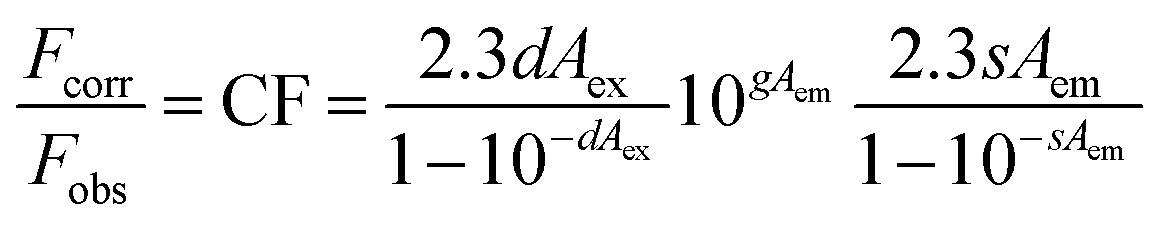DOI:
10.1039/D0PY01292K
(Paper)
Polym. Chem., 2020,
11, 7421-7428
Facile C–H iodination of electron deficient benzodithiophene-S,S-tetraoxide for the development of n-type polymers†
Received
9th September 2020
, Accepted 22nd October 2020
First published on 4th November 2020
Abstract
A facile functionalization of the electron-poor building block, benzo[1,2-b:4,5-b0] dithiophene-1,1,5,5-tetraoxide (BDTT) by C–H iodination reaction has been reported. This functionalization opens a new avenue to incorporate this electron-deficient moiety into conjugated systems for organic semiconductors (OSCs). Two novel donor–acceptor conjugated polymers (PBDTT-Th and PBDTT-BDT) were synthesized at high yields by incorporating this functionalized BDTT moiety as an acceptor unit, and thiophene and benzodithiophene as donor units respectively. Though both polymers showed similar LUMO energy levels, the HOMO level of PBDTT-BDT (−5.47 eV) was higher than PBDTT-Th (−5.74 eV), due to the presence of the strong electron donating nature of benzodithophene as compared to thiophene. Both polymers showed electron accepting behavior, which was probed by performing fluorescence quenching experiments with the donor material P3HT. Furthermore, the space charge limited current (SCLC) method using Mott–Gurney law was carried out to determine the electron mobility of PBDTT-Th and PBDTT-BDT, which were found to be 6.26 × 10−4 and 7.67 × 10−4 cm2 V−1 s−1 respectively.
1. Introduction
The ability to tailor chemical structures to achieve desired optoelectronic properties makes organic semiconductors (OSCs) an attractive material in the modern technology world. Additionally, OSCs offer the opportunity to fabricate low cost, lightweight and flexible electronics, and optoelectronic devices which otherwise are inaccessible using conventional semiconductor materials such as silicon.1,2 Due to these advantages, in recent decades OSCs have attracted considerable attention from the scientific community for their application in organic light-emitting diodes,3–5 organic photovoltaics,6–8 organic field-effect transistors,9–11 and sensors.12 OSCs, including both small molecules and polymers, are commonly classified as p-type and n-type, in which the primary charge carrier is hole and electron, respectively. While p-type OSCs have seen a dramatic rise in performance over the last decade, n-type OSCs, in general, still lag behind p-type OSCs.13,14 This lack of high performance n-type OSCs is mainly related to the stability and difficulties in the synthesis of these materials and the low availability of electron-deficient building blocks that can be used for making n-type OSCs.15,16 Both types of OSCs with high performance are required in order to fabricate high-performance complementary circuits or p–n junction devices. Thus, there is a critical need for the exploration of new building blocks for making novel n-type OSCs with improved electron mobility, material stability both under processing and operational conditions, and ease of synthesis.
Benzodithiophenes (BDT) have been extensively explored as high-performance p-type OSCs.17,18 However, their oxidized counterpart benzo[1,2-b:4,5-b0] dithiophene-1,1,5,5-tetraoxide (BDTT), has been less utilized as a building block for developing n-type OSCs, despite being an electron-poor heterocycle with promise for the development of n-type materials.19–23 One likely reason for the underuse of BDTT to make n-type OSCs is the difficulty involved in functionalizing BDTT to make a precursor for carbon–carbon cross-coupling reactions. Usually, the functionalized BDTT building blocks are derived from the oxidation of already functionalized BDT moieties resulting in low yield,21,23,24 and there are only few reports on direct functionalization of BDTT moiety. For example, Punzi et al. have reported solvent-free Pd-catalyzed C–H arylation of BDTT,25 and our group has also developed an efficient copper-catalyzed direct arylation reaction for the regioselective functionalization of BDTT,22 which provides an opportunity to generate extended heteroaromatic conjugated systems. However, in order to incorporate BDTT moiety as an acceptor in a donor–acceptor (D–A) polymer system efficiently, it is desirable to have some functionality of BDTT that can directly be utilized for polymerization. Therefore, finding a better way to functionalize BDTT building blocks at good yield is of critical importance for its efficient utilization in making OSCs.
In this paper, we report the successful functionalization of BDTT by a C–H iodination reaction. Stille cross-coupling conditions were employed for the incorporation of BDTT as the electron acceptor unit in a donor–acceptor (D–A) diad to generate two n-type polymers. The electron-accepting properties of these polymers were investigated and compared to the standard acceptor, phenyl-C60-butyric-acid-methyl-ester (PC60BM), by studying their fluorescence quenching properties against a prototypical donor material, regioregular poly(3-hexylthiophene) (rr-P3HT). Furthermore, the electron mobility of these polymers was also evaluated by space charge limited current (SCLC).
2. Experimental setup
2.1. Materials
Anhydrous toluene was obtained from a solvent purification system under ultrapure argon. 4,8-bis(decyloxy)benzo[1,2-b:4,5-b′]dithiophene 1,1,5,5-tetraoxide (BDTT) and (4,8-bis((2-ethylhexyl)oxy)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) were synthesized according to the previously reported procedure.19,26 Regioregular poly(3-hexylthiophene) (rr-P3HT) was purchased from American Dye Source, Inc. Unless stated otherwise, all other materials were purchased from Sigma-Aldrich and were used as received.
