
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneral approach to prepare polymers bearing pendant isocyanate groups†
Rodrigo
Navarro
 ,
Carolina
García
,
Juan
Rodríguez-Hernández
,
Carlos
Elvira
,
Angel
Marcos-Fernández
,
Alberto
Gallardo
and
Helmut
Reinecke
*
,
Carolina
García
,
Juan
Rodríguez-Hernández
,
Carlos
Elvira
,
Angel
Marcos-Fernández
,
Alberto
Gallardo
and
Helmut
Reinecke
*
Institute of Polymer Science and Technology (ICTP-CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain. E-mail: hreinecke@ictp.csic.es; Fax: +34-91-5644853; Tel: +34-91-2587557
First published on 28th July 2020
Abstract
A versatile synthetic approach for the easy preparation, under smooth reaction conditions, of diverse classes of linear polymers bearing aliphatic or aromatic isocyanate groups in the side chains is described. The procedure consists in the transformation of primary amine groups present in the polymer into isocyanates using equimolar amounts of diphosgene or triphosgene and a soluble tertiary amine as the acid scavenger. The transformation of all amine groups takes place quasi-simultaneously and instantaneously as shown by the invariability of the chain length of the polymer and the absence of crosslinked products. The number of isocyanate groups per molecule that can be achieved by this approach corresponds to the number of primary amine groups of the starting polymer and is up to three orders of magnitude higher than in any other non-crosslinked molecule described so far. The isocyanate functionalized polymers can be used to anchor a large variety of molecules to the polymer chains. The approach is in detail demonstrated with poly(vinyl chloride) (PVC) carrying aromatic amine groups but has also been confirmed by using a number of other polymer types bearing both aromatic and aliphatic primary amines. Furthermore, it has been used to prepare a novel low molecular weight compound, tris(2-cyanatoethyl)amine.
Introduction
The preparation of polymers bearing isocyanate groups in the side chains is of great interest due to the high reactivity of this functional group towards alcohols, amines, thiols or other groups carrying active hydrogen atoms. As a consequence, these polymers are easily modified, crosslinked or used for the grafting of biomolecules or other polymeric chains. Due to the enormous versatility of such materials, scientists started to work in this field right from the birth of polymer science. There are basically two synthetic strategies that may lead to polymers bearing isocyanate groups: (a) a bottom-up approach that consists in the use of a monomer with this functionality and subsequent homo- or co-polymerization, (b) chemical modification of polymers bearing side chain primary amines using phosgene or its derivatives.The bottom-up strategy was studied in a number of papers1–13 published on this subject as early as in the fifties. These authors used vinylic monomers bearing an isocyanate moiety such as vinylisocyanate or isopropenylisocyanate1–9 and studied their homo- and copolymerization. Other copolymers bearing isocyanate groups were studied by Butler and Monroe,10 Graham11 and Vollmert12 who used allyloxyethyl isocyanate,10 9-decenyl isocyanate,10O-isocyanatoethyl methacrylate11 or N-(6-isocyanato)-hexylacrylamide12 as the co-monomers able to introduce a NCO-functionality into the respective polymer. Liebermann, finally,13 used p-isocyanatostyrene copolymerized with styrene or acrylonitrile to prepare copolymers with up to 15 mol% of isocyanate groups in the side chains.
In most of the cited cases, due to side reactions during the polymerization caused by the isocyanate group, the obtained polymers were of low molecular weight, partially crosslinked and/or had a low content of isocyanate groups. For these reasons, these materials never had practical utility and have never again been mentioned in recent literature.
The second general approach to prepare polymers bearing isocyanate groups is the chemical modification of a polymer bearing primary amine groups. This, however, has never been attempted successfully for the following reason: as the target functionality NCO is highly reactive toward amines, isocyanate groups formed in an early stage of the reaction are liable to react with amines that have not yet been transformed into an isocyanate group and consequently crosslinked and undefined products should be obtained.
The most common way to transform amine groups into isocyanates is the use of phosgene and its derivatives. Phosgene is an extremely versatile and useful agent for the preparation of a number of important substances. However, particularly academic scientists were looking for safer alternative substitutes for this highly toxic gaseous compound. In 1976 Kurita et al.14 proposed the use of diphosgene for this purpose, a dense liquid of 128 °C boiling point at atmospheric pressure that decomposes under certain conditions into two phosgene molecules. Later, in 1987, H. Eckert et al.15,16 proposed the use of triphosgene as an alternative to phosgene as it is a solid (m.p. 80 °C, b.p. 206 °C) and, thus, even easier to transport and store than diphosgene. These authors brought different amines, alcohols and carbon acids to reaction with equimolar amounts of triphosgene obtaining good to excellent yield of the expected products. It was further shown that each molecule reacted like three phosgene molecules. Interestingly the authors describe that the same reactions using gaseous phosgene were not successful as “even the use of a several-fold excess gave only very moderate yields”.
