
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational synthesis of epoxy-functional spheres, worms and vesicles by RAFT aqueous emulsion polymerisation of glycidyl methacrylate†
Fiona L.
Hatton‡
 *,
Matthew J.
Derry§
and
Steven P.
Armes
*
*,
Matthew J.
Derry§
and
Steven P.
Armes
*
Dainton Building, Department of Chemistry, University of Sheffield, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: f.hatton@lboro.ac.uk; s.p.armes@sheffield.ac.uk
First published on 10th September 2020
Abstract
The rational synthesis of epoxy-functional diblock copolymer nano-objects has been achieved via RAFT aqueous emulsion polymerisation of glycidyl methacrylate (GlyMA; aqueous solubility ∼22 g dm−3 at 50 °C) by utilising relatively mild conditions (pH 7, 50 °C) to preserve the epoxy groups. High monomer conversions were achieved within 1 h when using a poly(glycerol monomethacrylate) chain transfer agent with a mean degree of polymerisation (DP) of 28, with GPC analysis indicating relatively narrow molecular weight distributions (Mw/Mn < 1.40) when targeting PGlyMA DPs up to 80. A phase diagram was constructed to identify the synthesis conditions required to access pure spheres, worms or vesicles. Transmission electron microscopy, dynamic light scattering and small-angle X-ray scattering (SAXS) studies indicated the formation of well-defined worms and vesicles when targeting relatively long PGlyMA blocks. These epoxy-functional nano-objects were derivatised via epoxy-thiol chemistry by reaction with L-cysteine in aqueous solution. Finally, an in situ SAXS study was conducted during the RAFT aqueous emulsion polymerisation of GlyMA at 50 °C to examine the nucleation and size evolution of PGMA48-PGlyMA100 diblock copolymer spheres using a bespoke stirrable reaction cell.
Introduction
Block copolymer self-assembly in solution can produce various types of organic nanoparticles that in principle can offer a range of potential applications.1 Traditionally, self-assembly has been achieved via post-polymerisation processing by lowering the degree of solvation for one of the blocks, typically by adding a non-solvent or by adjusting the temperature.2,3 Spheres or vesicles tend to be the most commonly observed morphologies, but worms, rods,4 toroids,5 bicontinuous structures or lamellae have also been reported.6 Block copolymer worms can be used as viscosity modifiers or gelators,7 while block copolymer vesicles can be used for the encapsulation of nanoparticles, proteins or enzymes.8–10Polymerisation-induced self-assembly (PISA) involves chain extension of a soluble homopolymer using a suitable second monomer that, once polymerised, becomes insoluble in the reaction media, thereby driving in situ self-assembly to form diblock copolymer nanoparticles.11–13 Reversible addition–fragmentation chain transfer (RAFT) polymerisation14 has proven to be a particularly popular technique for PISA syntheses, owing to its versatility in enabling the facile preparation of a wide range of functional diblock copolymers.15,16 Hawkett and co-workers reported the first example of PISA via RAFT aqueous emulsion polymerisation.17,18 Other notable pioneers in this field included Charleux,19 Rieger,20 D'Agosto and Lansalot.21,22 Since these seminal studies, PISA has been extended to include RAFT dispersion polymerisation conducted in water,23 polar organic solvents24 and non-polar media.25 For such formulations, many pseudo-phase diagrams (or morphology maps) have been constructed that enable the reproducible targeting of pure spheres, worms or vesicles.26–29 The formation of spheres is generally favored by using relatively long steric stabiliser blocks and/or by working at lower copolymer concentrations.26,30 In contrast, vesicles are typically obtained at higher copolymer concentrations when utilising relatively short steric stabiliser blocks and targeting long core-forming blocks. Generally, the phase space occupied by pure worms is relatively narrow hence this is usually the most elusive morphology.31,32 Although there are many literature examples of kinetically-trapped spheres,26,33–35 it is usually possible to prepare well-defined worms or vesicles for most RAFT dispersion polymerisations, given sufficient synthetic effort.36–39
Although some notable exceptions are known in the literature,40–49 the majority of RAFT aqueous emulsion polymerisation formulations only yield kinetically-trapped spheres when utilising various water-immiscible monomers such as styrene,50,51 methyl methacrylate,52,53n-butyl methacrylate,54,55n-butyl acrylate,17,18,56 benzyl methacrylate57 or 2,2,2-trifluoroethyl methacrylate.58,59 Charleux and co-workers reported the first PISA synthesis of block copolymer ‘nanofibers’ (or worms) via RAFT aqueous emulsion polymerisation when chain-extending a water-soluble statistical copolymer precursor comprising poly(ethylene glycol) methyl ether acrylate (PEGA) and acrylic acid with styrene.40,41 Well-defined spheres, worms and vesicles could be obtained using the analogous all-methacrylic statistical copolymer as a stabiliser block.60,61 In addition, D'Agosto and co-workers recently reported that copolymerisation of a relatively small amount of PEGA with N-acryloylmorpholine also resulted in the formation of vesicles with polystyrene membranes under certain conditions.43 Furthermore, Hawkett and coworkers reported the synthesis of a range of block copolymer nano-objects using either amphiphilic AB diblock or ABA triblock copolymers as steric stabiliser precursors.45,46 It is currently not understood why this rather small subset of RAFT aqueous emulsion polymerisation formulations can provide access to higher order morphologies, whereas the majority of such PISA syntheses only yield kinetically-trapped spheres. However, it is perhaps noteworthy that most of the counter-examples involve anionic statistical copolymer stabilisers, for which self-assembly is likely to be affected by both solution pH and ionic strength.
Recently, we reported that chain extension of a poly(methacrylic acid) stabiliser via RAFT aqueous emulsion polymerisation of 4-hydroxybutyl methacrylate (HBMA) at pH 5 produced an unusual ‘monkey nut’ morphology.62 We hypothesised that the relatively high aqueous solubility of HBMA (20 g dm−3 at the synthesis temperature of 70 °C) enabled more effective plasticisation of the core-forming block, which in turn facilitated fusion of the monomer-swollen spheres to produce ‘monkey nuts’, rather than kinetically-trapped spheres. Similarly, 2-methoxyethyl methacrylate (MOEMA) exhibits an aqueous solubility of 19.6 g dm−3 at 70 °C and also enables access to worms and vesicles as well as spheres.47 Glycidyl methacrylate (GlyMA) has a comparable aqueous solubility to that of HBMA and MOEMA.63 However, in our initial studies we found that the RAFT aqueous emulsion polymerisation of GlyMA using a water-soluble poly(glycerol monomethacrylate) (PGMA) macromolecular chain transfer agent (macro-CTA) only produced sterically-stabilised PGMA-PGlyMA spheres.64 Subsequently, we reported that PGMA-PGlyMA worms could be prepared using a highly convenient one-pot methodology provided that the steric stabiliser block was relatively short (DP = 25).48 In both cases, it was essential to use relatively mild conditions (pH 4–7, 50 °C) and short reaction times (1 h) to minimise ring-opening of the pendent epoxy groups by reaction with water. The resulting spheres and worms could be derivatised using epoxy-amine chemistry.65–67 In closely related work, Tan and co-workers recently reported that the RAFT aqueous emulsion polymerisation of GlyMA at 25–50 °C using a low-temperature redox initiator also enables preservation of epoxy functionality.49 More specifically, when targeting a PGlyMA DP of 150 using a non-ionic water-soluble precursor, spheres were obtained when conducting GlyMA polymerisations at 20 °C, spheres and worms were formed at 37 °C and vesicles were produced at 50 °C. Few other literature reports of RAFT aqueous emulsion polymerisation of GlyMA exist and are limited to seeded emulsion polymerisations using photo-PISA68 and enzyme-initiated PISA.69
Herein we report the synthesis of epoxy-functional diblock copolymer spheres, worms and vesicles via RAFT aqueous emulsion polymerisation of GlyMA under mild conditions (50 °C, pH 7). A pseudo-phase diagram is constructed to ensure reproducible targeting of these nano-objects and to examine the effect of varying the copolymer concentration on the final copolymer morphology. Selected examples of spheres, worms and vesicles are characterised using small angle X-ray scattering (SAXS). We demonstrate that aqueous dispersions of epoxy-functional diblock copolymer worms can be derivatised via epoxy-thiol chemistry by reaction with L-cysteine, which influences their aqueous electrophoretic behaviour. Finally, the RAFT aqueous emulsion polymerisation of GlyMA was monitored in situ using SAXS to monitor the nucleation and growth of PGMA48-PGlyMA100 diblock copolymer spheres.
Experimental
Materials
Glycerol monomethacrylate (GMA) was donated by GEO Specialty Chemicals (Hythe, UK) and used without further purification. Glycidyl methacrylate (GlyMA; 97%), 4,4′-azobis(4-cyanopentanoic acid) (ACVA; 99%), 2,2′-azobisisobutyronitrile (AIBN; 98%), hexane (HPLC grade; ≥97%), sodium hydroxide (NaOH; 98%), and L-cysteine (97%) were purchased from Sigma-Aldrich (UK) and were used as received. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044; ≥97%). 2-Cyano-2-propyl dithiobenzoate (CPDB) was purchased from Strem Chemicals Ltd (Cambridge, UK) and was used as received. Deuterated solvents were purchased from Goss Scientific Instruments Ltd (Cheshire, UK). All other solvents and concentrated hydrochloric acid (32%) were purchased from Fisher Scientific (Loughborough, UK) and used as received. Deionised water was used for all experiments.Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). Chromatograms were analysed using Agilent GPC/SEC software provided by the manufacturer.
θ/λ and a Ravonix MX-170HS CCD detector. A flow-through capillary set-up was used as the sample holder, with a glass capillary of 2 mm diameter. Scattering data were reduced using standard routines provided by the beamline and were further analysed using Irena SAS macros for Igor Pro.70 Water was used for the absolute intensity calibration. Data were recorded for 1.0% w/w aqueous dispersions of PGMA28-PGlyMAn nano-objects originally prepared at 10% w/w, PGMA28-PGlyMAn nano-objects originally prepared at 20% w/w solids (where n = 25, 40, 80), and the corresponding PGMA28-P(GlyMA-cys)n copolymers after derivatisation where n = 25, 40, 80.