2.2. Instrumentation
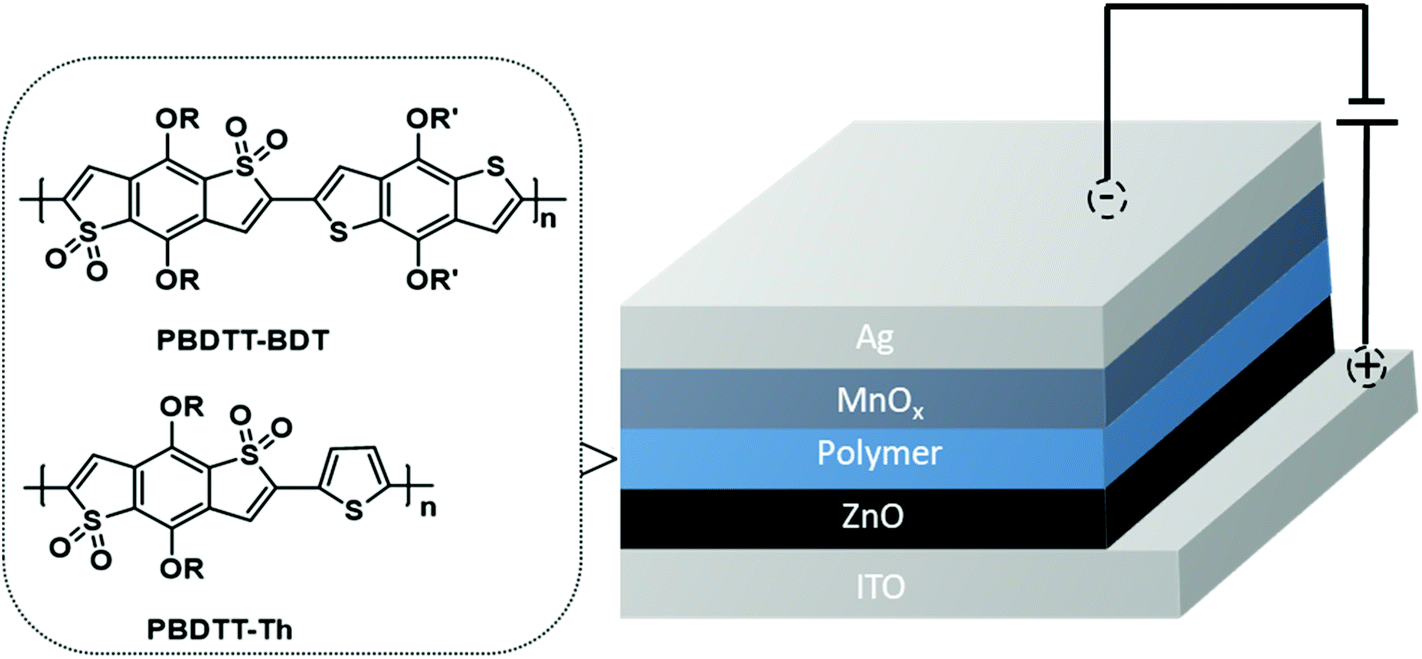
1H and 13C NMR spectra were acquired on a Bruker Avance 400 MHz instrument with TMS as the internal reference. Molecular weights of the polymers were obtained on a Waters gel permeation chromatography using a Waters pump with a Waters 2410 refractive index detector, with THF as the solvent at 35 °C with a flow rate of 1.0 mL min−1. Polystyrene standards were used for calibration. UV–visible and fluorescence spectra were recorded on a Cary 5000 UV–VIS–NIR spectrophotometer and a Cary Eclipse fluorescence spectrophotometer respectively. UV–visible and fluorescence measurements were obtained using polymer solutions in CHCl3, and thin films were drop-casted from these solutions. Cyclic voltammetry (CV) was performed on a BASI CV-50 W Version instrument with 0.1 M tetrabutylammonium hexafluorophosphate as the supporting electrolyte in dry acetonitrile using a platinum working electrode, a platinum wire as a counter electrode, and Ag/Ag+ as a reference electrode with a scan rate of 100 mV s−1. The CV experiment was performed by drop-casting a thin film of the polymer on the platinum working electrode. Thermal gravimetric analysis (TGA) measurements were performed using a high-resolution thermogravimetric analyzer TA instrument Model Q-50, within the temperature interval of 30–900 °C, with a rate of 20 °C min−1 under continuous nitrogen flow. Differential scanning calorimetry (DSC) was performed using a TA Instruments Model Q-2000 from −30 °C to 200 °C at a heating rate of 3 °C min−1 in a modulated mode. The second thermal cycle (cooling cycle) was chosen to observe the thermal transitions in polymers. The electron mobility of these two polymers was determined by SCLC measurements by using an electron-only device with the configuration of indium tin oxide (ITO)/ZnO/PBDTT-Th or PBDTT-BDT/oxides of manganese (MnOx)/Ag. The device configuration is shown in Fig. 1. The measurements of SCLC were performed on a cascade probe station equipped with Keithley 4200 systems. Each device has an active area of 10 mm2. The thickness of the film was measured with a XP1 Stylus Profilometer. Charge carrier mobility was measured in the SCLC regime according to the Mott–Gurney law: 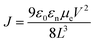 , where ε0 is the permittivity of free space (8.8542 × 10−12 F m−1), εr is the relative permittivity of semiconducting materials (which is used as 3.5 here), μe is electron mobility, V is applied voltage, and L is the thickness. All runs were performed under nitrogen.
, where ε0 is the permittivity of free space (8.8542 × 10−12 F m−1), εr is the relative permittivity of semiconducting materials (which is used as 3.5 here), μe is electron mobility, V is applied voltage, and L is the thickness. All runs were performed under nitrogen.
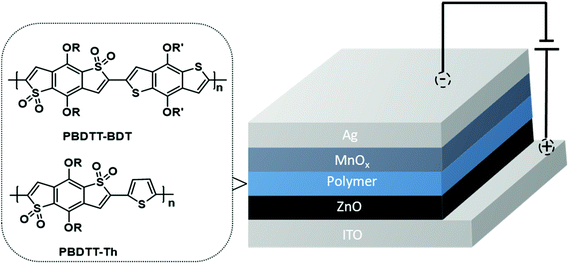 |
| | Fig. 1 Device configuration for electron mobility measurements. | |
2.3. Synthesis of 4,8-bis(dodecyloxy)-2,6-diiodobenzo[1,2-b:4,5-b′]dithiophene 1,1,5,5-tetraoxide (2)
Iodopentafluorobenzene (188 mg, 0.64 mmol), and potassium t-butoxide (9.0 mg, 0.08 mmol) were added to a solution of 4,8-bis(dodecyloxy)benzo[1,2-b:4,5-b′]dithiophene 1,1,5,5-tetraoxide (BDTT) (100 mg, 0.16 mmol) in toluene (0.2 M), and the mixture was stirred at room temperature for 30 minutes. After the reaction was completed, the solution was filtered through Celite, concentrated and subjected to chromatography on a silica gel with DCM and hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as eluent. Product 2 was isolated as a yellow solid (104 mg, 86%). Mp. 100–103 °C. 1H NMR (400 MHz, CDCl3) δ 7.59 (s, 2H), 4.43 (t, J = 6.5 Hz, 4H), 1.86 (p, J = 6.6 Hz, 4H), 1.48 (m, 4H), 1.38–1.27 (m, 32H), 0.88 (t, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 144.48, 132.97, 130.34, 129.49, 94.28, 31.92, 29.87, 29.65, 29.63, 29.57, 29.50, 29.35, 29.25, 25.67, 22.69, 14.13. HRMS (ESI) [M]+ calcd for C34H52I2O6S2m/z 874.1295, m/z found 874.1307.
:2) as eluent. Product 2 was isolated as a yellow solid (104 mg, 86%). Mp. 100–103 °C. 1H NMR (400 MHz, CDCl3) δ 7.59 (s, 2H), 4.43 (t, J = 6.5 Hz, 4H), 1.86 (p, J = 6.6 Hz, 4H), 1.48 (m, 4H), 1.38–1.27 (m, 32H), 0.88 (t, J = 6.5 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 144.48, 132.97, 130.34, 129.49, 94.28, 31.92, 29.87, 29.65, 29.63, 29.57, 29.50, 29.35, 29.25, 25.67, 22.69, 14.13. HRMS (ESI) [M]+ calcd for C34H52I2O6S2m/z 874.1295, m/z found 874.1307.