Another interesting work related to the results of the present paper is authored by Peerlings, Versteegen and Meijer17,18 who describe the successful use of di-tert-butyltricarbonate for the quantitative conversion of almost any primary monoamine into its corresponding isocyanate in less than 5 minutes at room temperature releasing two equivalents of CO2 and tert-butanol. The authors claim furthermore, that said compound is the reagent par excellence for the synthesis of multi-isocyanates, since the formation of cyclic ureas is suppressed. The approach is demonstrated on a propylene imine dendrimer of five generations.
Different standard procedures for the preparation of isocyanate groups have been described so far.19 In industry, the most widely used method is the transformation of a primary amine group into an isocyanate using phosgene at elevated temperature. In the case of mono- or diamines the corresponding isocyanates are usually obtained in excellent yields far above 90% and secondary by products, mainly ureas formed between newly formed isocyanate and non reacted amine can be easily eliminated by distillation or recrystallization. However, in a poly-aminated compound these urea bonds formed between different polymeric chains lead to crosslinked products that make this approach useless. For this reason, an aminated polymer can only be transformed into a multi-isocyanate compound if two conditions are met: (a) the polymer is at a concentration below its critical overlap concentration and (b) the reaction between the amine and the phosgenation agent is much faster than the reaction between amine and newly formed isocyanate. Thus, all amine groups involved must be transformed quasi-simultaneously and instantaneously into the corresponding isocyanate.
In the present work we have studied the use of phosgene and its derivatives for the quantitative transformation of aminated linear polymers into the corresponding macromolecules carrying isocyanate groups. As will be described, this strategy allows for the fabrication of functional polymers with a precisely controlled amount of reactive isocyanate functional groups.
Experimental part
Materials
Commercial bulk polymerized PVC was obtained from Rio Rodano Industries, Spain. The average molecular weights determined by GPC were MW = 112![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and Mn = 48000 g mol−1. The tacticity measured by 13C-NMR was syndio = 30.6%, hetero = 49.8% and iso = 19.6%.
000 g mol−1 and Mn = 48000 g mol−1. The tacticity measured by 13C-NMR was syndio = 30.6%, hetero = 49.8% and iso = 19.6%.
Tris(2-aminoethyl)amine was purchased from Aldrich and used as it is.
THF was dried over sodium and distilled. Methylene chloride and triethylamine were dried over CaH2 and distilled.
NMR spectra were obtained in CDCl3 solutions using a 300 MHz Varian FTIR measurements were carried out on a PerkinElmer Spectrum One spectrometer using an ATR device with a resolution of 2 cm−1.
Gel permeation chromatography (GPC) analyses were carried out with Styragel (300 × 7.8 mm, 5 mm nominal particle size) Water columns. DMF with LiBr (0.1 w/w) was used as a solvent. Measurements were performed at 70 °C at a flow rate of 0.7 mL min−1 using a RI detector. Molecular weights of polymers were referenced to PS standards.
Preparation of PVC modified with a tailored content of aromatic amine groups
The synthesis of PVC carrying primary aromatic amines (PVC-o-NH2) consists of chemically modifying PVC in cyclohexanone (CH) or methyl ethyl ketone (MEK) solution using sodium thiolate salts of 2,3 or 4-Aminothiophenol as described elsewhere.20 These thiolates are prepared by neutralization of the corresponding commercially available aromatic thiol with equimolar amounts of sodium hydroxide in ethanol.Preparation of PVC carrying NCO groups
1 g (14.4 mmol) of PVC-o-NH2-8% was dissolved in 20 mL of dry THF and 80 mL of methylene chloride and 0.33 mL (2.4 mmol) of triethylamine were added. This solution was poured at room temperature to a stirred solution of 125 mg (0.42 mmol) triphosgene in 25 mL methylene chloride. After five minutes reaction the mixture was washed twice with ice/water in order to eliminate the quaternary salts formed, the organic phase was dried over anhydrous MgSO4 and the solution filtered and precipitated in hexane. After drying 950 mg of PVC-NCO-8% were obtained.Preparation of PVC-NHCON(C2H5)2-8%
950 mg of PVC-NCO-8% were dissolved in 50 mL of dry THF, 0.12 mL (1.2 mmol) diethylamine were added and the mixture stirred for 1 hour at 40 °C. Then, the mixture was precipitated in methanol and the obtained polymer purified by three solution-precipitation cycles in THF-methanol. 1 g polymer whose 1H-NMR spectrum is shown in Fig. 2 was obtained.Preparaction of tris(2-isocyanatoethyl)amine
To a solution of 1.05 g (3.5 mmol) of Triphosgene in 400 ml of CH2Cl2 a mixture of 515 mg (3.5 mmol) tris(2-aminoethyl)amine and 2.9 ml (21 mmol) NEt3 in CH2Cl2 is added at room temperature. After 5 minutes the solvent is eliminated and the residue extracted with hot heptane. After elimination of heptane 380 mg (yield 95%) of the triisocyanate are obtained.Results
In this paper, we have studied the specific conditions under which this transformation is possible. For this purpose we have used PVC which has been shown to be a polymer that can selectively and in a clean manner be modified with a controlled number of primary aromatic amine groups.20In order to favor the main goal and reduce or avoid crosslinking between different chains, in this study all reactions were carried out under highly diluted conditions, below the critical overlap concentration of c* = 0.12 mol L−1 of the PVC/solvent system (determined according to ref. 21). PVC carrying 8 mol% of primary aromatic amine groups situated in ortho position to the sulfur anchor atom (Scheme 1a) was used as the starting polymer. As the phosgenating agents, 15 wt% solution of phosgene in toluene, diphosgene and triphosgene were tested using different stoichiometries with respect to the number of amine groups in the polymer. Furthermore, the type and quantity of the base used for scavenging the hydrochloride formed during the reaction were varied. The detailed reaction conditions are summarized in Table 1.
