850 to 350 nm.
0), the amounts of residual Ti4O7 in the product were calculated by the standard curves.44 More structural information on the products was obtained via characterization with high resolution transmission electron microscopy (HRTEM) and selected area electron diffraction (SAED), taking the raw materials and critical sample as references. For the purpose of (a) observing whether there was a clear phase interface and (b) characterizing the orientation relationships, the product synthesized in the presence of 15 mol% KClO4 was chosen. To compare the structures, information from the zone axis [1 0 0]T was carefully extracted. The content of oxygen in the products was measured via thermo-gravimetric analysis (TGA) at a rate of 10° min−1 from room temperature to 950 °C. UV-visible diffuse reflection spectroscopy (DRS) was used to compare the different light absorption capacities and band gaps as the amount of KClO4 increased. The scanning speed was 600 nm min−1, and the analysis range went from 1850 to 350 nm.
Photo-thermal experiment
Under simulated light irradiation, the photo-thermal response of the materials was evaluated by monitoring the temperature change in the compressed sample discs with time. These sample discs (m = 0.05 g, Φ = 8 mm) were compressed under 5 MPa. When testing, the UV region of the simulated light was cut off with a 400 nm filter to measure the non-intrinsic light-absorptions and heat-conversion abilities. The discs were placed in the middle of the light beam irradiation at an invariant distance (17 cm). The temperature of the discs’ top faces increased when illuminated for 4 min, and decreased when the light was turned off. In this study, simulated light was produced using a Xenon Lamp (CEL-HXF300, AULIGHT, Beijing) with a voltage of 14 V and a current of 20 A.Results and discussion
To some extent, there are structural connections between the Magnéli phase Ti4O7 and the rutile phase TiO2, as shown in Fig. S1.† Here, the structural models were downloaded from the ICSD database (ICSD 6098 for Ti4O7, ICSD 88625 for rutile TiO2). Structurally, Magnéli Ti4O7 and reduced rutile TiO2 can be reversibly transformed, which can be strictly controlled by an oxidation or reduction process by changing the O/Ti ratio (Fig. 1a). The projections of [TiO6]8− octahedral arrays have octagonal shapes, when viewed along the [1 1]R axis of rutile TiO2 or [0 1 0]T axis of the Magnéli phase Ti4O7 (Fig. 1c). Due to the difference in the unit cell selection, the subscripts ‘R’ and ‘T’ after the Miller index represent rutile TiO2 and Ti4O7 in the following discussion, respectively. From the projection in Fig. 1c, the structural similarities between rutile TiO2 and Ti4O7 are very clear. For contrast, the differences between the two structures are colored in orange, while the parts that show similarities are colored in pale blue. The coloring method of octahedrons in the Ti4O7 structure is similar to that of a chess board, in comparison with the projections along the [0 0 1]R and [3 2 1]T directions (Fig. S1d†). There is a certain corresponding relationship in these coordinate systems.47 In Fig. S1b and S1c,† the triclinic coordinate system of Ti4O7 were overlapped to the rutile structure. As a result, the (1 2 1)R CS plane is related to the (0 0 1)T plane. Actually, the structures with two different colors in Ti4O7 are the same, but with a mere shift in position. After the slip, the octahedrons in the Ti4O7 structure are now coplanar, while the octahedrons in the rutile structure only share points and edges.45 Along the [4 ]T direction, the co-plane joints of [TiO6]8− octahedrons are marked with Ti–Ti bonds, which form the intersection between (1 0 4)T (Fig. 1c) and (0 4)T (Fig. S5b†). It was the loss of oxygen in these joint parts that caused the slip to happen, as shown in Fig. 1c. According to the above description, the serrated shape of the (1 0 )T plane can be decomposed into alternating units of two rutile slabs and one corundum slab. As a result, the corundum slab needs oxygen supplementation. KClO4, which could be replaced by Na2O2, KMnO4 or crystal water,44 was adopted as the gas creator to supply the missing oxygen elements in this study. It is predicted that the amount of KClO4 used can determine the degree of reaction in our closed vessel (Fig. 1b). After incomplete oxidation of Ti4O7 using a controlled amount of solid atmosphere creator, black TiO2−x can be obtained.