2.4. General procedure for the Stille polycondensation reactions
4,8-Bis(dodecyloxy)-2,6-diiodobenzo[1,2-b:4,5-b′]dithiophene 1,1,5,5-tetraoxide (100 mg, 0.11 mmol), Pd2(dba)3 (6.2 mg, 2 mol%) and tri-tert-butylphosphine (8.2 mg, 8 mol%) were placed in a 10 mL oven-dried Schlenk flask and put under vacuum for five minutes, then purged with argon. This process was repeated three times. After adding the bis(trimethylstannyl)-substituted monomer (0.11 mmol) and chlorobenzene (3 mL) inside the glove box, the reaction flask was taken outside, purged with nitrogen for 15 minutes and then heated at reflux for two days. The reaction mixture was cooled to room temperature and precipitated in methanol (20 mL) to give black precipitates. The resulting precipitate was filtered through a Soxhlet thimble, which was then subjected to successive methanol, hexane, acetone and chloroform extractions. Finally, the chloroform extraction was evaporated to dryness and re-precipitated from methanol to yield black polymers.
2.4.1.
PBDTT-Th
.
Black powder (66 mg, 87% yield), 1H NMR (400 MHz, CDCl3) δ 7.71 (b, 2H), 7.32 (b, 2H), 4.52 (b, 4H), 1.93 (b, 4H), 1.27 (b, 36H), 0.88 (b, 6H). Mn = 10.7 kDa and Đ = 3.6.
2.4.2.
PBDTT-BDT
.
Black powder (93 mg, 82% yield), 1H NMR (400 MHz, CDCl3) δ 8.09 (b, 2H), 7.68 (b, 2H), 4.55 (b, 4H) 4.18 (b, 4H), 2.02 (b, 6H), 1.82–1.26 (b, 44H), 1.12–1.04 (b, 6H), 0.88 (b, 12H). Mn = 14.4 kDa and Đ = 2.6.
3. Results and discussion
3.1. Synthesis and characterization
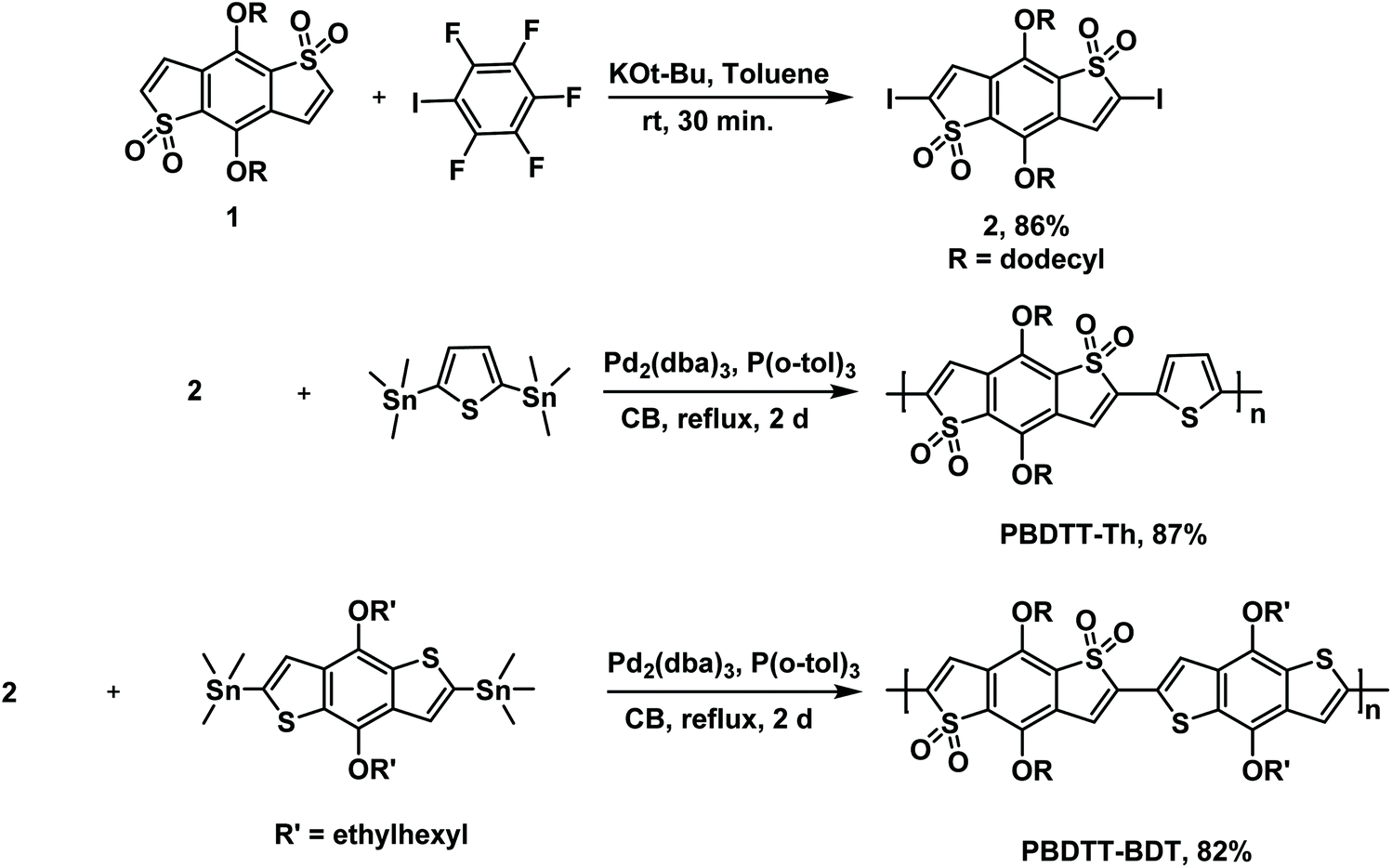
The synthetic routes for monomer 2 and polymers PBDTT-Th and PBDTT-BDT are depicted in Scheme 1. Compound 1 was successfully functionalized at high yield by utilizing the C–H iodination reaction of electron-deficient heterocycles recently developed by Q. Shi et al.27 This C–H iodination reaction was modified such that four equivalents of iodopentafluorobenzene and 0.5 equivalents of potassium tert-butoxide at room temperature afforded the diiodinated product 1 an 86% yield.
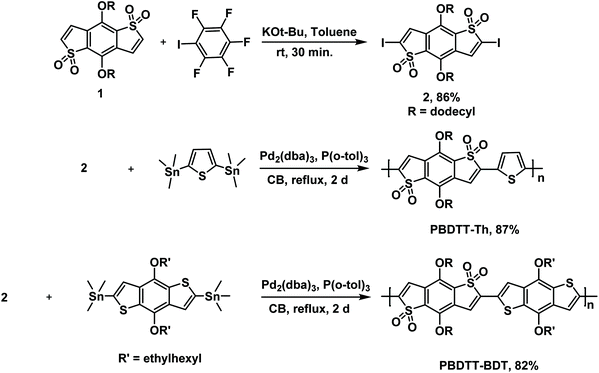 |
| | Scheme 1 Synthesis of monomers and polymers. | |
The PBDTT-Th and PBDTT-BDT copolymers were then synthesized by reacting compound 2 with 2,5-bis(trimethylstannyl)thiophene and (4,8-bis((2-ethylhexyl)oxy)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)bis(trimethylstannane) respectively under Stille copolymerization conditions at high yields of 87% and 82%. The average molecular weight (Mn) and polydispersity index (Đ) values of these polymers are given in Table 1. After Soxhlet extraction and reprecipitation, PBDTT-Th and PBDTT-BDT were obtained with moderate molecular weights of 10.7 and 14.4 kDa, respectively. Both of these polymers were soluble in common solvents like THF, chloroform, and chlorobenzenes, with PBDTT-BDT being highly soluble due to the presence of alkyl chains on the electron-donating unit (benzodithiophene).
Table 1 Structural and thermal properties of polymers
| Polymer |
M
na (kDa) |
M
wa (kDa) |
Đ
|
T
db (°C) |
T
gc (°C) |
|
Determined by GPC in THF using polystyrene standards.