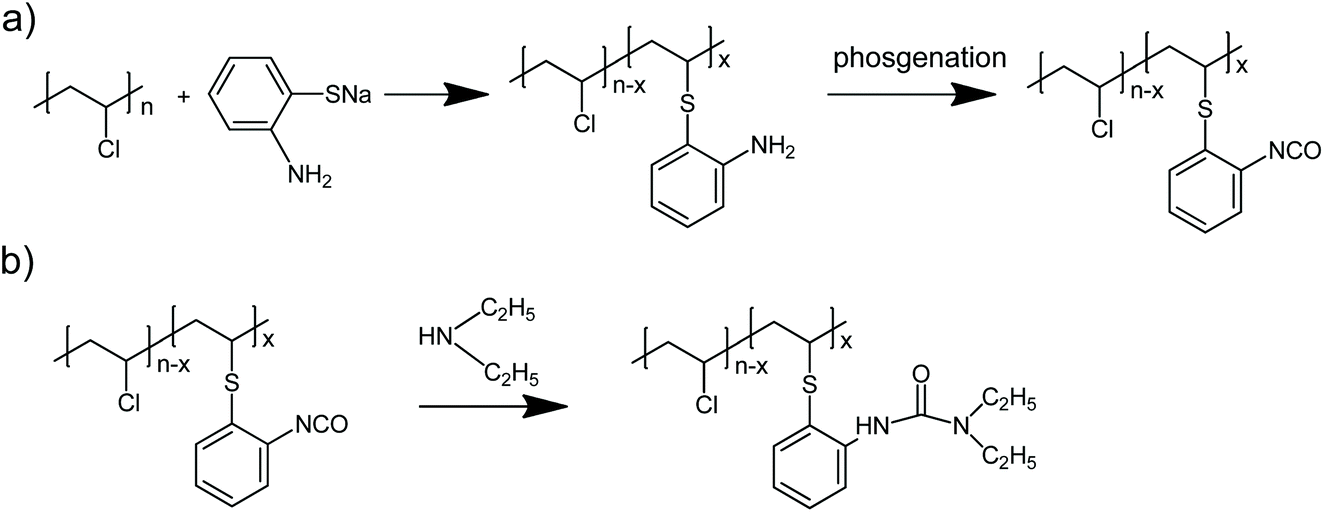 | ||
| Scheme 1 a) preparation of aminated PVC (PVC-o-NH2-8%) and transformation into PVC bearing isocyanate groups (PVC-NCO-8%), (b) formation of PVC bearing urea groups (PVC-NHCON(C2H5)2-8%). | ||
| Entry | Phosgenating Agent | Phosgenating agent per mol amine | Base | Base per mol amine | Observation |
|---|---|---|---|---|---|
| DIPEA: N,N-Diisopropylethylamine, NEt3: triethylamine, DABCO: 1,4-diazabicyclo[2.2.2]octane. | |||||
| 1 | Phosgene solution (15%) | 1.03 mol | NEt3 | 2.2 mol | Incomplete reaction and crosslinking |
| 2 | Phosgene solution (15%) | 2.0 mol | NEt3 | 2.2 mol | Incomplete reaction and crosslinking |
| 3 | Phosgene solution (15%) | 3.0 mol | NEt3 | 2.2 mol | Incomplete conversion, insoluble polymer |
| 4 | Triphosgene | 0.35 mol | — | — | Incomplete reaction and crosslinking |
| 5 | Triphosgene | 0.35 mol | NEt3 | 1.8 mol | Incomplete reaction and crosslinking |
| 6 | Triphosgene | 0.35 mol | NEt3 | 1.9 mol | Incomplete reaction and crosslinking |
| 7 | Triphosgene | 0.35 mol | NEt3 | 2.0 mol | Complete conversion, soluble polymer |
| 8 | Triphosgene | 0.35 mol | NEt3 | 2.1 mol | Complete conversion, soluble polymer |
| 9 | Triphosgene | 0.35 mol | DIPEA | 2.1 mol | Complete conversion, soluble polymer |
| 10 | Triphosgene | 0.5 mol | DABCO | 2.1 mol | Complete conversion, soluble polymer |
| 11 | Triphosgene | 0.35 mol | Na2CO3 | 2.1 mol | Incomplete reaction and crosslinking |
| 12 | Diphosgene | 0.5 mol | NEt3 | 2.1 mol | Complete conversion, soluble polymer |
Phosgenations were carried out in dry THF–CH2Cl2 1:4 mixtures. In order to avoid immediate crosslinking it is decisive to add the polymer/base solution to the phosgenating agent and not vice versa. After the addition of the polymer solution and base to the phosgenating agent the mixture was diluted with CH2Cl2 and extracted twice with ice-cold water in order to eliminate the quaternary ammonium salts formed during the reaction between the base and the hydrochloride formed. The organic phase was subsequently dried and precipitated in hexane. The dried precipitated polymers were analyzed by FTIR and, if soluble, by 1H-NMR spectroscopy and GPC.