The XRD results (Fig. 2a) of the products revealed that Magnéli Ti4O7 gradually transformed into rutile TiO2 when the amount of KClO4 was continuously increased. The diagram for the calculated ratio of residual Ti4O7versus the mixing ratio of KClO4 in Fig. 2b helps visualize this concept. From the linear fitting results in Fig. 2b, the critical mixing ratio of KClO4 with respect to Ti4O7 is around 17.5 mol%, after which rutile TiO2−x with different subscripts “x” can be obtained. The changed color in the products during the reaction verified the gradual process, and pictures of the samples are presented in Fig. 2b. The calculated O/Ti ratio for the 17.5 mol% KClO4 sample with the rutile phase is 1.925, which is close to that for pure rutile TiO1.933 reported by Alcock, et al.41,48 Based on our existing knowledge, it is difficult to keep the rutile structure unchanged at such a high oxygen deficit, but rather the Magnéli phase (e.g. TinO2n−1, n ≥ 12) should be the expected product. After comparison, we found that the temperatures for the syntheses of the two were different. Namely, the as-prepared products were obtained at 600 °C for 2 h in this study, while the Magnéli phases are usually synthesized at over 850 °C.45 In this case, the product obtained using 15 mol% KClO4 was reheated at 950 °C for 2 h in a vacuum container. The phases of the sample after furnace cooling were identified to be Ti8O15 (PDF# 50-0790, major phase) and anatase TiO2 (PDF# 71-1169, second phase), as shown in Fig. S2,† while the main rutile phase TiO2 in this study(treated at 600 °C) disappeared. There are two problems in using Ti as a raw material. One is that the particle size will affect the oxidation degree, and the other is a mixture of rutile and anatase TiO2 (Fig. S3†) will be obtained as the product. Because of the similar structure between the Ti4O7 and rutile TiO2 (Fig. 1c), it was prior to producing rutile TiO2 when the Ti4O7 was incompletely oxidized at 600 °C. In this case, the O/Ti ratio of rutile TiO2 obtained is lower than 2, and this metastable structure can be rearranged to a Magnéli phase at high temperatures.
 | ||
| Fig. 2 (a) The XRD patterns for products with different molar ratios of KClO4. When the amount of KClO4 exceeded 17.5 mol%, Ti4O7 almost disappeared. (b) The residual content of Ti4O7 was calculated by peak area comparison.44 The product colors changed from black to grey after controllable oxidation, integrated in (b). | ||
On account of the structural similarity between Ti4O7 and rutile TiO2, the structural rearrangement along the CS plane can be reversed when using a solid atmosphere creator KClO4. According to TEM and SAED results (Fig. 3), the reverse slip was monitored in this study. Fig. 3a–c correspond to Ti4O7 and Fig. 3d–f correspond to samples prepared with 15 mol% KClO4. Single grains were selected for high resolution studies (Fig. 3a and d). In Fig. 3b, a long-period structure emerged in Magnéli Ti4O7, which originated from rutile TiO2 formation. The long-period structure, also known as super lattice, is