5% weight loss temperature by TGA under N2.
Data from second scan reported, heating rate 3 °C min−1 under N2.
|
|
PBDTT-Th
|
10.7 |
38.5 |
3.6 |
335 |
— |
|
PBDTT-BDT
|
14.4 |
34.5 |
2.4 |
325 |
— |
3.2. Thermal characterization
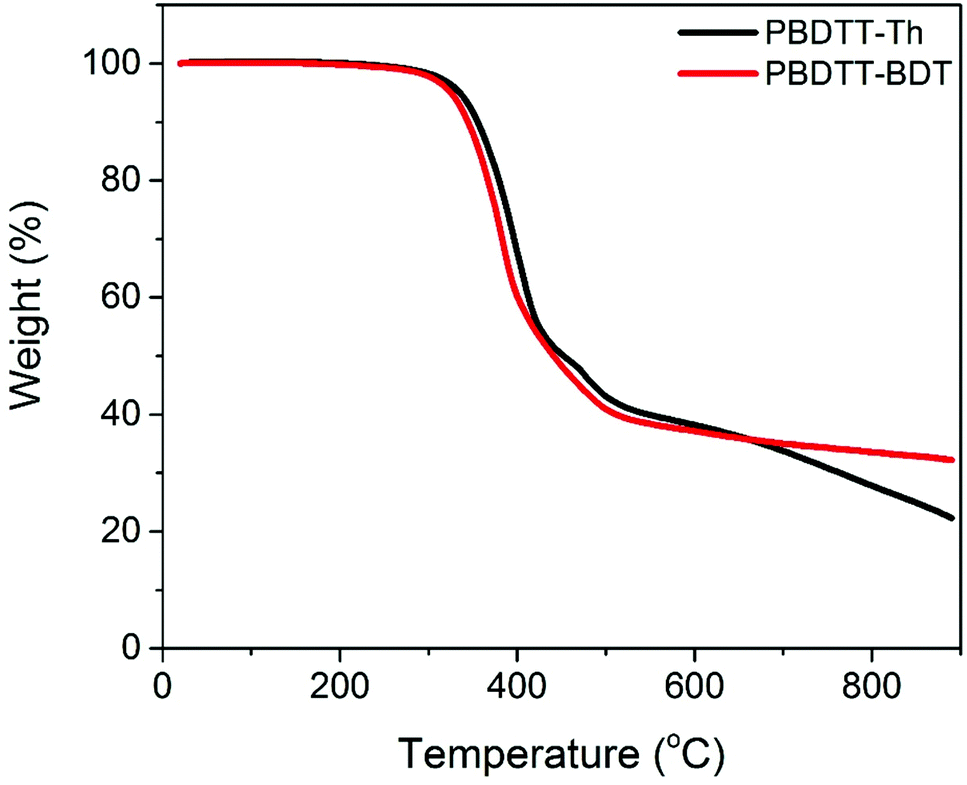
The thermal properties of the polymers were investigated by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA curves are shown in Fig. 2, and the data are summarized in Table 1. Both polymers showed excellent thermal stability with a decomposition temperature above 300 °C for both. DSC scans of the second cycle (cooling cycle) for the polymers are shown in Fig. S6.† In DSC thermograms, neither crystallization or melting transitions, nor glass transition temperatures, were observed in the scanned temperature range (−30 to 250 °C), suggesting the amorphous nature of the polymers.
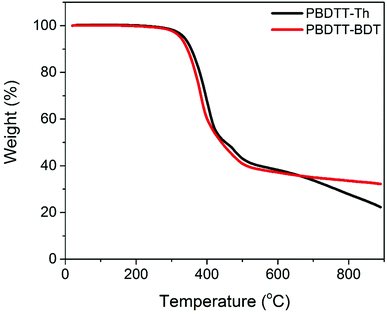 |
| | Fig. 2 TGA curves of PBDTT-Th and PBDTT-BDT. | |
3.3. Optical and electronic properties
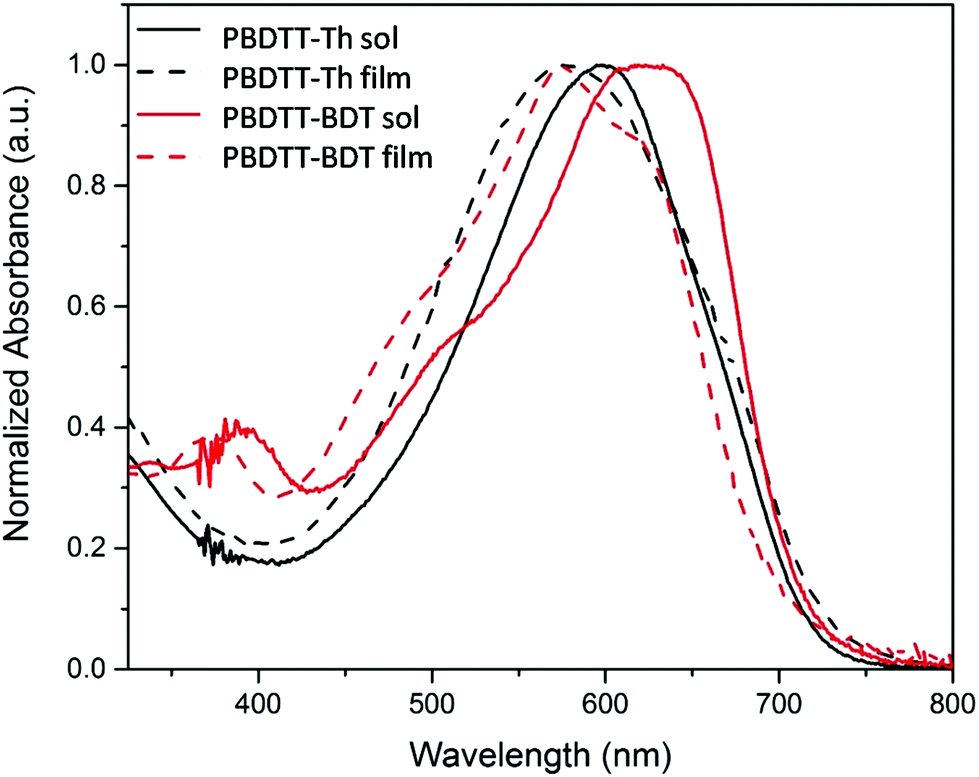
The optical properties of the polymers were investigated using UV-Vis absorption spectroscopy both in CHCl3 solutions and as drop cast films from a CHCl3 solution. The normalized absorption spectra of the polymers are shown in Fig. 3, and the results are summarized in Table 2. Both of these polymers showed broad absorption ranging from 400 nm to 725 nm with a high molar extinction coefficient (5 × 105 M−1). For PBDTT-Th, the absorption maxima of 595 nm and 577 nm were observed in solution and film, whereas for PBDTT-BDT the absorption maxima were 619 nm and 574 nm in solution and film respectively. Hypsochromic-shifted spectra were observed for both polymers upon film formation compared to those in solution, which was more pronounced in PBDTT-BDT. This spectral shift is consistent with the literature reporting similar polymer structure, suggesting that the packing manner of the main chains in these polymers may contain H-aggregation on film24,28 and may also have some pre-aggregates in solution due to the strong intermolecular attraction of the large fused and rigid conjugated systems.29
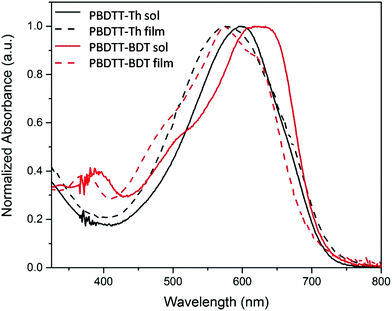 |
| | Fig. 3 Absorption spectra of polymers in solution (CHCl3, solid lines) and thin film (cast from CHCl3, dashed lines). | |
Table 2 The optical and electronic properties of the polymers
| Polymers |
Media |
λ
absa (nm) |
εa (M−1) |
E
oxb (V) |
E
reb (V) |
HOMOc (eV) |
LUMOd (eV) |
E
ecge (eV) |
|
Measured in dilute chloroform.