000 g mol−1). Chromatograms were analysed using Agilent GPC/SEC software provided by the manufacturer.
θ/λ and a Ravonix MX-170HS CCD detector. A flow-through capillary set-up was used as the sample holder, with a glass capillary of 2 mm diameter. Scattering data were reduced using standard routines provided by the beamline and were further analysed using Irena SAS macros for Igor Pro.70 Water was used for the absolute intensity calibration. Data were recorded for 1.0% w/w aqueous dispersions of PGMA28-PGlyMAn nano-objects originally prepared at 10% w/w, PGMA28-PGlyMAn nano-objects originally prepared at 20% w/w solids (where n = 25, 40, 80), and the corresponding PGMA28-P(GlyMA-cys)n copolymers after derivatisation where n = 25, 40, 80.
For in situ SAXS experiments conducted during the RAFT aqueous emulsion polymerisation of GlyMA, SAXS patterns were collected at a synchrotron source (Diamond Light Source, station I22, Didcot, UK) using monochromatic X-ray radiation (wavelength, λ = 0.124 nm, with q ranging from 0.002 to 0.23 Å−1, where q = 4π·sinθ/λ is the length of the scattering vector and θ is one-half of the scattering angle) and a 2D Pilatus 2 M pixel detector (Dectris, Switzerland). The reactions were conducted as previously described.47 A bespoke stirrable reaction cell was used as the sample holder, with Kapton film windows of 0.075 mm thickness. All reagents were purged with nitrogen gas for 30 min before a known volume of the deoxygenated solution was transferred into the reaction cell, which had been previously purged with nitrogen. The reaction cell was then sealed to prevent oxygen ingress before being placed in the X-ray beam along with a magnetic stirrer unit. The reaction cell was heated to 50 °C using a water circulating bath. SAXS patterns were collected every 3 s for 7.5 min, then every 10 s for the following 27.5 min, then every 100 s for 30 min, or until no further evolution in the scattering pattern was observed. Scattering data were reduced using standard routines from the beamline and were further analysed using Irena SAS macros for Igor Pro.70 Water was used for the absolute intensity calibration. SAXS patterns were recorded for 1 h during the in situ synthesis of PGMA48-PGlyMA100 spheres at 10% w/w. The final PGMA48-PGlyMA100 spheres were diluted to 1.0% w/w with deionised water and a SAXS pattern was recorded using a flow-through capillary set-up as the sample holder (glass capillary diameter = 2 mm).
Synthesis of PGMA28via RAFT solution polymerisation of GMA in ethanol
The PGMA precursor used in this study was prepared via RAFT solution polymerisation of GMA in ethanol as previously described.26,64 The target mean DP was 31 and the [GMA]:[CPDB]:[ACVA] relative molar ratios were 31:1:0.25. Briefly, CPDB (2.79 g, 12.6 mmol, assuming a RAFT CTA efficiency of 80%), GMA (50.0 g, 0.312 mol) and ethanol (80.2 g, 60 wt%) were weighed into a round-bottomed flask. ACVA (0.706 g, 2.52 mmol) was added and the reaction mixture was cooled in an ice bath and degassed with N2 gas for 40 min. After degassing, the flask was immersed in an oil bath set at 70 °C and the polymerisation was quenched after 160 min after the GMA conversion had reached 63%. The crude PGMA precursor was diluted with methanol and precipitated into dichloromethane, redissolved in methanol and precipitated once more to yield the final purified PGMA28 precursor. Its mean DP was confirmed by end-group analysis using 1H NMR spectroscopy in CD3OD. DMF GPC analysis indicated an Mn of 8300 g mol−1 and a Đ of 1.15.
Synthesis of PGMA28-PGlyMAn diblock copolymer nano-objects by RAFT aqueous emulsion polymerisation
The synthesis of PGMA28-PGlyMA100 vesicles at 10% w/w using a macro-CTA/initiator ratio of 4.0 is representative of the general PISA protocol. PGMA28 macro-CTA (0.25 g, 0.053 mmol), and deionised water (9.09 g) were weighed into a sample tube. VA-044 initiator (4.30 mg, 0.013 mmol) was added and the pH was adjusted to 7.0–7.5 by addition of 0.01 M NaOH. GlyMA (0.756 g, 5.32 mmol) was added and the reaction mixture was sealed with a rubber septum and immersed in an ice bath and degassed with N2 for 30 min, before being placed in an oil bath set at 50 °C. The polymerisation was quenched after 1 h by removing the reaction vessel from the oil bath and exposing its contents to air, followed by 1H NMR, DLS, GPC and TEM analysis. When targeting different PGlyMA DPs the PGMA, GlyMA and VA-044 molar ratios were adjusted accordingly, maintaining a consistent solids content.Synthesis of PGlyMA by RAFT solution polymerisation
The RAFT solution polymerisation of GlyMA was conducted in chloroform using CPDB as the RAFT agent and AIBN initiator. First, GlyMA (5.02 g, 35.2 mmol) and CPDB (0.078 g, 0.352 mmol; target DP = 100) were weighed into a 25 mL round-bottomed flask, and CHCl3 (7.64 g) was added to produce a final monomer concentration of 40% w/w. AIBN (0.012 g, 0.073 mmol; CPDB/AIBN ∼5.0) was added to the reaction flask, which was immersed in an ice bath and the reaction mixture was degassed using a stream of N2 gas for 30 min. The flask was then sealed and placed in an oil bath set at 60 °C for 19 h. The GlyMA polymerisation was quenched by cooling to 20 °C, exposing the reaction solution to air and dilution with CHCl3. The GlyMA conversion was 95% as determined by 1H NMR analysis in CDCl3. The crude PGlyMA was precipitated into excess n-hexane (three times), isolated by filtration and dried in a vacuum oven. Residual n-hexane (detected by 1H NMR analysis) was removed by dissolving the purified PGlyMA in acetone and concentrating by rotary evaporation (three times), and subsequently dried using a high vacuum manifold. DMF GPC analysis indicated an Mn of 17100 g mol−1 and an Mw/Mn of 1.17, while helium pycnometry measurements indicated a solid-state density of 1.25 ± 0.01 g cm−3 at 20 °C.
Estimation of the aqueous solubility of GlyMA at 50 °C
Deionised water (10.0 g) was added to a pre-weighed vial equipped with a magnetic flea. This vial was placed in an oil bath set at 50 °C and allowed to equilibrate for 20 min. GlyMA (∼1.5 g) was added to a pre-weighed vial and then added dropwise to the water at 50 °C. After addition of each drop of GlyMA, the aqueous GlyMA mixture was stirred at 50 °C for 1–2 min. The point at which the GlyMA monomer droplets no longer fully dissolved as judged by visual inspection was noted and the vial containing GlyMA monomer was reweighed. Hence the total mass of added GlyMA was determined and its aqueous solubility at 50 °C was calculated using the following equation: aqueous solubility = (mass of GlyMA/mass of water) × 100. This solubility experiment was performed in triplicate and the mean aqueous solubility of GlyMA at 50 °C was found to be 2.2% w/w, or 22.0 g dm−3. This is in good agreement with data previously reported by Ratcliffe et al., who determined the aqueous solubility of GlyMA to be 1.4–1.5% w/w at 21 °C and 2.4–2.5% w/w at 80 °C.63In situ SAXS studies during RAFT aqueous emulsion polymerisation of GlyMA
The PGMA48 precursor used for the in situ SAXS studies was prepared according to a previous literature protocol.64 This PGMA48 precursor (0.14 g, 0.018 mmol) was weighed into a sample tube along with deionised water (3.44 g). VA-044 initiator (4.43 μmol; 0.10 mL of a 0.044 M stock solution) was added and the solution pH was adjusted to 7.0–7.5 by adding 0.01 M NaOH. GlyMA (0.252 g, 1.77 mmol; target DP = 100) was added and the reaction mixture was then sealed with a rubber septum, immersed in an ice bath and degassed with N2 for 30 min, before being placed in the stirrable reaction cell for the in situ SAXS experiment. After 1 h at 50 °C, the reaction cell was removed from the beam line and the GlyMA polymerisation quenched by exposure to air. Postmortem analysis of the reaction mixture was conducted using 1H NMR, DLS, GPC and TEM.Results and discussion
Recently, we reported that the RAFT aqueous emulsion polymerisation of GlyMA using a PGMA45 chain transfer agent only led to the formation of kinetically-trapped spheres.64 However, the aqueous solubility of GlyMA (22 g dm−3 at 50 °C) is comparable to that of HBMA or MOEMA,47,63 hence access to epoxy-functional worms or vesicles might be expected for such PISA formulations. Drawing on our prior experience,26,30,71–74 we decided to revisit the RAFT aqueous emulsion polymerisation of GlyMA to examine whether utilising a shorter PGMA precursor as the steric stabiliser block might enable access to such higher order morphologies.First, a kinetic study of the RAFT solution polymerisation of GMA in ethanol using 2-cyano-2-propyl dithiobenzoate (CPDB) at 70 °C was conducted by sampling the reaction mixture periodically, see Fig. S1.† First-order kinetics and a linear increase in Mn with conversion were observed when targeting a degree of polymerisation (DP) of 31.26 A dithiobenzoate-capped PGMA28 precursor (Mn = 8300 g mol−1, Đ = 1.15) was prepared on a 30 gram scale and used for all subsequent RAFT aqueous emulsion polymerisation syntheses. Chain extension of this PGMA28 steric stabiliser block with GlyMA under mild conditions (50 °C, pH 7) yielded a range of epoxy-functional diblock copolymer nano-objects (see Scheme 1).