parallel to the (0 0 2)T plane with a d-spacing of 6.13 Å. The SAED pattern shows that there are four proportional planes in this super lattice,41 which are demarcated as (0 0 2)T, (0 0 4)T, (0 0 6)T and (0 0 8)T in Fig. 3c. When treated with 15 mol% KClO4, the long-period structure disappeared, as the alternating units of two rutile slabs and one corundum slab are destroyed by the generated oxygen gas. The oxidation process is incomplete, so some slips happened on the transient period between Ti4O7 and rutile TiO2. The diffraction points of the super lattice were replaced by a series of parallel white lines (Fig. 3f), oriented at about 45°. The contrast lines in HRTEM images were repainted deconvoluted, obtaining the graph for the distribution of intensity with distance (Fig. 3g and h), where the unified long period for Ti4O7 and varied long period for 15 mol% KClO4 products are clearly presented. The existence of the (0 0 8)T planes with different periodicities can partly explain the result, as the rearrangement happened along the (1 2 1)R CS plane (Fig. 1c). Rutile TiO2 was produced in situ after the aforementioned process, and the diffraction spots of the (0 0 7)T or (1 2 1)R plane are clearly visible among the white lines (Fig. 3f and Table S1†). In order to facilitate a comparison, the calibration results of diffraction points in Fig. 3f were accomplished remodeled in the Ti4O7 coordinate system. XRD results in Fig. 2a revealed that the main phase in 15 mol% KClO4 oxidation product is rutile TiO2. As a consequence, we compared the similar structure of Ti4O7 and rutile TiO2 in Fig. S5,† viewing along the [1 0 0]T and [ 0 1]R directions, respectively. Despite the different structures after slipping, there were still some connections between Ti4O7 and rutile TiO2, such as (0 4)T to (0 2 0)R, (0 2 4)T and (0 2 3)T to (1 0 1)R. The theoretical diffraction points (Fig. S5c and S5f†) calculated with the given models indicated a slight change in the angle of base vectors when oxidizing Ti4O7 to rutile TiO2, as shown in Table S2.† What's more, the split points in Fig. 3f can be attributed to the reordering of the (0 4)T plane after oxidation.42 In spite of the difficulty of phase discrimination caused by the similarities of these structures, we could determine that the oxidation process was not just a surface reaction, but involved many complex processes such as slips and structural rearrangements. The circuitous, two-step approach from white TiO2 to Ti4O7 and then to black TiO2−x, was observed to introduce planar defects into the crystal. For example, periodic disordered structures were formed by the slip along the CS plane (Fig. 3e). In Fig. S6,† the existence of twin structures and crystal mismatching were attributed to the stacking fault that happens after rearrangement.
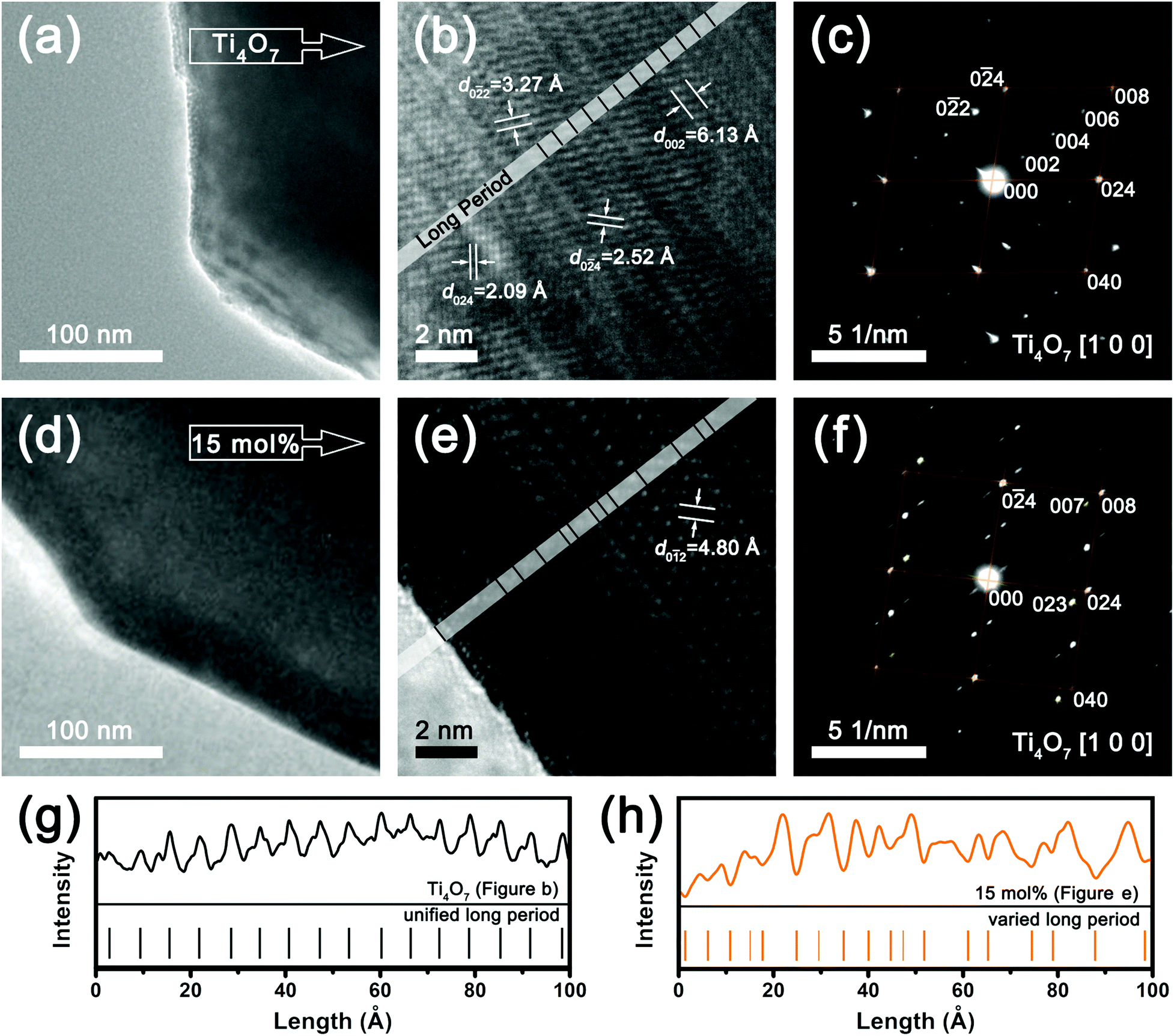 | ||