Measured from the onset of oxidation wave and reduction wave respectively.
Calculated from the onset of the first oxidation using the equations EHOMO (eV) = −[Eonsetox − E1/2(Fc/Fc+) + 4.8] where E1/2 (Fc/Fc+) is cell correction.
Calculated from the onset of the first reduction using the equation ELUMO (eV) = −[Eonsetred − E1/2(Fc/Fc+) + 4.8].
Calculated using the equation Eecg (eV) = HOMO–LUMO.
|
|
PBDTT-Th
|
CHCl3 |
595 |
4.99 × 105 |
|
|
|
|
|
| Film |
581 |
|
1.07 |
−0.95 |
−5.74 |
−3.72 |
2.02 |
| |
|
PBDTT-BDT
|
CHCl3 |
610 |
5.06 × 105 |
|
|
|
|
|
| Film |
584 |
|
0.80 |
−1.07 |
−5.47 |
−3.60 |
1.87 |
The electronic properties of PBDTT-Th and PBDTT-BDT were determined by cyclic voltammetry by depositing thin films of these polymers on the working electrode; the results are summarized in Table 2, and the voltammograms are shown in Fig. S5.† devices. The incorporation of the electron-deficient unit in the conjugated system helps to lower the energy of the LUMO level which is important to improve the air stability of the n-type polymer, as it could prevent the electron from being captured by water or oxygen in the air.30 However, the n-type OSCs cannot have a LUMO energy level lower than −4 eV because such polymers could be air-doped, resulting in the loss of their semiconducting properties.31 Therefore, n-type OSCs with a LUMO energy level between −4 to −4.5 eV are desired. Both of these polymers have lower LUMO energy levels, and as expected, both PBDTT-Th and PBDTT-BDT have quite similar LUMO energy levels of −3.72 eV and −3.60 eV respectively. But the HOMO energy level of PBDTT-BDT (−5.47 eV) is higher than that of PBDTT-Th (−5.74 eV). This is because both of these polymers have the same electron-poor unit with different electron-rich units. Polymers that have alternating electron-rich and electron-poor conjugated repeat units typically have HOMO energy levels that resemble the HOMO level of the electron-rich unit, while the LUMO resembles the electron deficient unit. The HOMO energy level of PBDTT-BDT is higher than that of PBDTT-Th, due to the presence of the comparatively high electron-rich unit benzodithiophene, as compared to the thiophene unit in PBDTT-Th. This also causes a decrease in the electronic bandgap of the PBDTT-BDT polymer (1.87 eV) as compared to PBDTT-Th (2.02 eV) as the high electron-rich unit increases the HOMO, thus making the bandgap less in PBDTT-BDT.