From the experiments (entries 1–3) it was concluded that it was not possible to achieve soluble products using phosgene solutions, not even with a threefold molar excess with respect to the number of amine groups, a result that confirms observations made by Eckert on reactions with low molecular weight amines.19,20 On the other hand, 0.5 mol diphosgene per mol amine as a precursor of two equivalents of phosgene or 0.35 mol per amine of triphosgene22,23 as a precursor of three equivalents of phosgene were able to quantitatively transform all amine groups of the polymer into isocyanates when at least two equivalents of a soluble acid scavenger were present in the reaction solution. It is noteworthy that even a slight defect of base (entries 5 and 6 in Table 1) or the use of an insoluble base like Na2CO3 (entry 11) that is not immediately available for the scavenging process, lead to the formation of crosslinked systems.
In Fig. 1, the IR-spectra of pure PVC, aminated PVC (PVC-o-NH2-8%) and polymer from entry 8 (PVC-NCO-8%) after quantitative phosgenation are compared. The aminated PVC is characterized by two N–H valence bonds (according to associated and non-associated amines) around 3400 cm−1, an N–H deformation band at 1610 cm−1 and the aromatic C–C valence bonds at 1570 and 1480 cm−1. After the phosgenation reaction under the chosen reaction conditions N–H valence and NH-deformation bonds have completely disappeared and instead a strong NCO valence bond at 2250 cm−1 has formed.
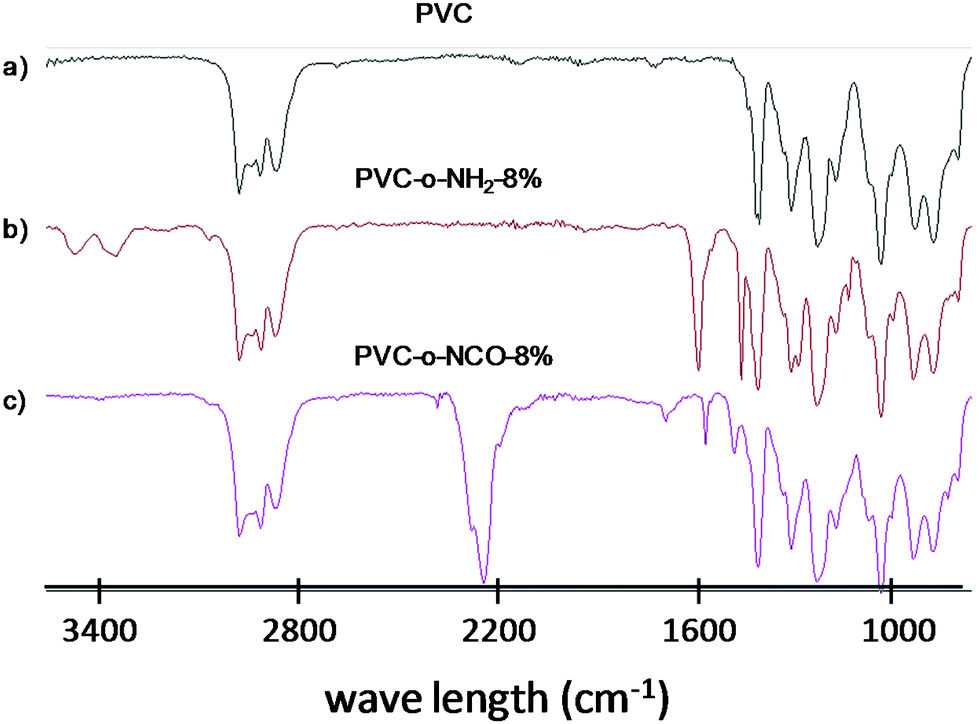 | ||
| Fig. 1 FTIR-ATR spectra of (a) pure PVC, (b) PVC-o-NH2-8% and (c) PVC-NCO-8%. | ||
Although it is possible to isolate and dry the obtained PVC-NCO, it is recommendable to carry out subsequent reactions on this highly reactive polymer in situ, directly after the formation of the isocyanate groups. In this way, its reaction with traces of humidity that would lead to the formation of CO2 and amine groups that in turn would form a crosslinked material via formation of urea bridges due to its reaction with isocyanate groups, is avoided.