| Fig. 3 The TEM and SAED results for Ti4O7 (a–c) and 15 mol% KClO4 products (d–f). For the ease of comparison, auxiliary lines in orange represent the grids made up of base vectors. The yellow green lines in (f) show that the emerging points are associated with rutile TiO2, where (0 0 7)T and (0 2 3)T correspond to (1 2 1)R and (1 0 1)R, respectively (Fig. S5†). The long periods of the CS plane were determined by contrast change, (g) for Ti4O7 and (h) for 15 mol% KClO4 sample. | ||
The loss of oxygen in the products was measured via TG analysis, as shown in Table S3 and Fig. S7.† The samples were heated in air from room temperature to 1000 °C. After that, they were all completely oxidized into TiO2. The mass change in the samples can be divided into three stages. In stage I, the weight of the products decreased due to the loss of residual water. In fact, the products (denoted as TiO1.9 and TiO1.925) were washed using DI water and then freeze-dried to reduce the amount of residual KCl. In stage II, the main oxidation process occurred, and the mass sharply increased. In stage III, the rate of oxidation slowed down because of the diffusion and redistribution of oxygen between inner and outer layers. According to the results of stage II and III (Table S3†), the subscript “x” in our products (TiO2−x) was calculated, which was close to the theoretical value. That is to say, the mixing ratio of KClO4 indeed determined the O/Ti ratio in these products. The solid atmosphere creator KClO4 is much easier to control if compared to gas flow systems. As a result, black TiO2−x with a specific subscript “x” can be prepared in this way.
High concentrations of defects can improve the light absorption of TiO2, which was verified by UV-visible diffuse reflection spectroscopy (Fig. 4). With the increase in the KClO4 content for Ti4O7, the subscript “x” in the as-prepared TiO2−x decreased (Table S3 and Fig. S7†). In this process, oxygen defects can be effectively supplemented, resulting in the Fermi level falling from the conduction band of Ti4O7 to the forbidden band of rutile TiO2.49,50 Meanwhile, the light absorption ability in the range from 600 to 1800 nm decreased and absorption shifted to lower wavelengths (inner plot in Fig. 4a), which implies a change in the band structure. The calculated band gap (Eg) values of the as-prepared samples were consistent with above conclusion (Fig. 4b). There seems to be a relationship between the phase purity and light absorption, which means that the degree of oxidation needs to be controlled to ensure that the desired product is black rutile TiO2−x. We think that this is possible with the reverse slipping strategy, after combining the results of XRD and DRS. The control of the reaction degree ultimately becomes the weight control of the solid atmosphere creator, such as KClO4 in this study. Moreover, Ti4O7 was designed to be totally oxidized (Fig. S8†) and the Eg of the white product was calculated to be 3.03 eV, which further verified the TG results. As for the absorption results of the mixture (mixing ratios less than 17.5 mol% KClO4) in Fig. S9,† there seems to be an overall effect that consists of the combined metallic absorption of Ti4O7 and the semiconductor absorption of rutile TiO2 (Fig. S8†).