Neither of these polymers showed any significant emission. This gave an opportunity to illustrate the electron-accepting behavior of these polymers by performing fluorescence quenching experiments with the prototypical donor material, regioregular poly(3-hexylthiophene) (rr-P3HT).32 The fluorescence quenching experiments with rr-P3HT were performed with varying concentrations of PBDTT-Th and PBDTT-BDT. Due to the absorbance of polymers PBDTT-Th and PBDTT-BDT at both the excitation (360 nm) and emission wavelengths, the inner filter effect correction was carried out to get the corrected fluorescence spectra of rr-P3HT. The fluorescence intensity of P3HT was corrected using the following equation (eqn (1)),33
| | 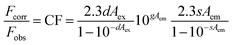 | (1) |
where
s is the excitation beam thickness (0.10 cm),
g is the distance from the edge of the sample beam to the edge of the cuvette (0.40 cm), and
d is the width of the cuvette (1.0 cm).
Aex and
Aem are the absorbances of the solution at the excitation and emission wavelengths (units of cm
−1).
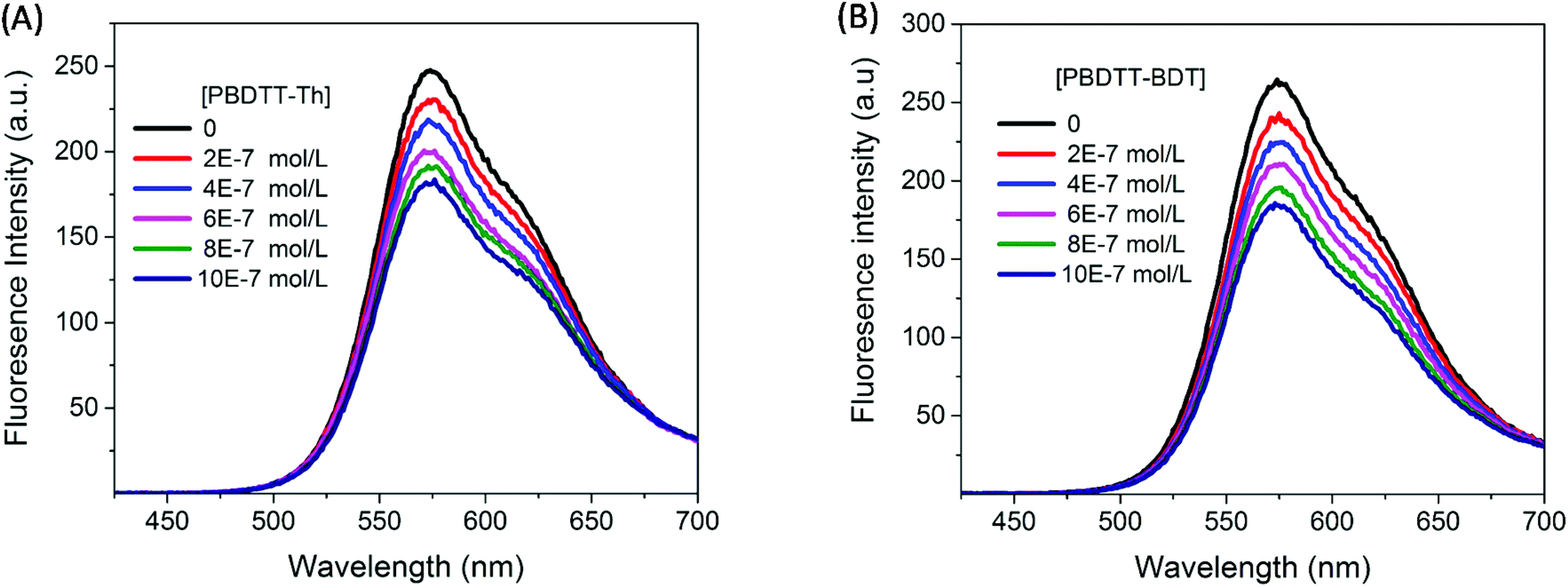
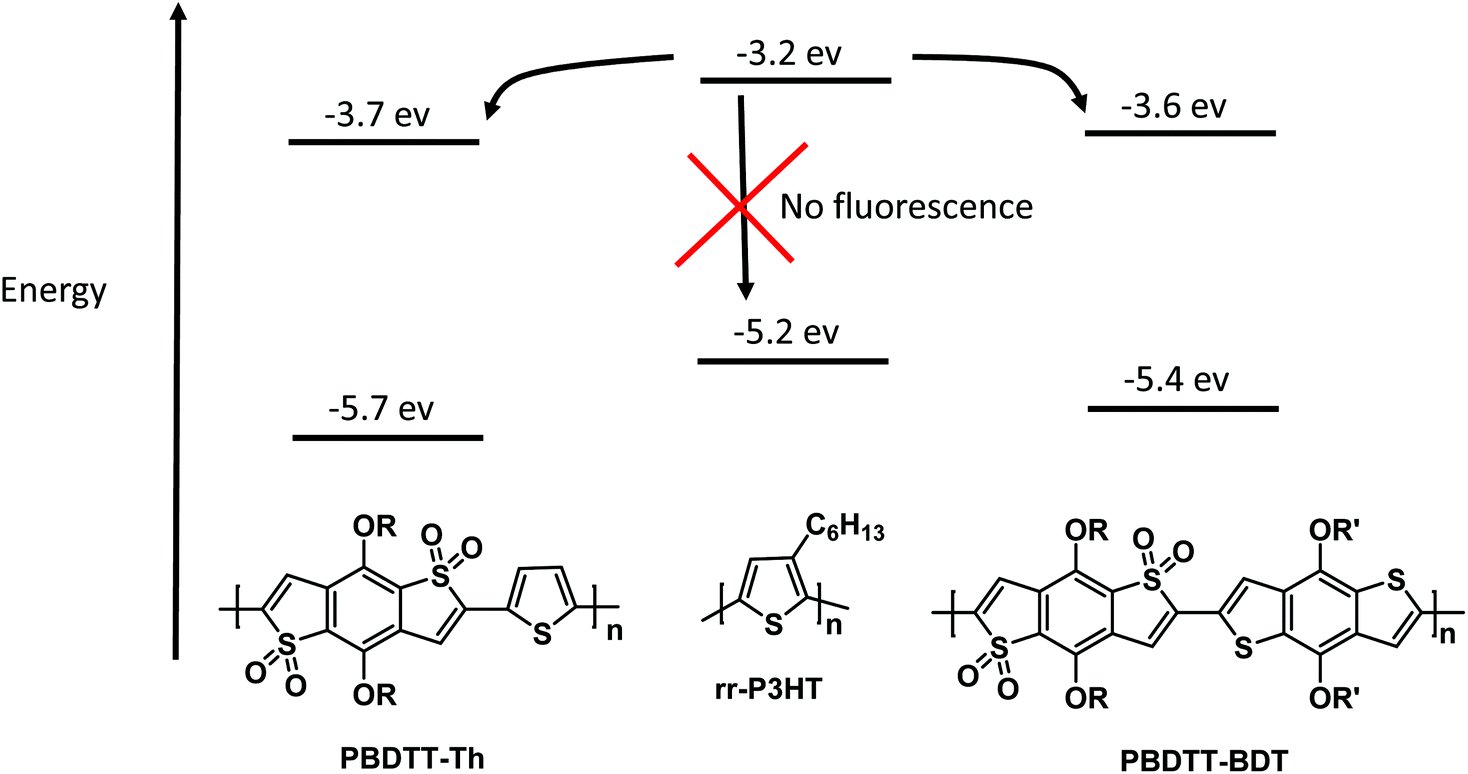
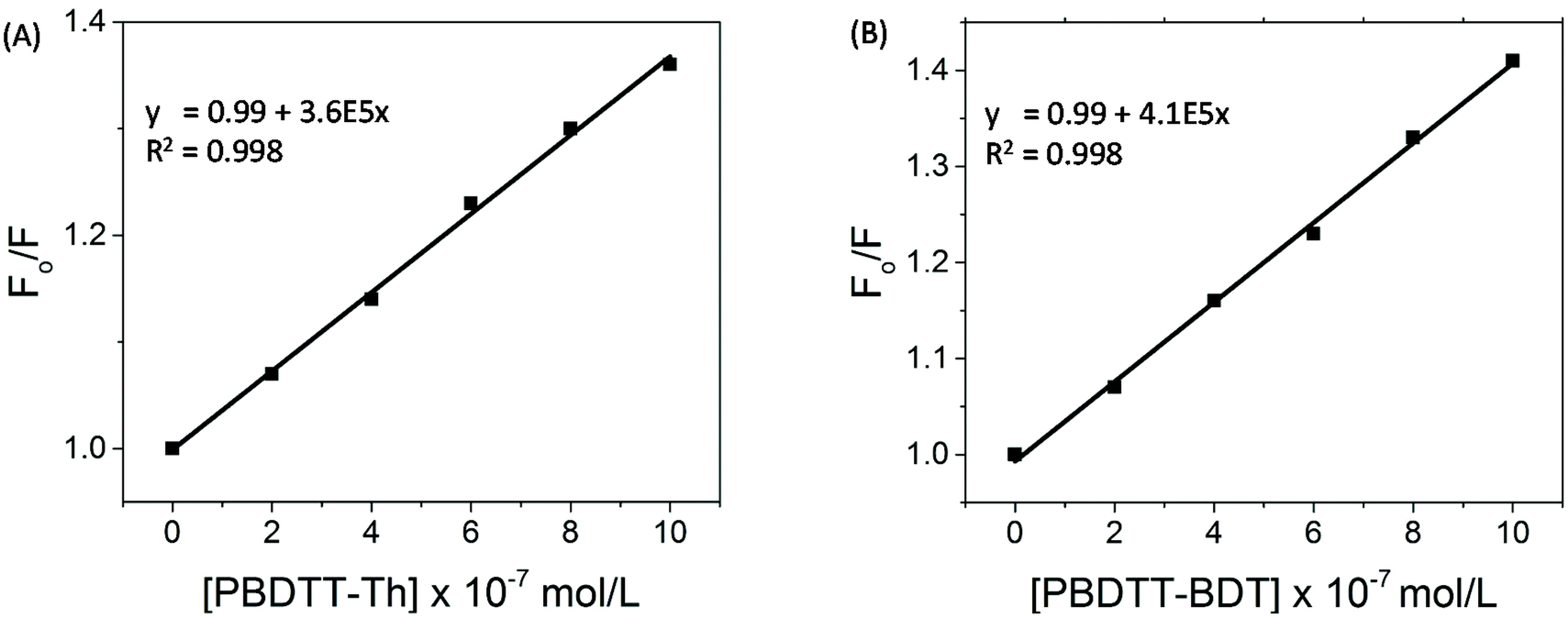
The correction factor (CF) was calculated for each wavelength, and the largest CF was observed for high concentration of both polymers (10−6 mol L−1). The greatest CF was 2.1 for PBDTT-Th found at 585 nm, and 2.3 for PBDTT-BDT found at 610 nm which were within the acceptable range (CF < 3).33 The corrected fluorescence spectra of rr-P3HT with varying concentrations of PBDTT-Th and PBDTT-BDT are shown in Fig. 4A and B respectively. The observed fluorescence intensity of rr-P3HT was markedly reduced upon addition of PBDTT-Th and PBDTT-BDT, indicating excellent quenching behavior of these polymers. The quenching behavior of these polymers is explained by a general electron-transfer mechanism as depicted in Fig. 5, which involves the energetically favorable transfer of electrons from the LUMO of rr-P3HT (3.2 eV) to the LUMO of the acceptor polymers PBDTT-Th (3.7 eV) and PBDTT-BDT (3.6 eV). Furthermore, we analyzed the quenching data using the Stern–Volmer equation which showed a linear fitting (see Fig. 6A and B) and gave a quenching constant (KSV) of 3.7 × 105 mol−1 L and 4.1 × 105 mol−1 L for PBDTT-Th and PBDTT-BDT respectively, similar to that of PC60BM, and indicating the strong electron-accepting behavior of these polymers.