Such a subsequent reaction was carried out using equimolar amounts of diethylamine. At a temperature of 40 °C the reaction reached complete conversion after thirty minutes as it is shown in Fig. 2 where the 1H-NMR spectrum in CDCl3 of the isolated and purified polymer is compared with that of the PVC-o-NH2 starting material. In the spectrum of the aminated polymer, additionally to the PVC chain proton peaks at 2.2 and 4.5 ppm, four signals between 6.6 and 7.3 ppm (aromatic ring protons) are observed while the -NH2 protons are partially overlapped by the –CH–Cl protons at 4.5 ppm. Proton peaks from modified chain segments –CH–S– appear at 3.6 ppm. After transformation of the aromatic amine groups into isocyanates and subsequent reaction of the latter with diethylamine the four aromatic proton peaks shifted considerably towards higher ppm values and a new signal related to the urea protons appeared at 6.8 ppm. Additionally, new peaks at 3.4 ppm and 1.2 ppm corresponding to the ethyl groups of the newly formed ureas developed. The integral values indicated in the NMR spectra are normalized with respect to one modifier molecule. This means that the value 26 of the CH2-protons of PVC (4.5 ppm) indicates that per modifier molecule there are 13 monomeric PVC units or, in other words: the degree of modification is 1/13 = 0.078 = 7.8 mol%. The urea product obtained from this polymer is formed using diethylamine HN(C2H5)2 which reacts with each modifier molecule (via previous transformation to the isocyanate). Consequently the expected integrals of the corresponding proton peaks are 4 (at 3.4 ppm) and 6 (at 1.2 ppm).
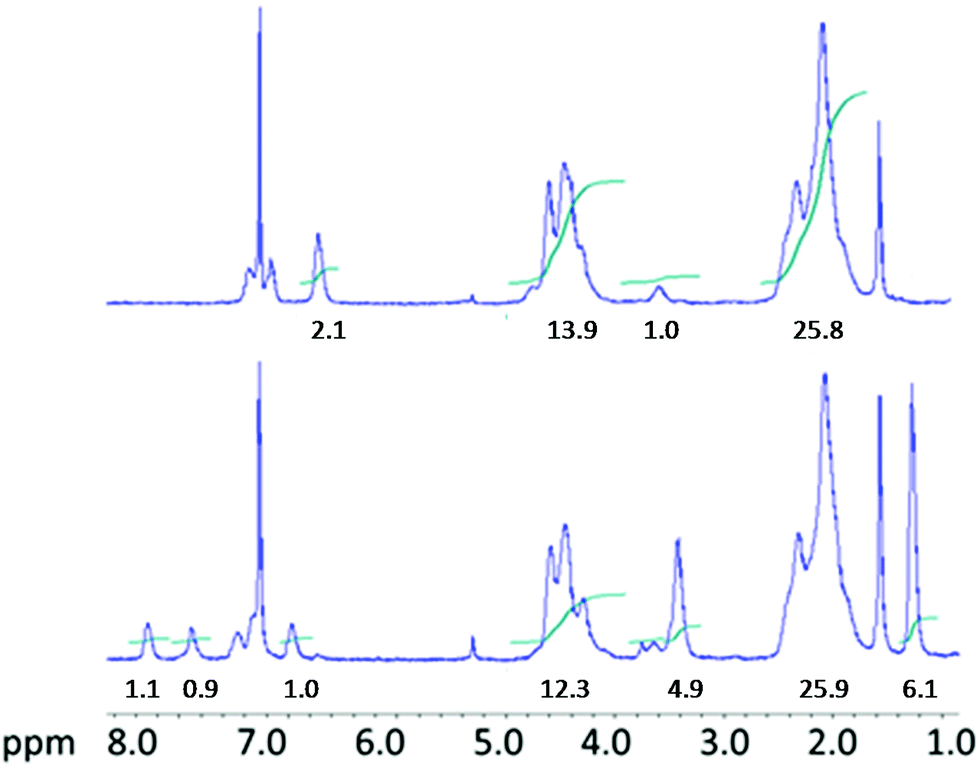 | ||
| Fig. 2 H-NMR spectra in CDCl3 of PVC-o-NH2-8% (top) and PVC-NHCON(C2H5)2-8% (bottom). | ||
In order to check whether the phosgenation reaction could have led to branching with its corresponding increase in molecular weight, GPC traces in DMF of aminated PVC and that of the polymer after reaction of its isocyanates with diethylamine were compared (it was not possible to carry out GPC on the polymers with NCO groups due to their high reactivity with protons from the column). The GPC traces depicted in the ESI (Fig. S1†) show that both the aminated polymer and PVC-NHCON(C2H5)2-8% are eluted at the same time indicating that aminated and isocyanated polymers show virtually the same degree of polymerization. This confirms that no chain extension side reactions or even crosslinking took place during phosgenation under the experimental conditions chosen.