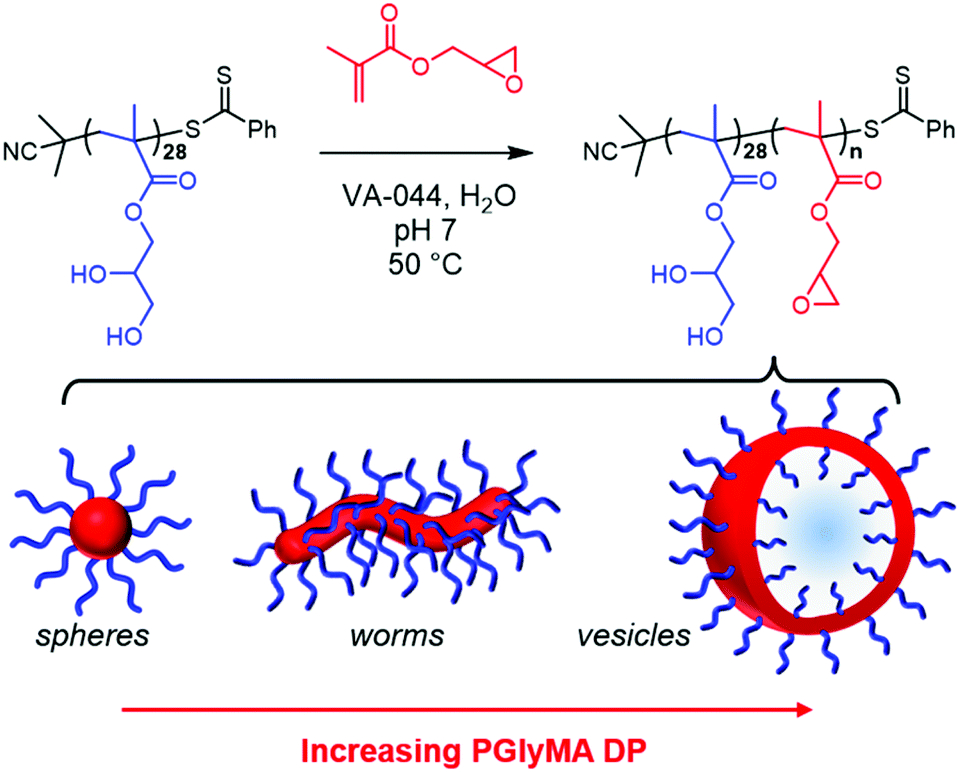 | ||
| Scheme 1 Schematic representation of the RAFT aqueous emulsion polymerisation of GlyMA at 50 °C using a PGMA28 precursor at neutral pH to form sterically-stabilised PGMA28-PGlyMAn spheres, worms or vesicles. Notably, only kinetically-trapped spheres were obtained with longer PGMA45 and PGMA48 stabiliser blocks. | ||
Kinetic studies confirmed that high GlyMA conversions were achieved within 1 h at 50 °C with a linear evolution of Mn with conversion (see Fig. S2†). It is also worth emphasising that these polymerisations were conducted at neutral pH in order to prevent premature loss of the epoxy groups via ring-opening side-reactions with water. Copolymer morphologies were initially assigned by transmission electron microscopy (TEM) in order to construct a phase diagram, see Fig. 1 and Table S1.† These assignments were subsequently confirmed for selected PGMA28-PGlyMAn nano-objects using small-angle X-ray scattering (SAXS).
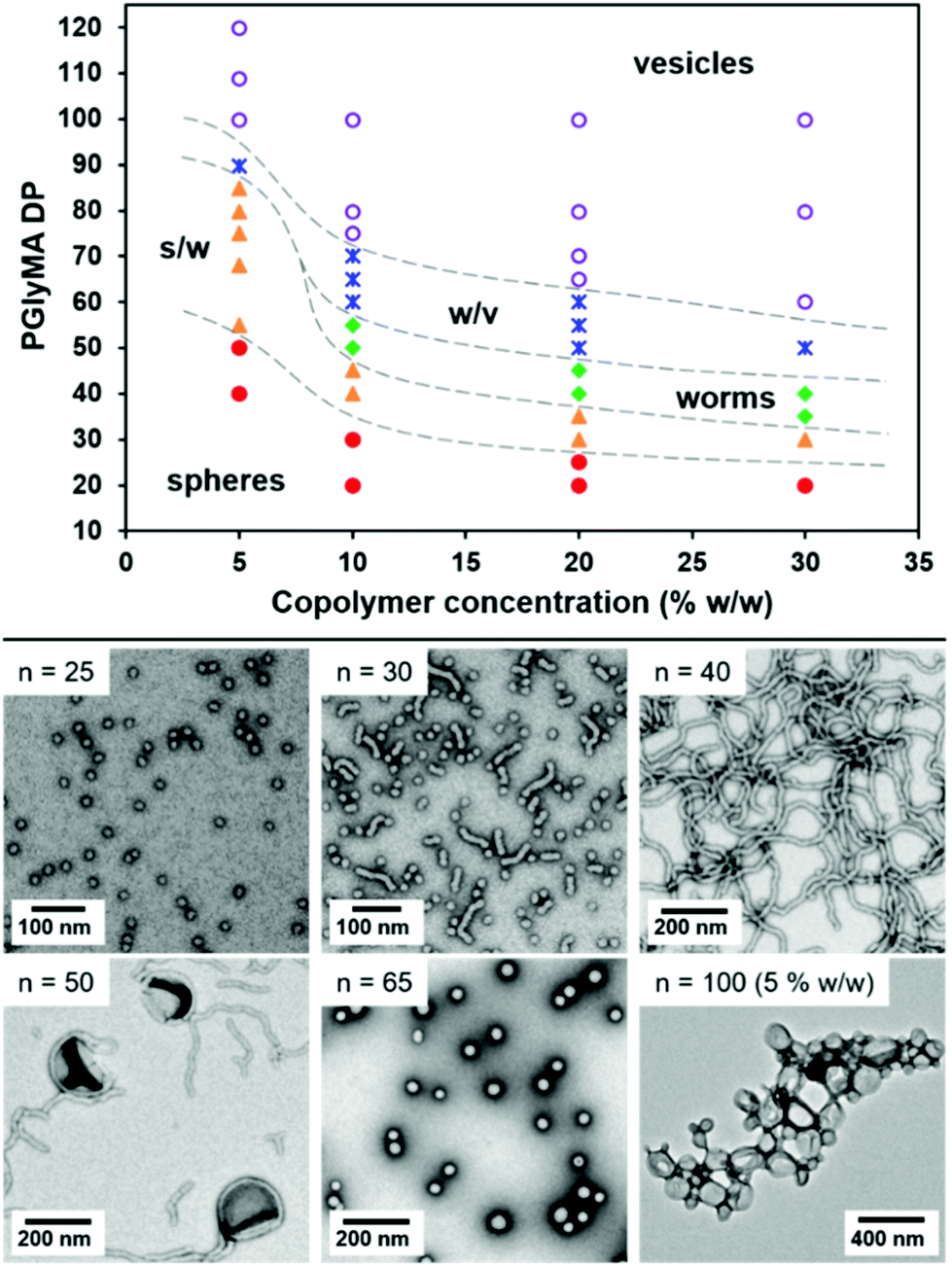 | ||
| Fig. 1 Phase diagram constructed for a series of PGMA28-PGlyMAn diblock copolymer nano-objects synthesised by RAFT aqueous emulsion polymerisation of GlyMA using a PGMA28 precursor at copolymer concentrations of 10–30% w/w [s/w denotes a mixed phase of spheres and worms, while w/v denotes a mixed phase of worms and vesicles]. Representative TEM images are shown for selected nano-objects prepared at 20% w/w (where n indicates the mean PGlyMA DP). | ||
DLS studies indicated the formation of well-defined spheres of with z-average diameters (Dz) of 15–26 nm when targeting short core-forming blocks (e.g. for DP = 25 at 20% w/w and up to DP = 50 at 5% w/w). For copolymer concentrations of 10–30% w/w, increasing the core-forming block DP initially afforded a mixed phase of spheres and short worms followed by a pure worm phase, with higher concentrations being required for a lower PGlyMA DP to access the latter morphology. The mean cross-sectional worm core diameter estimated from TEM images was comparable to the mean sphere diameter. For example, PGMA28-PGlyMA30 spheres prepared at 10% w/w had a mean diameter of 15.6 ± 1.5 nm, while the cross-sectional diameter for PGMA28-PGlyMA50 worms was estimated to be 16.6 ± 1.5 nm (see Fig. S3†). This is consistent with worm formation via the stochastic 1D fusion of multiple spheres.73 Targeting longer PGlyMA DPs produced a mixed phase of worms and vesicles for PISA syntheses conducted at copolymer concentrations of 10–30% w/w. Representative TEM images showing the various PGMA28-PGlyMAn nano-objects obtained at 10% w/w are shown in Fig. S3.† Based on TEM studies alone, relatively small spheres with Dz ranging from 45 to 90 nm are apparently obtained when targeting PGlyMA DPs of 75–100 at such copolymer concentrations. However, this tentative morphology assignment proved to be erroneous: subsequent SAXS studies confirmed that these ‘spheres’ were in fact unusually small vesicles (see below for further details). For PISA syntheses performed at 5% w/w, larger vesicles were observed when targeting PGMA28-PGlyMA100 and PGMA28-PGlyMA110, while increasing the PGlyMA DP up to 120 resulted in vesicle aggregates (DLS studies indicated a Dz of 1588 nm and a polydispersity of 0.97, while visual inspection confirmed that particle sedimentation occurred over time). These experiments clearly demonstrate that the relatively high aqueous solubility of GlyMA monomer (∼22 g dm−3 at 50 °C) provides convenient access to higher order morphologies via RAFT aqueous emulsion polymerisation, thus avoiding the well-known problem of kinetically-trapped spheres reported in the literature.50–59
DMF GPC studies of a series of PGMA28-PGlyMAn diblock copolymers indicated relatively narrow, unimodal molecular weight distributions, see Fig. 2. These observations are comparable with our previous observations for PGMA45-PGlyMAn spheres, where dispersities remained below 1.30 when targeting PGlyMA DPs up to 100.61 In the present study, the broader molecular weight distributions observed when targeting higher PGlyMA DPs (Table S1†) are attributed to low levels of intermolecular branching, which leads to the formation of higher molecular weight species (Fig. 2). Interestingly, in some cases narrower dispersities were obtained when targeting the same diblock copolymer compositions at higher solids. For example, PGMA28-PGlyMA100 prepared at 20% w/w had a dispersity of 1.36, while the same diblock copolymer prepared at 10% w/w and 5% w/w exhibited dispersities of 1.43 and 1.70, respectively. It is hypothesised that reaction of a minor fraction of epoxy groups, first with water and then with the hydroxyl-functional monomer that is generated in situ (i.e. glycerol monomethacrylate) generates a small amount of dimethacrylate impurity.63 The faster rates of reaction achieved at higher copolymer concentration means that the GlyMA monomer is more quickly converted into less reactive PGlyMA chains, which reduces the propensity for this side-reaction to occur. Moreover, the ensuing intermolecular branching that occurs during GlyMA polymerisation only becomes evident when targeting higher DPs.75