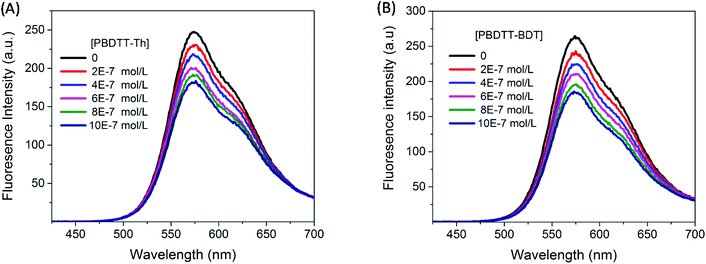 |
| | Fig. 4 Fluorescence spectrum of P3HT (4.0 × 10−7 M) in chloroform with varying concentrations of the electron acceptor (A) PBDTT-Th and (B) PBDTT-BDT. | |
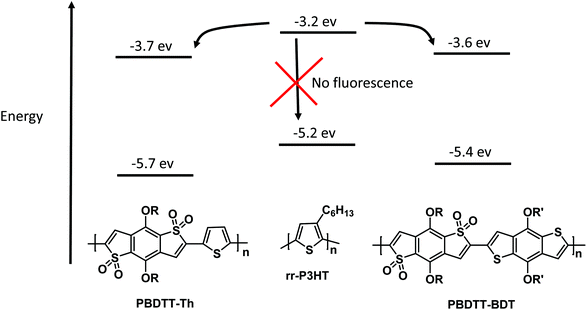 |
| | Fig. 5 Illustration of the electron transfer mechanism for fluorescence quenching of rr-P3HT. | |
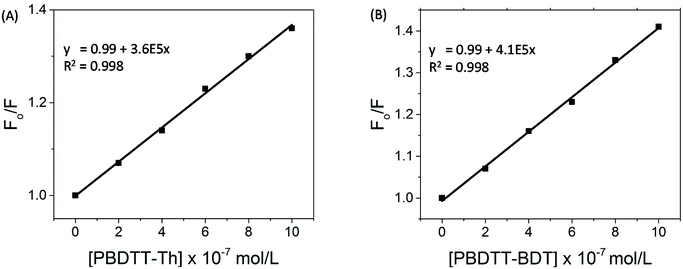 |
| | Fig. 6 (A) Stern–Volmer plots for the data from Fig. 3A and (B) Stern–Volmer plots for the data from Fig. 3B. | |
3.4. Electron mobility
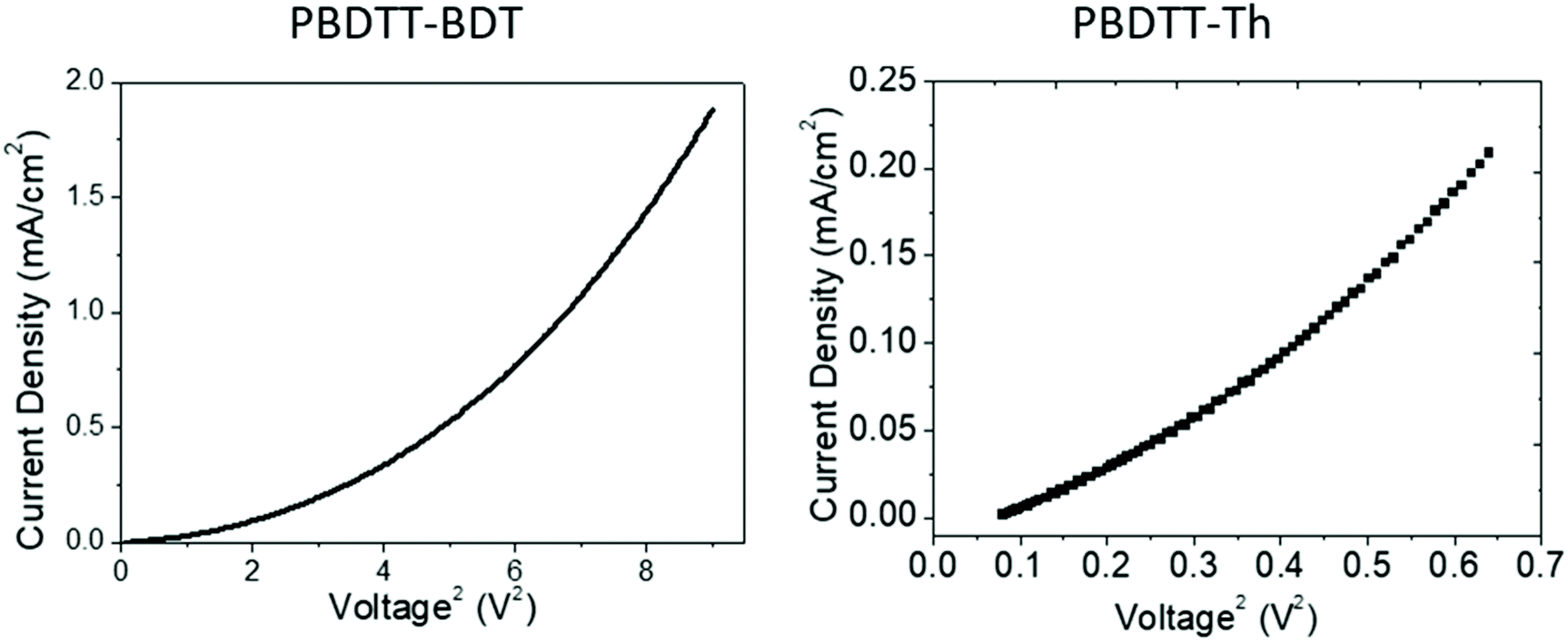
After investigation of the electronic properties and electron accepting behavior of these polymers, the electron mobilities of these polymers were probed and outlined in Table 3. The SCLC method using the Mott–Gurney law was carried out to determine the electron mobility of PBDTT-Th and PBDTT-BDT, and the curves were linearly fitted in the space charge region as shown in Fig. 7. The synthesized polymers were tested in the configuration of ITO/ZnO/PBDTT-Th or PBDTT-BDT/MnOx/Ag. The calculated mobilities for PBDTT-Th and PBDTT-BDT were determined to be 6.26 × 10−4 and 7.67 × 10−4 cm2 V−1 s−1, respectively.