Other systems studied
In order to check the versatility of the above approach a number of different polymers (in particular polystyrene (PS), polyepichlorohydrine (PECH) and polyvinylpyrrolidone (PVP), Fig. 3) containing primary aromatic or aliphatic amine groups, were subjected to the phosgenation treatment successfully used in the case of aminated PVC, that is 0.33 mol of triphosgene and at least 2 mol triethylamine per mol of primary amine and polymer/solvent concentrations below the critical overlap concentration. In all cases the amine groups were transformed quantitatively into isocyanate groups. This was demonstrated, on the one hand, by the fact that soluble, non-crosslinked products were obtained and, on the other hand, that subsequent reactions with different alcohols or amines gave the corresponding polymers quantitatively with the expected structures as shown by GPC, IR and 1H-NMR spectroscopy for some specific cases (ESI, Fig. S2–5†).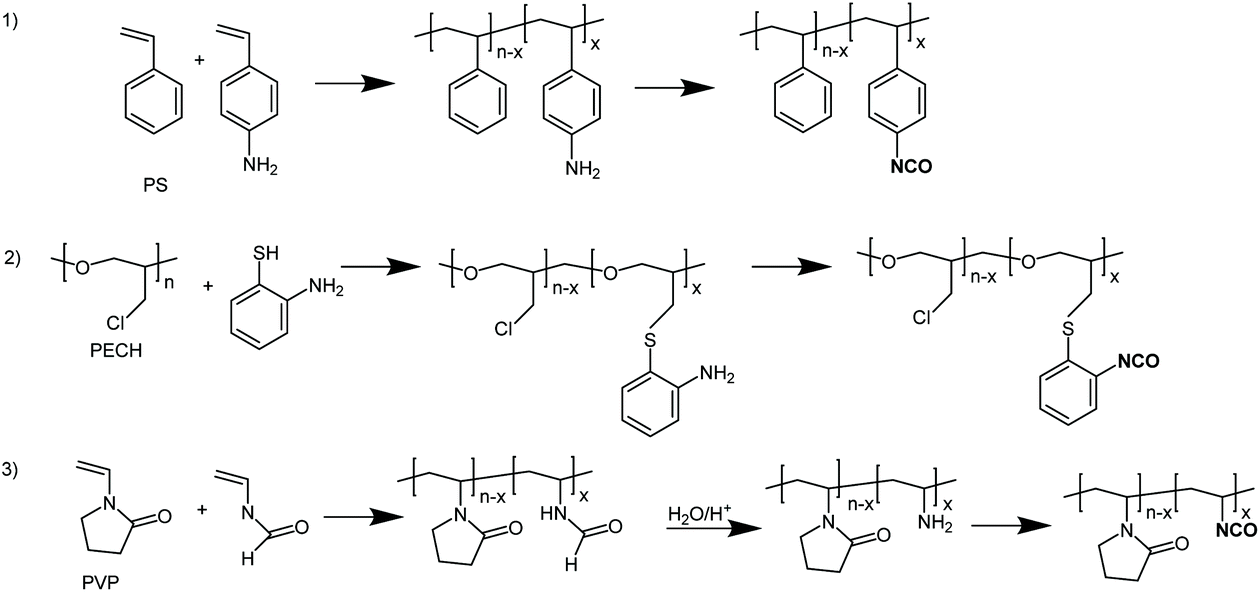 | ||
| Fig. 3 The phosgenation approach using condensed forms of phosgene may be carried out in different classes of aminated polymers. | ||
We have furthermore used the approach to prepare a novel triisocyanate of low molecular weight that, to our knowledge has never been prepared before. The starting compound was tris(2-aminoethyl)amine (Fig. 4).
 | ||
| Fig. 4 Preparation of tris(2-cyanatoethyl)amine from tris(2-Aminoethyl)amine. | ||
In this case, due to the high density of amine groups in the solution the reaction mixture has to be highly diluted. The critical concentration for complete conversion is c = 0.02 mol amine groups per liter. The obtained triisocyanate is highly reactive and it is recommended to be used for subsequent reactions directly from the crude CH2Cl2 solution.
The molecular weight of the new compound with a formula of C9H12N4O3 was determined by mass spectroscopy: 224.09 g mol−1. IR, 1H-NMR and 13C-NMR are shown below (Fig. 5 and 6).