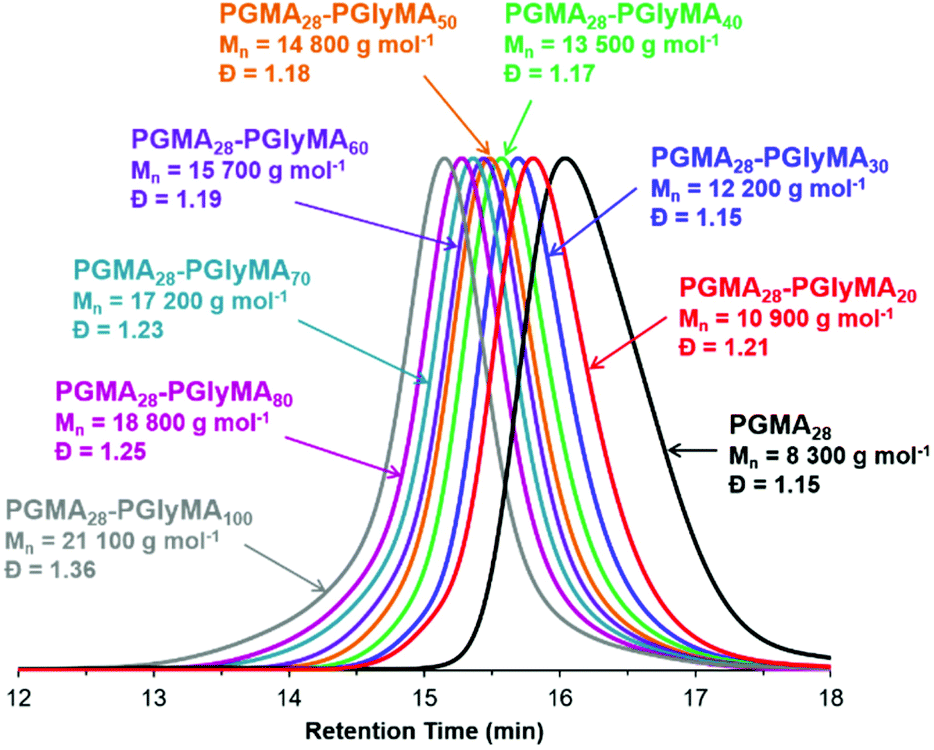 | ||
| Fig. 2 Overlaid DMF GPC traces recorded for PGMA28-PGlyMAn diblock copolymers prepared by RAFT aqueous emulsion polymerisation of GlyMA at 20% w/w solids (conditions: pH 7, 50 °C, 1 h) for n = 20, 30, 40, 50, 60, 70, 80 or 100. Molecular weight data are expressed relative to a series of near-monodisperse poly(methyl methacrylate) calibration standards. | ||
Small angle X-ray scattering studies
In order to confirm the copolymer morphologies assigned by TEM (see Fig. 1), SAXS patterns were recorded for 1.0% w/w aqueous dispersions of three types of PGMA28-PGlyMAn nano-objects originally prepared at 10% w/w solids. Radially-integrated scattering patterns obtained for PGMA28-PGlyMA30, PGMA28-PGlyMA50 and PGMA28-PGlyMA75 are shown in Fig. 3, where q is the scattering vector and I(q) is the X-ray scattering intensity. These scattering patterns were fitted using a PGlyMA homopolymer density of 1.25 ± 0.01 g cm−3 as determined by helium pycnometry. The PGlyMA scattering length density (ξPGlyMA = 11.34 × 1010 cm−2) is comparable to that of the PGMA stabiliser block (ξPGMA = 11.94 × 1010 cm−2; ξwater = 9.42 × 1010 cm−2) so the scattering from each component is comparable. To minimise the number of adjustable parameters when fitting these scattering patterns, the solvent volume fraction within the PGlyMA cores (xsol) was taken to be zero. This assumption is reasonable given the relatively hydrophobic nature of these chains. Moreover, when xsol was allowed to vary during preliminary attempts to model the data, this parameter always tended to zero. A detailed description of the scattering models and fitting parameters utilised to analyse these SAXS patterns is provided in the ESI (see SAXS section and Table S2†).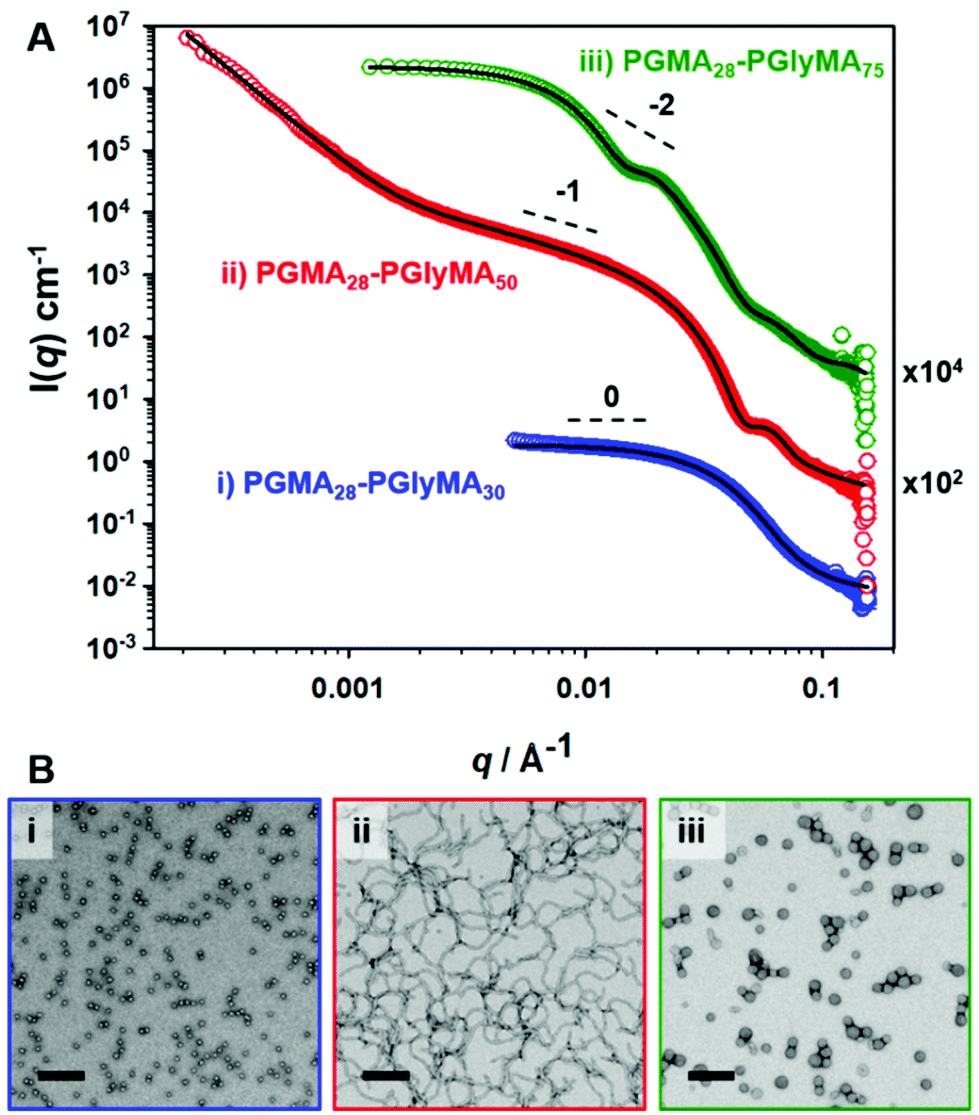 | ||
| Fig. 3 (A) SAXS patterns recorded for 1.0% w/w aqueous dispersions of PGMA28-PGlyMAn nano-objects prepared at 10% w/w solids for (i) n = 30, (ii) n = 50 and (iii) n = 75. Experimental data points are denoted by open circles and solid black lines indicate the data fits. For clarity, the red and green curves are offset by arbitrary factors of 102 and 104, respectively. (B) Representative TEM images recorded for the corresponding (i) PGMA28-PGlyMA30, (ii) PGMA28-PGlyMA50 and (iii) PGMA28-PGlyMA75 nano-objects. Scale bars represent 200 nm in each case. | ||
The SAXS pattern obtained for PGMA28-PGlyMA30 could be satisfactorily fitted using a spherical micelle model.76 A low q gradient of approximately zero was obtained, which is consistent with the spherical morphology indicated by TEM studies, see Fig. 3B(i). The volume-average sphere diameter, Ds, calculated from this model was 15.5 nm. As expected, this is lower than the z-average sphere diameter, Dz, of 21 nm reported by DLS. The SAXS pattern recorded for PGMA28-PGlyMA50 had a gradient of approximately −1 at low q, which indicates a highly anisotropic morphology.77 Again, this is consistent with the corresponding TEM image, which reveals a pure worm phase. The upturn at low q (below q ∼ 0.02 Å−1) suggests either some degree of worm branching (for which there appears to be some TEM evidence) or inter-worm interactions. The volume-average cross-sectional worm diameter, Dw, was calculated to be 17.8 ± 1.7 nm by fitting the SAXS pattern to a worm-like micelle model.76 This is in good agreement with the number-average worm width estimated by TEM (16.6 ± 1.5 nm). Perusal of the PISA literature indicates that well-defined worms are seldom reported for RAFT aqueous emulsion polymerisation formulations, which tend to produce kinetically-trapped spheres. Even in the few cases where anisotropic worms (sometimes described as nanofibres) are reported, close inspection of TEM images usually indicates the presence of minor populations of either spheres or vesicles.40,44 Although the PGMA28-PGlyMA75 nanoparticles were initially assigned as spheres by TEM, a spherical micelle model could not be fitted to the scattering pattern recorded for this dispersion (see Fig. S4†). However, satisfactory data fits could be obtained when using a vesicle model.78 Moreover, the volume-average vesicle diameter (Dv) of 53 nm calculated using this latter model was consistent with the corresponding DLS diameter, Dz, of 64 nm (see Table S2†). The mean vesicle membrane thickness, Tm, was calculated to be 8.2 nm, which is relatively large relative to the vesicle diameter. Thus, these rather small vesicles are much more resistant to deformation under ultrahigh vacuum than the larger vesicles commonly reported in the PISA literature,26 which makes their unambiguous morphological assignment using TEM alone somewhat problematic. This example serves to highlight the importance of using a statistically robust scattering technique such as SAXS for structural characterisation, rather than simply relying on TEM observations. SAXS patterns recorded for other PGMA28-PGlyMAn nanoparticles (where n = 20, 55, 80 or 100) prepared at 10% w/w can be found in Table S2† and Fig. S5 (see ESI†).