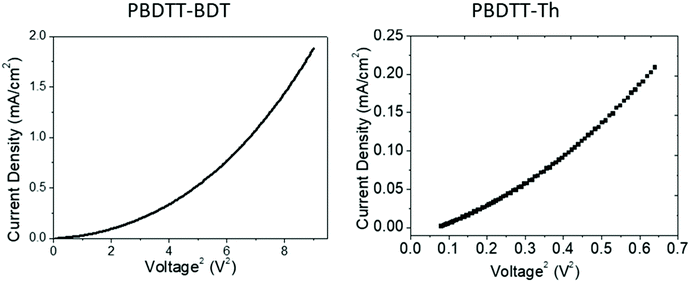 |
| | Fig. 7
J–V curve of PBDTT-BDT and PBDTT-Th measured on the probe station using SCLC model. | |
Table 3 Electron mobility characterization of PBDTT-BDT
| Polymers |
μ
e (cm2 V−1 s−1) |
Thickness (nm) |
Spin rate (rpm) |
Solvent |
|
PBDTT-BDT
|
7.67 × 10−4 |
40 |
2000 |
Chlorobenzene |
|
PBDTT-TH
|
6.26 × 10−4 |
45 |
2000 |
Chlorobenzene |
4. Conclusion
In conclusion, we have reported the successful diiodination of the electron-poor building block, BDTT, by a C–H iodination reaction at high yield. The diiodinated BDTT core was incorporated as an acceptor unit for the synthesis of donor–acceptor conjugated polymers at good yield. The resulting polymers showed electron accepting behavior which was probed by performing fluorescence quenching experiments with the donor material P3HT. It has been found that both of these polymers are excellent quenchers with quenching constants (KSV) of 3.7 × 105 L mol−1 and 4.1 × 105 L mol−1 for PBDTT-Th and PBDTT-BDT respectively, similar to that of PC60BM. Furthermore, the SCLC method using the Mott–Gurney law was carried out to determine the electron mobility of PBDTT-Th and PBDTT-BDT, which were found to be 6.26 × 10−4 and 7.67 × 10−4 cm2 V−1 s−1 respectively.
Author contributions
Dr Adhikari synthesized all starting materials, polymers, characterized the polymers’ structural, optical and electronic properties and wrote the sections related to the synthesis and characterization. Dr Ren performed the electron mobility studies and wrote the charge mobility section of the manuscript. Dr Stefan supervised the charge mobility studies and wrote the section. Dr Nelson supervised the whole research project and wrote the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We gratefully acknowledge support for this work from the National Science Foundation under the Center for Chemical Innovation in Selective C–H Functionalization (CHE-1205646), and Oklahoma State University Division of Institutional Diversity. Mihaela C. Stefan gratefully acknowledges the financial support from Welch Foundation (Grant AT-1740), National Science Foundation (CHE-1609880 and CHE-1566059). Mihaela C. Stefan also acknowledges the generous endowed chair support from the Eugene McDermott Foundation.
References
- T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413–4422 CrossRef CAS.
-
A. Rockett, Organic Semiconductors, in The Materials Science of Semiconductors, Spinger, Boston, MA, 2008, pp. 395–453 Search PubMed.
- R.-P. Xu, Y.-Q. Li and J.-X. Tang, J. Mater. Chem. C, 2016, 4, 9116–9142 RSC.
- K. A. N. Sachinthani, N. Kaneza, R. Kaudal, E. Manna, M. A. Eastman, B. Sedai, S. Pan, J. Shinar, R. Shinar and T. L. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 125–131 CrossRef CAS.
- N. Thejo Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2012, 16, 2696–2723 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Chem. Rev., 2017, 117, 796–837 CrossRef CAS.
-
C. Brabec, U. Scherf and V. Dyakonov, Organic Photovoltaics, Wiley–VCH Verlag GmbH & Co. KGaA, 2014 Search PubMed.
- J. D. Myers and J. Xue, Polym. Rev., 2012, 52, 1–37 CrossRef CAS.
- L. Torsi, M. Magliulo, K. Manoli and G. Palazzo, Chem. Soc. Rev., 2013, 42, 8612–8628 RSC.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- Y. Yamashita, Sci. Technol. Adv. Mater., 2009, 10, 024313 CrossRef.
-
D. A. Bernards, G. G. Malliaras and R. M. Owens, Organic Semiconductors in Sensor Applications, Springer, Berlin, Heidelberg, 2008 Search PubMed.
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS.
- J. T. E. Quinn, J. Zhu, X. Li, J. Wang and Y. Li, J. Mater. Chem. C, 2017, 5, 8654–8681 RSC.
- B. Shan and Q. Miao, Tetrahedron Lett., 2017, 58, 1903–1911 CrossRef CAS.
- W. Chen, M. Xiao, L. Han, J. Zhang, H. Jiang, C. Gu, W. Shen and R. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 19665–19671 CrossRef CAS.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS.
- M. Nandakumar, J. Karunakaran and A. K. Mohanakrishnan, Org. Lett., 2014, 16, 3068–3071 CrossRef CAS.
- I. H. Jung, W.-Y. Lo, J. Jang, W. Chen, D. Zhao, E. S. Landry, L. Lu, D. V. Talapin and L. Yu, Chem. Mater., 2014, 26, 3450–3459 CrossRef CAS.
- T. M. Pappenfus, D. T. Seidenkranz, M. D. Lovander, T. L. Beck, B. J. Karels, K. Ogawa and D. E. Janzen, J. Org. Chem., 2014, 79, 9408–9412 CrossRef CAS.
- D. P. Khambhati, K. A. N. Sachinthani, A. L. Rheingold and T. L. Nelson, Chem.
Commun., 2017, 53, 5107–5109 RSC.
- S. Zhen, S. Wang, S. Li, W. Luo, M. Gao, L. G. Ng, C. C. Goh, A. Qin, Z. Zhao, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1706945 CrossRef.
- T. Lei, R. Peng, X. Fan, Q. Wei, Z. Liu, Q. Guan, W. Song, L. Hong, J. Huang, R. Yang and Z. Ge, Macromolecules, 2018, 51, 4032–4039 CrossRef CAS.
- A. Punzi, M. A. M. Capozzi, S. Di Noja, R. Ragni, N. Zappimbulso and G. M. Farinola, J. Org. Chem., 2018, 83, 9312–9321 CrossRef CAS.
- J. Hou, M.-H. Park, S. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Q. Shi, S. Zhang, J. Zhang, V. F. Oswald, A. Amassian, S. R. Marder and S. B. Blakey, J. Am. Chem. Soc., 2016, 138, 3946–3949 CrossRef CAS.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. Tang and E. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- M. Neophytou, D. Bryant, S. Lopatin, H. Chen, R. K. Hallani, L. Cater, I. McCulloch and W. Yue, Macromol. Rapid Commun., 2018, 39, 1700820 CrossRef.
- Y. Sui, Y. Deng, T. Du, Y. Shi and Y. Geng, Mater. Chem. Front., 2019, 3, 1932–1951 RSC.
- J. Choi, H. Song, N. Kim and F. S. Kim, Semicond. Sci. Technol., 2015, 30, 064002 CrossRef.
- J. D. Wood, J. L. Jellison, A. D. Finke, L. Wang and K. N. Plunkett, J. Am. Chem. Soc., 2012, 134, 15783–15789 CrossRef CAS.
- T. D. Gauthier, E. C. Shane, W. F. Guerin, W. R. Seitz and C. L. Grant, Environ. Sci. Technol., 1986, 20, 1162–1166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01292k |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b and
Toby L.
Nelson
b and
Toby L.
Nelson
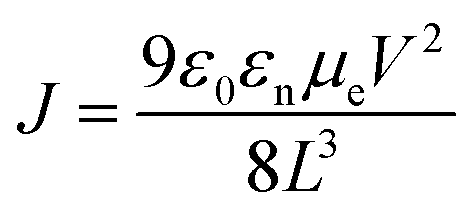 , where ε0 is the permittivity of free space (8.8542 × 10−12 F m−1), εr is the relative permittivity of semiconducting materials (which is used as 3.5 here), μe is electron mobility, V is applied voltage, and L is the thickness. All runs were performed under nitrogen.
, where ε0 is the permittivity of free space (8.8542 × 10−12 F m−1), εr is the relative permittivity of semiconducting materials (which is used as 3.5 here), μe is electron mobility, V is applied voltage, and L is the thickness. All runs were performed under nitrogen.