 | ||
| Fig. 4 (a) UV-visible diffuse reflection spectrum of the as-prepared rutile TiO2−x. With the increase in the KClO4 content, the absorption in the range of 600 to 1800 nm decreased and absorption edge shifted to lower wavelengths (inner figure). (b) Calculation of the band gap (Eg) of the as-prepared samples using the formula: (αhν)n = A(hν − Eg), in which n = 2 for the direct absorption of rutile TiO2.51,52 With the increase in the KClO4 content, the Eg gradually increased until close to Eg of rutile TiO2. | ||
To further illustrate the ability of light absorption, a simple photo-thermal response experiment was conducted. The surface temperature of the as-prepared products, which was monitored using an IR sensor, showed a convex exponential growth characteristic (Fig. S10†), when illuminated with a Xenon lamp simulator for 4 min. In order to carry out control experiments, 0.05 g of different samples were all compressed into 8 mm discs under 5 MPa. For defective TiO2−x, we were concerned with the photo-thermal properties in the visible and infrared regions. With a 400 nm filter, the real-time temperature data were monitored and fitted in Fig. 5a. When the lamp was turned on, the curves increased rapidly at first, then slowed down, and finally they reached equilibrium. The equations were similar to Newton's cooling formula,53,54 assuming that the heating rate (dT/dt) is proportional to the temperature difference, as shown in the following formula:

 | ||
| Fig. 5 (a) Real-time temperature change (scatter points) of the as-prepared samples under irradiation of simulative light with a 400 nm filter. The fitting curves (lines) are added with the fitting formula (b). Related parameters are given, such as (b) equilibrium temperature (Tmax, Tmin), equilibrium time (t0.99, time to reach 0.99Tmax), (c) time constant (τon, τoff) and (d) instantaneous velocity (dT/dt|t=0 s) of temperature when the simulative light was turned on or off. | ||
The Tmax values were determined by the built-in fitting formula, which was obtained by solving the above differential equation. With the increase in the mixing ratio, Tmax values decreased gradually (Fig. 5b), consistent with results of light absorption (Fig. 4a). With a 400 nm filter, the Tmax values for samples obtained with 15 mol% and 17.5 mol% KClO4 can reach 70.7 °C and 66.8 °C within one minute, respectively, which were higher than 38.7 °C for white TiO2 in three minutes (Fig. 5b). A high oxygen deficiency in TiO2−x may introduce more relaxation levels in the band structure, with which the absorbed light energy can convert into heat energy.54 The proportionality coefficient bon represents the speed of heating up,54 and its reciprocal τon has the dimension of time. In Fig. 5c, the integrated time constants showed that 15 mol% and 17.5 mol% KClO4 samples had the fastest temperature changes among the samples, almost five times higher than pristine rutile TiO2. For comparative purposes, the time required to reach 0.99Tmax was denoted as t0.99. With the parameter t0.99, we can comprehensively estimate the photo-to-thermal speed. For products made at the critical mixing ratio of 17.5 mol% KClO4, the t0.99 value is 51.8 s. By contrast, the t0.99 value is 182.8 s for pristine TiO2, which is 3.5 times than that for the 17.5 mol% KClO4 sample. As a result, 17.5 mol% KClO4 samples spend the least time achieving balance, exhibiting better photo-to-thermal response. It is not particularly ideal to use only one formula to fit the total data, as shown in Fig. 5a. Therefore, we used the linear fitting method to calculate the start-up speed, and the calculated results are presented in Fig. 5d. The linear fitting value also showed that the initial response speed decreased with the increase of mixing ratio, which was consistent with the above formula.