Long-term stability of epoxy groups for aqueous dispersions of PGMA28-PGlyMA55 worms
The chemical stability of PGMA28-PGlyMA55 worms was assessed during the long-term storage of a 10% w/w aqueous dispersion for 12 weeks at 20 °C. It is well-known that epoxy groups are susceptible to nucleophilic ring-opening by water, or by neighbouring hydroxyl groups.63 Nevertheless, 1H NMR spectroscopy studies indicated that 90% of the original epoxy groups remained intact after 6 weeks at pH 7, although only 74% were retained after ageing for 12 weeks (Fig. 4A). These observations are in good agreement with the gradual loss of epoxy functionality previously reported for PGMA45-PGlyMA100 spheres.64 Moreover, a concomitant increase in dispersity was observed when analyzing the aged PGMA28-PGlyMA55 chains by DMF GPC (Fig. 4B). This suggests that the hydroxyl groups that are generated via ring-opening of the epoxy groups by reaction with water subsequently react with adjacent epoxy groups, leading to intermolecular branching. Given the relatively low degree of hydration of such diblock copolymer nano-objects indicated by SAXS studies, this chemical degradation presumably involves initial reaction of epoxy groups located at the near-surface of the PGlyMA cores. Interestingly, rheological studies of the 10% w/w worm gel revealed a significant reduction in the critical strain (γc) from 10% to 2.4%, suggesting that gel embrittlement occurs over time (Fig. 4C).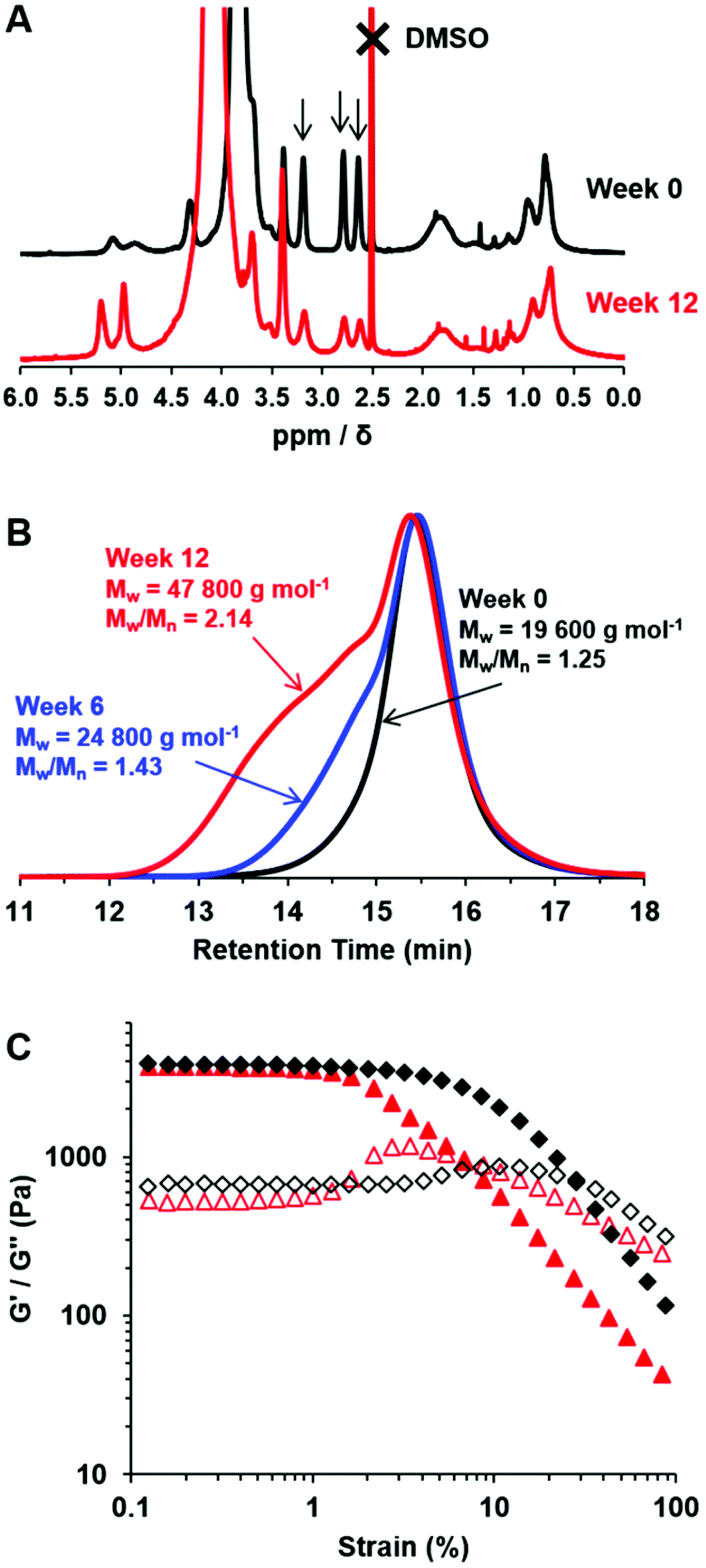 | ||
| Fig. 4 Evaluation of the chemical stability of the epoxy groups within a 10% w/w aqueous dispersion of PGMA28-PGlyMA55 worms during their long-term storage at 20 °C and pH 7. (A) 1H NMR spectra confirm that ring-opening of epoxy groups occurs gradually over time as a result of nucleophilic attack by water. (B) DMF GPC traces recorded for the molecularly-dissolved diblock copolymer chains at various time points, indicating light branching caused by intermolecular ring-opening side reactions. (C) Rheological strain sweeps showing G′ (closed symbols) and G′′ (open symbols) for fresh PGMA28-PGlyMA55 worms (black diamonds) and the same worms after ageing for 12 weeks (red triangles). The yield strain of this worm gel is reduced significantly after long-term storage at 20 °C. | ||
Derivatisation of PGMA28-PGlyMAn spheres, worms and vesicles using L-cysteine
The inherent reactivity of the epoxy ring offers a convenient handle for post-polymerisation derivatisation of freshly-prepared PGMA-PGlyMA nano-objects. We, and other labs, have previously demonstrated that either homopolymerisation or statistical copolymerisation of GlyMA to form core-forming blocks64 enables the facile preparation of covalently-stabilised nanoparticles using various diamines,65,79,80 3-aminopropyltriethoxysilane66,81 or 3-mercaptopropyltrimethoxysilane as crosslinkers.82 Moreover, we recently reported the use of epoxy-functional stabiliser blocks to functionalise spherical nanoparticles via epoxy-thiol chemistry83 and also block copolymer worms via epoxy-amine chemistry.48,64 Here, we examine epoxy-thiol chemistry for the convenient derivatisation of PGMA28-PGlyMA25 spheres, PGMA28-PGlyMA40 worms and PGMA28-PGlyMA80 vesicles with L-cysteine at pH 8.5 to afford the corresponding PGMA28-P(GlyMA-Cys)n nano-objects directly in water, see Scheme 2.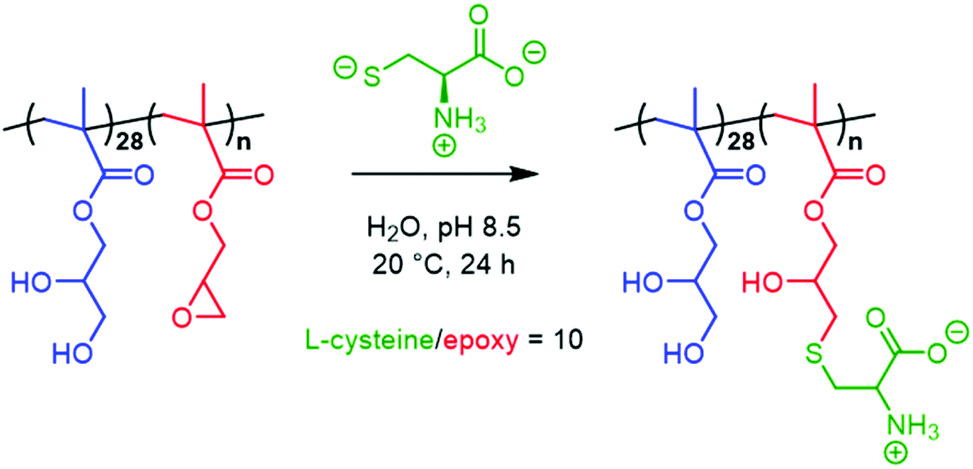 | ||
| Scheme 2 Schematic representation of the derivatisation of a 5% w/w aqueous dispersion of PGMA28-PGlyMAn nano-objects via epoxy-thiol chemistry using excess L-cysteine (conditions: L-cysteine/epoxy molar ratio = 10; pH 8.5; 20 °C, 24 h). | ||
An as-prepared 20% w/w aqueous dispersion of each type of PGMA28-PGlyMAn diblock copolymer nano-object was diluted to 5% w/w (primarily to allow efficient stirring in the case of the PGMA28-PGlyMA40 diblock copolymer worms). Excess L-cysteine (L-cysteine/epoxy molar ratio = 10) was added and allowed to react with the epoxy groups at pH 8.5, which is close to the pKa of its thiol group (pKa ≈ 8.2). Therefore, this amino acid reagent is present in its more reactive thiolate form, while its primary amine group remains protonated (pKa ≈ 10.3) to minimise epoxy-amine side-reactions. Following epoxy-thiol derivatisation for 24 h at 20 °C, the unreacted L-cysteine was removed by dialysis and the purified copolymer spheres, worms and vesicles were freeze-dried prior to elemental microanalyses (see Table S3†). Both the nitrogen and sulfur contents of these nano-objects increased significantly after L-cysteine derivatisation, from 0.23% and 1.04% in the original PGMA28-PGlyMA25 spheres up to 2.94% and 6.98%, respectively for PGMA28-P(GlyMA-cys)25. The latter values indicate a mean degree of derivatisation of 91%. Similarly, the nitrogen and sulfur contents increased from 0.16% and 0.70% for the original PGMA28-PGlyMA40 worms up to 3.42% and 8.02% for the PGMA28-P(GlyMA-cys)40 worms, which is equivalent to a mean degree of derivatisation of 91%. In contrast, a much lower mean degree of derivatisation of 44.5% was obtained for the PGMA28-P(GlyMA-cys)80 vesicles. Accordingly, a ten-fold excess of L-cysteine was added to a 5% w/w aqueous dispersion of PGMA28-PGlyMA80 vesicles and this reaction mixture was heated to 50 °C for 24 h. After exhaustive dialysis and freeze-drying, the mean degree of derivatisation indicated by elemental microanalysis was much higher (89%).