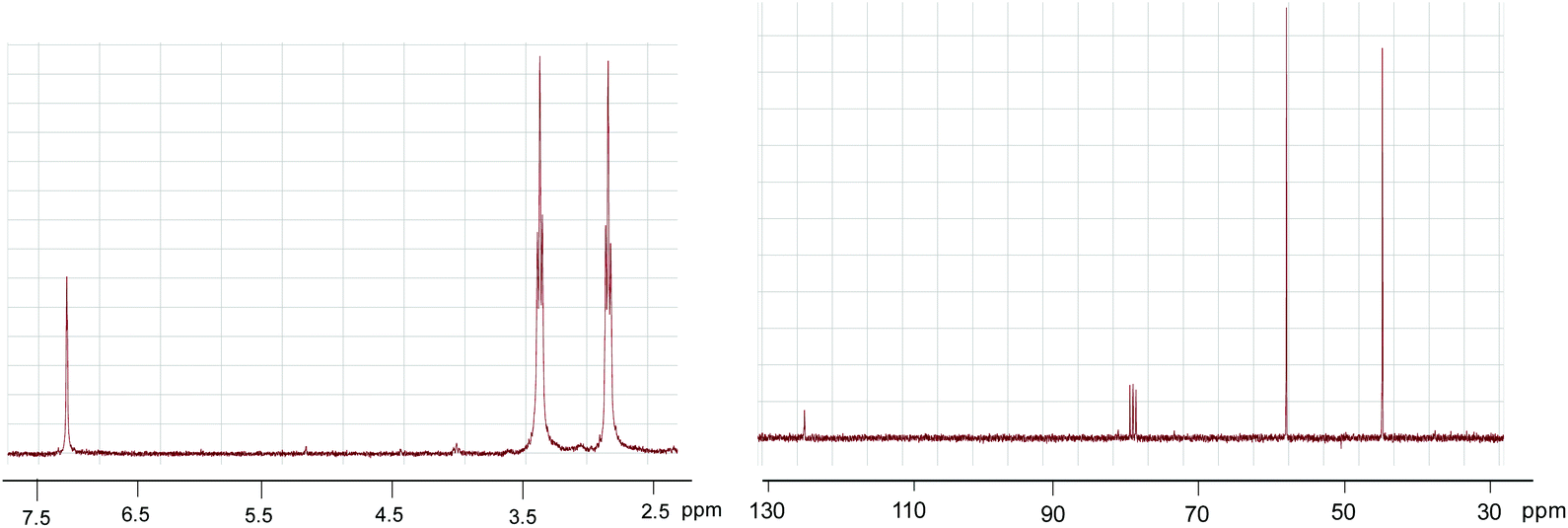 | ||
| Fig. 5 1H-NMR and 13C-NMR sprectra of tris(2-isocyanatoethyl)amine recorded in CDCl3. | ||
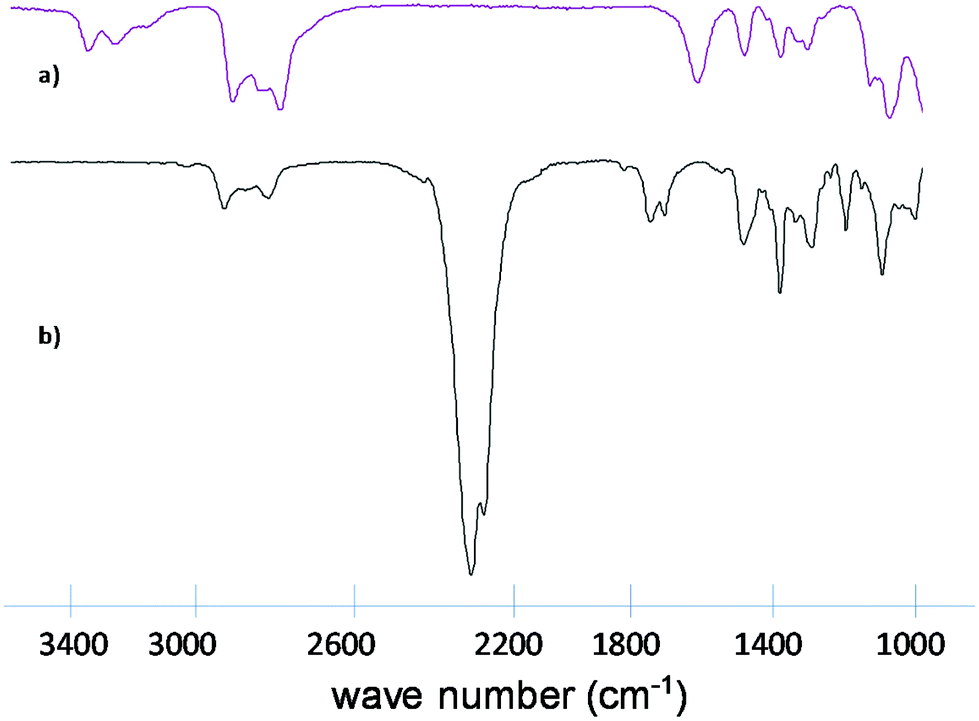 | ||
| Fig. 6 IR sprectra of tris(2-aminoethyl)amine (above) and tris(2-isocyanatoethyl)amine (below). | ||
Discussion
Up to date, the exact reaction mechanisms of diphosgene and triphosgene are not well understood. It is supposed19 that triphosgene, when attacked by a nucleophile (Nu), decomposes into phosgene and diphosgene-Nu derivative. Diphosgene, on the other hand, shows reactivity as a tricoordinated carbonic acid derivative being these reactions largely accelerated using catalysts like chlorine, and particularly basic nitrogenous compounds like pyridine, quinoline, tetramethyl urea, or tertiary amines that were reported to catalyze the quantitative decomposition of diphosgene into phosgene. However, the complete decomposition and transformation of di- and triphosgene into gaseous phosgene do not explain the selective and quantitative formation of multi-isocyanates without secondary reactions. Otherwise the use of stoichiometric amounts of phosgene in solution should give the same results as those obtained using its condensed versions. However, not even with a threefold excess of phosgene it was possible to obtain soluble polymers.Despite the impossibility to draw a mechanism from these results it is clear that, it is essential to completely remove the generated HCl with a base to avoid the formation of urea groups ultimately leading to crosslinking and that no urea groups are significantly formed (that would increase the molecular weight) under the experimental conditions as proved by the invariability of the molecular weight after the transformation of the amino groups in the polymer backbone.