Conclusion
Considering the structural similarities between Ti4O7 and rutile TiO2, a reverse slipping strategy is presented for the preparation of black TiO2−x with great solar absorption in this study. The serrated shape in the (1 0) T plane could be decomposed into alternating units of two rutile slabs and one corundum slab. As the corundum slab needs oxygen supplementation, solid KClO4 serves as the oxygen provider. Because of the similar slabs in structure, it was prior to producing rutile TiO2 when the Ti4O7 was incompletely oxidized at 600 °C. The slip was evidenced by SAED results. The super lattice diffractions were replaced with lines in the same position, which indicated the occurrence of a slip during the oxidation process. The color and band gap can be effectively controlled by adjusting the mixing ratios of KClO4. TG measurement further verified the obtained results. The as-prepared black TiO2−x with tunable band gaps showed a considerable photo-thermal response. Using a 400 nm filter, the Tmax values of 15 mol% and 17.5 mol% KClO4 samples can reach 70.7 °C and 66.8 °C within one minute, respectively, which are higher than the 38.7 °C of white TiO2 in three minutes. The response speeds of those two samples were about five times faster than pristine rutile TiO2. Moreover, the existence of oxygen defects has a great correlation with absorption and thermal dispersion of visible and infrared light. Further consideration should be taken into the essential determinations of the photo-thermal response speed and equilibrium temperature. Moreover, the controllable oxidation of the Magnéli phase oxides can be understood from the slip phenomenon, and the series of oxidants can be further expanded.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National key research and development program (Grant No. 2016YFB0901600), NSF of China (Grant No. 21871008, 21801247), Science and Technology Commission of Shanghai (Grant No. 17ZR1434400, 18YF1427200), Innovation Project of Shanghai Institute of Ceramics (Grant No. Y93ZC2120G) and sponsored by Shanghai Sailing Program (Grant 18YF1427200).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- J. Huang, Y. He, M. Chen, B. Jiang and Y. Huang, Sol. Energy, 2017, 155, 1225–1232 CrossRef CAS.
- Z. W. Seh, S. Liu, M. Low, S. Y. Zhang, Z. Liu, A. Mlayah and M. Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef CAS.
- Z. Wang and X. Lou, Adv. Mater., 2012, 24, 4124–4129 CrossRef CAS.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928–3934 CrossRef CAS.
- J. Xu, W. Ding, W. Zhao, W. Zhao, Z. Hong and F. Huang, ACS Energy Lett., 2017, 2, 659–663 CrossRef CAS.
- H. Luo, T. Takata, Y. Lee, J. Zhao, K. Domen and Y. Yan, Chem. Mater., 2004, 16, 846–849 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS.
- C. Burda, Y. Lou, X. Chen, A. C. S. Samia and J. L. Gole, Nano Lett., 2003, 3, 1049–1051 CrossRef CAS.
- G. Liu, J. Pan, L. Yin, J. T. Irvine, F. Li, J. Tan, P. Wormald and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 3233–3238 CrossRef CAS.
- P. V. R. K. Ramacharyulu, D. B. Nimbalkar, J. P. Kumar, G. K. Prasad and S.-C. Ke, RSC Adv., 2015, 5, 37096–37101 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Müller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309 CrossRef CAS.
- H. Lu, B. Zhao, R. Pan, J. Yao, J. Qiu, L. Li and Y. Liu, RSC Adv., 2013, 4, 1128–1132 RSC.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- Y. Guo, S. Chen, Y. Yu, H. Tian, Y.-L. Zhao, J.-C. Ren, C. Huang, H. Bian, M. Huang, L. An, Y. Li and R. Zhang, J. Am. Chem. Soc., 2019, 141, 8407–8411 CrossRef CAS.
- W. Zhou, W. Li, J.-Q. Wang, Y. Qu, Y. Yang, Y. Xie, K. Zhang, L. Wang, H. Fu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 9280–9283 CrossRef CAS.
- S.-T. Myung, M. Kikuchi, C. S. Yoon, H. Yashiro, S.-J. Kim, Y.-K. Sun and B. Scrosati, Energy Environ. Sci., 2013, 6, 2609–2614 RSC.
- T. Lin, C. Yang, Z. Wang, H. Yin, X. Lü, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2014, 7, 967 RSC.
- S. Hoang, S. P. Berglund, N. T. Hahn, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2012, 134, 3659–3662 CrossRef CAS PubMed.
- Y. Lu, K. Sagara, L. Hao, Z. Ji and H. Yoshida, Mater. Trans., 2012, 53, 1208–1211 CrossRef CAS.
- L. Hao, K. Miyazawa, H. Yoshida and Y. Lu, Mater. Res. Bull., 2018, 97, 13–18 CrossRef CAS.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin, X. Xie and M. Jiang, Energy Environ. Sci., 2013, 6, 3007–3014 RSC.