The PGMA28-P(GlyMA-cys)n copolymer nano-objects were characterised by TEM and DLS studies (see Fig. 5 and Table S4,† respectively). The former technique confirmed that the worm (Fig. 5E) and vesicle (Fig. 5F) morphologies were retained after L-cysteine derivatisation. However, the well-defined PGMA28-PGlyMA25 spheres (Fig. 5A; Dz = 18 nm, DLS polydispersity = 0.12) did not retain their original spherical morphology after derivatisation (Fig. 5D). DLS studies indicated that, although the Dz value was comparable (14 nm), the corresponding polydispersity had increased to 0.36 with a concomitant reduction in the scattered light intensity (count rate). This suggested that the originally hydrophobic PGlyMA chains had become sufficiently hydrophilic after derivatisation to cause partial molecular dissolution of the original spheres. This interpretation was confirmed by SAXS analysis of 1.0% w/w aqueous dispersions of the same spheres, worms and vesicles before and after derivatisation using L-cysteine, see Fig. 6.
 | ||
| Fig. 5 TEM images recorded for (A) PGMA28-PGlyMA25 spheres, (B) PGMA28-PGlyMA40 worms and (C) PGMA28-PGlyMA80 vesicles, (D) molecularly-dissolved PGMA28-P(GlyMA-cys)25 chains (derivatised using excess L-cysteine at 20 °C), (E) PGMA28-P(GlyMA-cys)40 worms (derivatised using excess L-cysteine at 20 °C) and (F) PGMA28-P(GlyMA-cys)80 vesicles (derivatised using excess L-cysteine at 50 °C). Scale bars represent 200 nm in each case. | ||
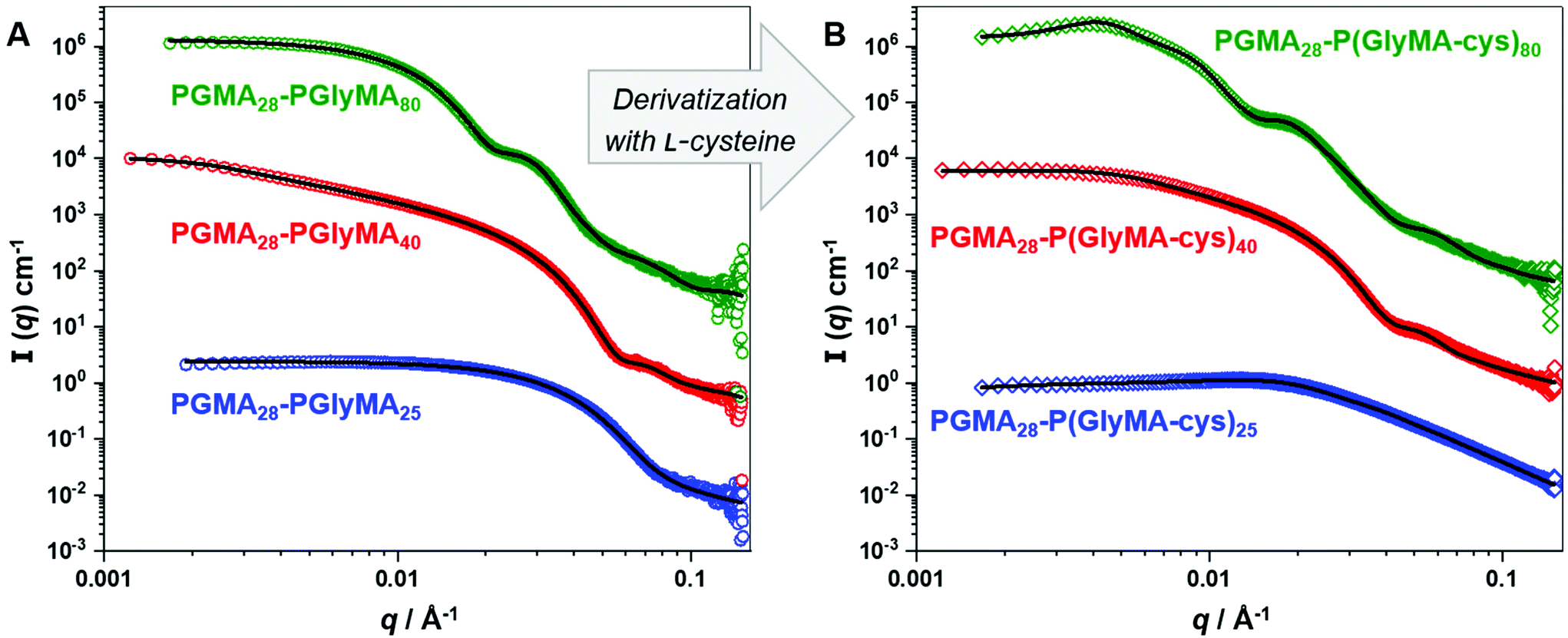 | ||
| Fig. 6 SAXS patterns recorded for 1.0% w/w aqueous dispersions of (A) precursor PGMA28-PGlyMA25 spheres, PGMA28-PGlyMA40 worms and PGMA28-PGlyMA80 vesicles originally prepared at 20% w/w solids, and (B) the corresponding molecularly-dissolved PGMA28-P(GlyMA-cys)25 chains, PGMA28-P(GlyMA-cys)40 worms and PGMA28-P(GlyMA-cys)80 vesicles obtained after derivatisation using excess L-cysteine at 20, 20 and 50 °C, respectively. Experimental data points are denoted by open symbols and solid black lines indicate data fits. | ||
SAXS patterns recorded for the as-synthesised PGMA28-PGlyMA25 spheres, PGMA28-PGlyMA40 worms and PGMA28-PGlyMA80 vesicles (Fig. 6A) were fitted using a spherical micelle model,76 a worm-like micelle model76 or a vesicle model,78 respectively, as previously described (see Table S5† for a summary of the fitting parameters). SAXS patterns were also recorded following derivatisation of these nano-objects with L-cysteine in aqueous solution (Fig. 6B). The scattering pattern obtained for PGMA28-P(GlyMA-cys)25 was consistent with that expected for molecularly-dissolved copolymer chains and could be fitted using a Gaussian coil model.84 However, despite the relatively low copolymer concentration of 1.0% w/w, a satisfactory data fit could only be achieved by incorporating an appropriate structure factor to account for the polyelectrolytic nature of the derivatised copolymer chains. More specifically, the Hayter–Penfold approximation for coulombic interactions85 was used to account for charge repulsion, with the particle charge being estimated from initial fittings. Owing to the zwitterionic nature of the cysteine-derivatised copolymers, the formal overall charge was not expected to deviate significantly from neutrality. Electrophoretic mobility studies performed under the same conditions as those studied by SAXS indicated a negative zeta potential at pH 7 (see Table S4†), which is consistent with the expected contribution from anionic carboxylate groups.
A satisfactory fit to the SAXS pattern recorded for the PGMA28-P(Gly-cys)40 worms was obtained using a worm-like micelle model76 provided that a minor population of molecularly-dissolved copolymer chains was included (fitted to a generalised Gaussian coil model using the Hayter–Penfold approximation,85 as previous described). Interestingly, SAXS analysis indicated that the mean cross-sectional diameter of the worm cores, Dw, increased from 15.3 nm to 17.8 nm after L-cysteine derivatisation. TEM analyses of the dried copolymer worms confirmed this change in dimensions: Dw increased from 13 nm for the precursor worms to 16 nm after derivatisation. Similarly, the SAXS pattern recorded for the PGMA28-P(Gly-cys)80 vesicles was fitted using a vesicle model78 by incorporating the Hayter–Penfold approximation.85 However, an additional population was required to account for the presence of a small number of large scattering objects, which suggests incipient aggregation. The volume-average diameter, Dv, increased from 45 nm for the precursor vesicles to 58 nm after derivatisation, with a corresponding increase in the vesicle membrane thickness, Tm, from 7.5 to 9.7 nm. DLS studies indicated that the PGMA28-PGlyMA80 precursor vesicles exhibited a z-average diameter, Dz, of 49 nm, whereas that for the final PGMA28-P(Gly-cys)80 vesicles was 81 nm. Interestingly, the vesicular morphology was much more clearly visualised by TEM after epoxy-thiol derivatisation, which suggests a stronger interaction of the ionic groups with the TEM staining agent (uranyl formate).
In summary, L-cysteine derivatisation introduces a significant amount of charge into the originally hydrophobic PGlyMA chains. This accounts for the sphere-to-unimer transition observed for PGMA28-PGlyMA25 spheres and also the increase in size for the PGMA28-PGlyMA40 worms and the PGMA28-PGlyMA80 vesicles.
In situ SAXS studies during RAFT aqueous emulsion polymerisation of GlyMA
We recently reported the first in situ SAXS studies during RAFT aqueous emulsion polymerisation of MOEMA at 10% w/w solids using a bespoke stirrable reaction cell.47 Here, we use the same experimental set-up to monitor the RAFT aqueous emulsion polymerisation of GlyMA and hence examine the nucleation and growth of PGMA48-PGlyMA100 spheres at 10% w/w solids. Such spheres were targeted for this in situ SAXS experiment because they were larger (Dz = 37 nm) than the spheres prepared using the PGMA28 precursor (Dz = 15–21 nm; Table S1†). Larger spheres are easier to image by postmortem TEM analysis, which was conducted to compare the PGMA48-PGlyMA100 spheres formed during this in situ SAXS experiment with the equivalent laboratory-scale formulation performed in the absence of any synchrotron radiation. 1H NMR spectroscopy studies confirmed that a high monomer conversion was achieved in both cases (>98% within 1 h at 50 °C). DMF GPC analysis indicated that the molecular weight distributions for the two diblock copolymers were comparable (Mn = 22800; Mw/Mn = 1.18 vs. Mn = 26800; Mw/Mn = 1.22 for the in situ SAXS and laboratory-scale syntheses, respectively), see Fig. S6A.† DLS measurements indicated that relatively narrow unimodal size distributions were obtained in both cases (Fig. S6B†). However, a Dz of 26 nm was observed for the nanoparticles prepared during the in situ SAXS experiment, which is somewhat lower than that for the nanoparticles (Dz = 37 nm) obtained from the laboratory-scale synthesis. Postmortem TEM images indicated the formation of well-defined spheres for both PISA formulations, see Fig. 7.
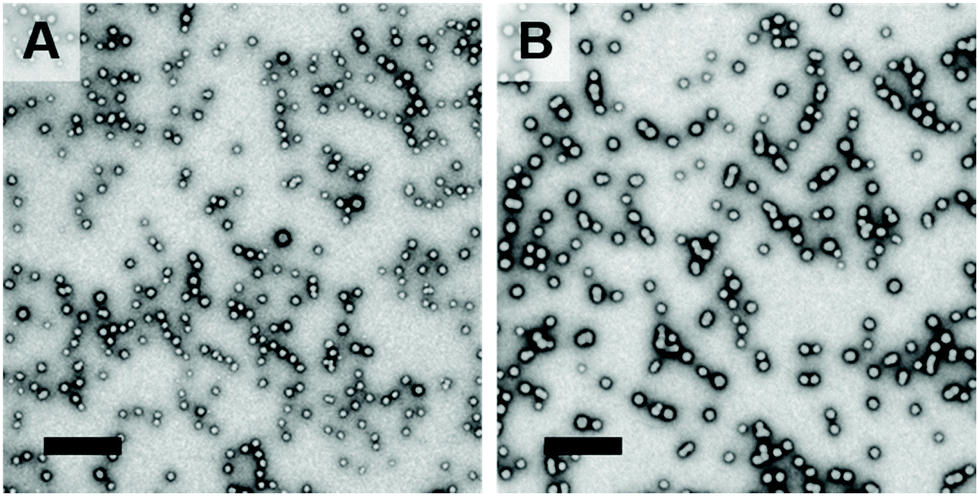 | ||
| Fig. 7 Representative TEM images recorded for the dried PGMA48-PGlyMA100 spheres prepared via RAFT aqueous emulsion polymerisation of GlyMA at 10% w/w solids at 50 °C: (a) after in situ SAXS experiments (b) after the equivalent laboratory-scale synthesis conducted in the absence of X-ray synchrotron radiation using the same PISA formulation. Scale bars represent 200 nm. | ||
SAXS patterns were collected every 2.5 min for 40 min, see Fig. 8A. As recently reported for the RAFT aqueous emulsion polymerisation of MOEMA, we focus on two key aspects of this PISA formulation: (i) the timescale for the onset of micellar nucleation and (ii) the timescale for cessation of the polymerisation. During the laboratory-scale experiment (Fig. 8B), the initial rate of polymerisation was relatively slow and the onset of nucleation was observed after 22 min; the instantaneous conversion was 25%, which corresponded to a critical PGlyMA DP of 25. This GlyMA polymerisation was essentially complete (>98% conversion) within 35 min at 50 °C. To assess the onset of micellar nucleation during the in situ SAXS experiment, the scattering intensity, I(q), at an arbitrary q of 0.01 Å−1 was plotted as a function of time, see Fig. 8C. An upturn was observed at 14 min owing to the formation of larger scattering objects, thus indicating the onset of micellar nucleation. No further change in the scattering patterns was discernible after 33 min, so the GlyMA polymerisation was judged to be complete on this timescale. Thus, both the onset of nucleation and cessation of the polymerisation occurred within slightly shorter timescales for the in situ SAXS experiment. This is attributed to a modest rate enhancement caused by the ionising nature of the high-energy X-rays which generates an additional radical flux;86,87 such observations are consistent with our prior in situ SAXS studies of other PISA formulations.47,88 The mean sphere core radius, Rs, can be estimated from the local minimum at q = 0.075 Å−1 that becomes discernible after nucleation, using the well-known relationship d = 4.49/q, where d is a real-space distance corresponding to Rs (the mean core radius in Å). This local minimum gradually shifts to lower q throughout the polymerisation, see Fig. 8D. More specifically, Rs increases from 5.80 to 8.16 nm, indicating a final volume-average spherical core diameter of 16.3 nm. The final PGMA48-PGlyMA100 diblock copolymer nanoparticles obtained after the in situ SAXS experiment were diluted to 1.0% w/w and a SAXS pattern was recorded. The data were fitted to a spherical micelle model,76 see Fig. S7,† to give a volume-average overall sphere diameter, Ds, of 23.1 nm, where Ds = 2Rs + 4Rg. Given that DLS is known to be more biased towards larger particles than SAXS, this is consistent with the corresponding Dz diameter of 26 nm. Parameters such as the mean sphere core radius (Rs) and mean radius of gyration of the PGMA48 stabiliser block (Rg) calculated from fitting this SAXS pattern enabled the 10% w/w final SAXS pattern recorded after 40 min to be analysed using a spherical micelle model by incorporating an appropriate structure factor to account for the higher nanoparticle concentration (Fig. S8†). This latter analysis afforded a comparable Ds of 23.3 nm. Fitting parameters and further information regarding these SAXS models can be found in the ESI and are summarised in Table S6.†
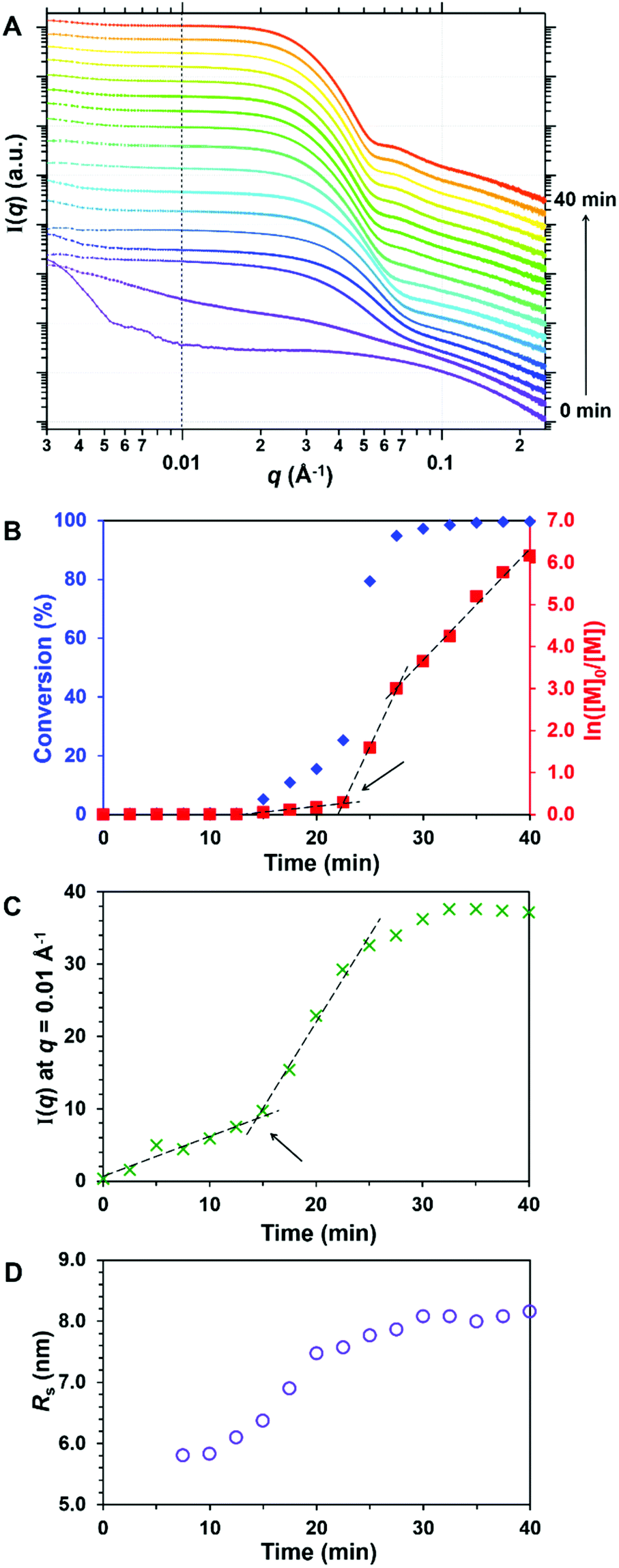 | ||
| Fig. 8 SAXS patterns recorded during the RAFT aqueous emulsion polymerisation of GlyMA targeting PGMA48-PGlyMA100 spheres at 10% w/w solids using a stirrable reaction cell. (A) SAXS patterns recorded every 2.5 min from 0 to 40 min. (B) Conversion vs. time curve and corresponding semi-logarithmic plot obtained for the equivalent laboratory-scale synthesis. (C) the I(q) at q = 0.01 Å−1 as a function of time. (D) Evolution of the mean sphere core radius, Rs, over time, calculated using Rs = 4.49/q, where q is the local minimum between 0.078–0.054 Å−1. Arrows indicate the onset of micellar nucleation. | ||
Conclusions
Using a sufficiently short non-ionic PGMA28 stabiliser block for the RAFT aqueous emulsion polymerisation of GlyMA under mild conditions (50 °C, pH 7) provides convenient access to epoxy-functional spheres, worms and vesicles. This is attributed to the relatively high aqueous solubility of GlyMA (∼22 g dm−3), which enables the restrictive paradigm of kinetically-trapped spheres observed for many such PISA formulations to be circumvented. High GlyMA conversions (>98%) can be obtained within 1 h and molecular weight distributions remained relatively narrow (Mw/Mn < 1.5) if the target PGlyMA DP remains below 100. A pseudo-phase diagram was constructed for the synthesis of PGMA28-PGlyMAn nano-objects at copolymer concentrations ranging from 5 to 30% w/w. This systematic approach is essential for the reproducible targeting of pure spheres, worms or vesicles. Each morphology was initially assigned on the basis of TEM studies and subsequently confirmed by SAXS analysis. However, long-term storage of a 10% w/w aqueous dispersion of PGMA28-PGlyMA55 worms under ambient conditions led to a 26% loss of the original epoxy groups over 12 weeks, as determined by 1H NMR spectroscopy. Concomitant GPC studies indicated that significant broadening of the molecular weight distribution occurred over the same time period. This suggests that the hydroxyl groups generated via ring-opening of the epoxy groups by reaction with water can themselves react with adjacent epoxy groups, leading to intermolecular branching. The epoxy-functional cores of aqueous dispersions of PGMA28-PGlyMAn diblock copolymer spheres, worms and vesicles can be conveniently derivatised by reacting with excess L-cysteine to afford zwitterionic copolymers. Elemental microanalyses indicate that high degrees of derivatisation (89–91%) can be achieved using such this epoxy-thiol chemistry. Finally, an in situ SAXS study was conducted during the RAFT aqueous emulsion polymerisation of GlyMA, targeting PGMA48-PGlyMA100 spheres. A modest rate enhancement was observed for this experiment, with both nucleation and cessation of the polymerisation occurring on somewhat shorter timescales compared to the equivalent laboratory-scale formulation owing to the ionising nature of the high-energy X-ray synchrotron beam. Postmortem TEM and DLS analysis confirmed that well-defined spheres were obtained in both cases and the evolution of the sphere core diameter over the time was monitored.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. P. A. thanks the ERC for an Advanced Investigator grant (PISA 320372) to support F. L. H. and the EPSRC for an Established Career Fellowship in Particle Technology (EP/R003009). The Leverhulme Trust is also thanked for post-doctoral funding of M. J. D. (RPG-2016-330). The authors wish to thank the European Synchrotron Radiation Facility and Diamond Light Source for providing synchrotron beamtime at ID02 station (beamtime proposal numbers SC4384 and SC4864) and I22 station (beamtime proposal number SM15933), respectively. Dr Svetomir Tzokov is thanked for carbon-coating the TEM grids.References
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef.