- G. Ou, Y. Xu, B. Wen, R. Lin, B. Ge, Y. Tang, Y. Liang, C. Yang, K. Huang and D. Zu, Nat. Commun., 2018, 9, 1302 CrossRef.
- A. Sinhamahapatra, J.-P. Jeon and J.-S. Yu, Energy Environ. Sci., 2015, 8, 3539–3544 RSC.
- J. Xu, Z. Tian, G. Yin, T. Lin and F. Huang, Dalton Trans., 2016, 46, 1047 RSC.
- M. Ye, J. Jia, Z. Wu, C. Qian, R. Chen, P. G. O'Brien, W. Sun, Y. Dong and G. A. Ozin, Adv. Energy Mater., 2017, 7, 1601811 CrossRef.
- X. Wang, R. Fu, Q. Yin, H. Wu, X. Guo, R. Xu and Q. Zhong, J. Nanopart. Res., 2018, 20, 89 CrossRef.
- L. Yi, S. Ci, S. Luo, P. Shao, Y. Hou and Z. Wen, Nano Energy, 2017, 41, 600–608 CrossRef CAS.
- G. Zhu, J. Xu, W. Zhao and F. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 31716–31721 CrossRef CAS.
- X. Zou, J. Liu, J. Su, F. Zuo, J. Chen and P. Feng, Chem. – Eur. J., 2013, 19, 2866–2873 CrossRef CAS.
- Q. Kang, J. Cao, Y. Zhang, L. Liu, H. Xu and J. Ye, J. Mater. Chem. A, 2013, 1, 5766–5774 RSC.
- S. Tominaka, Y. Tsujimoto, Y. Matsushita and K. Yamaura, Angew. Chem., 2011, 123, 7556–7559 CrossRef.
- S. Tominaka, Inorg. Chem., 2012, 51, 10136–10140 CrossRef CAS.
- G. Zhu, Y. Hao, C. Yang, H. Cui, W. Zhou, J. Xu, T. Lin and F. Huang, ChemCatChem, 2015, 7, 2614–2619 CrossRef CAS.
- G. Zhu, Y. Shan, T. Lin, W. Zhao, J. Xu, Z. Tian, H. Zhang, C. Zheng and F. Huang, Nanoscale, 2016, 8, 4705 RSC.
- M. Reece and R. Morrell, J. Mater. Sci., 1991, 26, 5566–5574 CrossRef CAS.
- E. Iguchi and F. Matsushima, J. Mater. Sci., 1986, 21, 1046–1050 CrossRef CAS.
- J. S. Anderson and R. J. D. Tilley, J. Solid State Chem., 1970, 2, 11 Search PubMed.
- L. A. Bursill, B. G. Hyde and D. K. Philp, Philos. Mag., 1971, 23, 1501–1513 CrossRef CAS.
- L. A. Bursill, B. G. Hyde, O. Terasaki and D. Watanabe, Philos. Mag., 1969, 20, 347–359 CrossRef CAS.
- J. Huang, J. Xu, X. Che and C. Huang, Chem. – Eur. J., 2019, 25, 10642–10649 CrossRef CAS.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- V. Eyert, U. Schwingenschlogl and U. Eckern, Chem. Phys. Lett., 2004, 390, 151–156 CrossRef CAS.
- M. Marezio and P. D. Dernier, J. Solid State Chem., 1971, 3, 340–348 CrossRef CAS.
- B. C. Alcock, B. C. H. Steele and S. Zador, Proc. Br. Ceram. Soc., 1967, 8, 231–245 Search PubMed.
- A. C. M. Padilha, A. R. Rocha and G. M. Dalpian, Phys. Rev. Appl., 2015, 3, 24009 CrossRef CAS.
- H. Li, Z. Zhang and L. P. Shi, J. Electron. Mater., 2016, 45, 1142–1153 CrossRef CAS.
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS.
- R. H. S. Winterton, Contemp. Phys., 1999, 40, 205–212 CrossRef CAS.
- J. Wang, Y. Li, L. Deng, N. Wei, Y. Weng, S. Dong, D. Qi, J. Qiu, X. Chen and T. Wu, Adv. Mater., 2017, 29, 1603730 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qi01042d |
| This journal is © the Partner Organisations 2020 |