- Y. Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef.
- W.-N. He and J.-T. Xu, Prog. Polym. Sci., 2012, 37, 1350–1400 CrossRef.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 CrossRef.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2019, 59, 2–27 Search PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 Search PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef.
- C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis and C. H. Such, Macromolecules, 2002, 35, 9243–9245 CrossRef.
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191–2204 CrossRef CAS.
- M. Manguian, M. Save and B. Charleux, Macromol. Rapid Commun., 2006, 27, 399–404 CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CAS.
- A. M. dos Santos, J. Pohn, M. Lansalot and F. D'Agosto, Macromol. Rapid Commun., 2007, 28, 1325–1332 CAS.
- I. Chaduc, W. J. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromol. Rapid Commun., 2011, 32, 1270–1276 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CAS.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Polym. Chem., 2016, 7, 851–859 RSC.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. Tan, Y. Bai, X. Zhang and L. Zhang, Polym. Chem., 2016, 7, 2372–2380 RSC.
- E. R. Jones, M. Semsarilar, A. Blanazs and S. P. Armes, Macromolecules, 2012, 45, 5091–5098 CAS.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 CAS.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS.
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881–5893 CAS.
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013–6023 CrossRef CAS.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883–5885 RSC.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CAS.
- S. Boisse, J. Rieger, K. Belal, A. Di-Cicco, P. Beaunier, M. H. Li and B. Charleux, Chem. Commun., 2010, 46, 1950–1952 RSC.
- S. Boisse, J. Rieger, G. Pembouong, P. Beaunier and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3346–3354 CrossRef CAS.
- X. L. Fan, X. J. Jia, H. P. Zhang, B. L. Zhang, C. M. Li and Q. Y. Zhang, Langmuir, 2013, 29, 11730–11741 CrossRef CAS.
- J. L. de la Haye, X. W. Zhang, I. Chaduc, F. Brunel, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2016, 55, 3739–3743 CrossRef.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 CrossRef CAS.
- B. T. T. Pham, D. Nguyen, V. T. Huynh, E. H. Pan, B. Shirodkar-Robinson, M. Carey, A. K. Serelis, G. G. Warr, T. Davey, C. H. Such and B. S. Hawkett, Langmuir, 2018, 34, 4255–4263 CrossRef CAS.
- D. Nguyen, V. Huynh, N. Pham, B. Pham, A. Serelis, T. Davey, C. Such and B. Hawkett, Macromol. Rapid Commun., 2019, 40, 1800402 CrossRef.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS.
- F. L. Hatton, A. M. Park, Y. R. Zhang, G. D. Fuchs, C. K. Ober and S. P. Armes, Polym. Chem., 2019, 10, 194–200 RSC.
- X. Dai, L. Yu, Y. Zhang, L. Zhang and J. Tan, Macromolecules, 2019, 52, 7468–7476 CrossRef CAS.
- N. P. Truong, M. V. Dussert, M. R. Whittaker, J. F. Quinn and T. P. Davis, Polym. Chem., 2015, 6, 3865–3874 RSC.
- W. J. Zhang, F. D'Agosto, O. Boyron, J. Rieger, B. Charleux, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584–7593 CrossRef CAS.
- L. Carlsson, A. Fall, I. Chaduc, L. Wagberg, B. Charleux, E. Malmstrom, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmstrom and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 CrossRef CAS.
- J. Rieger, F. Stoffelbach, C. Bui, D. Alaimo, C. Jerome and B. Charleux, Macromolecules, 2008, 41, 4065–4068 CrossRef CAS.
- J. Engström, F. L. Hatton, L. Wågberg, F. D'Agosto, M. Lansalot, E. Malmström and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- J. Rieger, G. Osterwinter, C. O. Bui, F. Stoffelbach and B. Charleux, Macromolecules, 2009, 42, 5518–5525 CrossRef CAS.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 CrossRef CAS.
- B. Akpinar, L. A. Fielding, V. J. Cunningham, Y. Ning, O. O. Mykhaylyk, P. W. Fowler and S. P. Armes, Macromolecules, 2016, 49, 5160–5171 CrossRef CAS.
- L. H. Guo, Y. J. Pang, T. Qiu, Y. Meng and X. Y. Li, Polymer, 2014, 55, 4601–4610 CrossRef CAS.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- W. J. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075–4084 CrossRef CAS.
- A. A. Cockram, T. J. Neal, M. J. Derry, O. O. Mykhaylyk, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, Macromolecules, 2017, 50, 796–802 CrossRef CAS.
- L. P. D. Ratcliffe, A. J. Ryan and S. P. Armes, Macromolecules, 2013, 46, 769–777 CrossRef CAS.
- F. L. Hatton, J. R. Lovett and S. P. Armes, Polym. Chem., 2017, 8, 4856–4868 RSC.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Langmuir, 2012, 28, 1196–1205 CrossRef CAS.
- N. J. W. Penfold, Y. Ning, P. Verstraete, J. Smets and S. P. Armes, Chem. Sci., 2016, 7, 6894–6904 RSC.
- J. Tan, D. Liu, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1700195 CrossRef.
- X. Dai, Y. Zhang, L. Yu, X. Li, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 955–961 CrossRef CAS.
- Q. Xu, Y. Zhang, X. Li, J. He, J. Tan and L. Zhang, Polym. Chem., 2018, 9, 4908–4916 RSC.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347–353 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef CAS.
- O. Glatter and O. Kratky, Small-angle X-ray Scattering, Academic Press, London, 1982 Search PubMed.
- J. Bang, S. M. Jain, Z. B. Li, T. P. Lodge, J. S. Pedersen, E. Kesselman and Y. Talmon, Macromolecules, 2006, 39, 1199–1208 CrossRef CAS.
- Y. Zhang, L. Yu, X. Dai, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1102–1109 CrossRef CAS.
- X. Li, J. Tan, Q. Xu, J. He and L. Zhang, Macromol. Rapid Commun., 2018, 39, 1800473 CrossRef.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS.
- N. J. W. Penfold, A. J. Parnell, M. Molina, P. Verstraete, J. Smets and S. P. Armes, Langmuir, 2017, 33, 14425–14436 CrossRef CAS.
- C. György, J. R. Lovett, N. J. W. Penfold and S. P. Armes, Macromol. Rapid Commun., 2018, 1800289 Search PubMed.
- B. Hammouda, Probing Nanoscale Structures - The SANS Toolbox, National Institute of Standards and Technology, Gaithersburg, 2008 Search PubMed.
- J. B. Hayter and J. Penfold, Mol. Phys., 1981, 42, 109–118 CrossRef CAS.
- S. J. Tseng, C. C. Chien, Z. X. Liao, H. H. Chen, Y. D. Kang, C. L. Wang, Y. Hwu and G. Margaritondo, Soft Matter, 2012, 8, 1420–1427 RSC.
- M. H. Qiao, F. Q. Yan, W. S. Sim, J. F. Deng and G. Q. Xu, Surf. Sci., 2000, 460, 67–73 CrossRef CAS.
- M. J. Derry, L. A. Fielding, N. J. Warren, C. J. Mable, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2016, 7, 5078–5090 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinetic data for RAFT solution polymerisation of GMA in ethanol; kinetic data for RAFT aqueous emulsion polymerisation of GlyMA, summary of copolymer characterisation data, GPC curves for selected diblock copolymers, SAXS patterns, summary tables of SAXS parameters from model fittings, elemental microanalytical data, and DLS analysis before and after L-cysteine derivatisation, further details of the SAXS models. See DOI: 10.1039/d0py01097a |
| ‡ Current address: Department of Materials, Loughborough University, Loughborough, LE11 3TU, UK. |
| § Current address: Aston Institute of Materials Research, Aston University, Birmingham, B4 7ET, UK. |
| This journal is © The Royal Society of Chemistry 2020 |