Conclusions
Linear polymers with different backbone types carrying a controllable number of isocyanate groups were prepared and isolated for the first time. These materials were obtained by chemical modification of aminated polymers using stoichiometric amounts of diphosgene or triphosgene as phosgenation agent in combination with stoichiometric quantities of a tertiary base soluble in the reaction medium. The use of phosgene in solution, even in excess, was not efficient leading to incomplete conversion and a crosslinked material. The use of a tertiary base in at least stoichiometrical amount was necessary to achieve a linear soluble polymer with complete conversion of amine to isocyanate groups. The synthetic approach could be applied to different types of polymers carrying either aliphatic or aromatic primary amine groups.The obtained functionalized polymers are soluble and highly reactive towards amines, alcohols and thiols that can be linked to the activated sites with quantitative conversions. This approach opens the door to the development of new systems that leverage the high reactivity of isocyanate groups and lead to the evolution of new, increasingly complex polymer materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from Grants MAT2013-42957-R, and RTI2018-096636-J-I00.References
- R. Hart and A. van Dormael, Bull. Soc. Chim. Belg., 1956, 65, 571–579 CrossRef CAS.
- R. C. Schulz and H. Hartmann, Makromol. Chem., 1962, 55, 227–229 CrossRef.
- C. G. Overberger, S. Ozaki and H. Mukamal, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 627 CrossRef.
- G. B. Butler and S. B. Monroe, J.Macromol.Sci., Chem., 1971, A5(6), 1063 CrossRef.
- Y. Iwakura, M. Sata, T. Tamikado and T. Mizoguchi, Kobunshi Kagaku, 1956, 13, 390–396 CrossRef CAS.
- G. Welzel and G. Greber, Makromol. Chem., 1959, 31, 230–232 CrossRef CAS.
- E.L. Kropa and S. A. Nyquist, U.S. Pat, 2606892, 1952 Search PubMed.
- N. Fujisaki and K. Shibata, Jap. Pat, 5145, 1958 Search PubMed.
- W. Mormann and K. Schmalz, Macromolecules, 1994, 27, 7115–7120 CrossRef CAS.
- G. B. Butler and S. B. Monroe, J.Macromol.Sci., Chem., 1971, A5(6), 1063 CrossRef.
- R. K. Graham, J. Polym. Sci., 1957, 24, 367–382 CrossRef CAS.
- B. Vollmert, Angew. Makromol. Chem., 1968, 3(1), 1–27 CrossRef.
- (a) A. Liebersohn and D. H. Kohn, J. Appl. Polym. Sci., 1976, 20(2), 411–420 CrossRef CAS; (b) A. Liebersohn and D. H. Kohn, J. Appl. Polym. Sci., 1976, 20, 1795–1802 CrossRef CAS; (c) A. Liebersohn and D. H. Kohn, J. Appl. Polym. Sci., 1979, 23, 3445–3448 CrossRef CAS.
- K. Kurita, T. Matsumura and Y. Iwakura, J. Org. Chem., 1976, 41(11), 2070–2071 CrossRef CAS.
- H. Eckert and B. Forster, Angew. Chem., Int. Ed. Engl., 1987, 26(9), 894–895 CrossRef.
- L. Cotarca and H. Eckert, Phosgenations –a Handbook, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003, ch. 2, ISBN: 3-527-29823-1 Search PubMed.
- H. W. I. Peerlings and E. W. Meijer, Tetrahedron Lett., 1999, 40(5), 1021–1024 CrossRef CAS.
- R. V. Versteegen, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 1999, 38, 2917–2019 CrossRef CAS PubMed.
- Houben-Weyl, Methoden der Organischen Chemie, Georg Thieme Verlag, 4th edn, 1982, vol. VIII, pp. 119–121 Search PubMed.
- H. Reinecke and C. Mijangos, Polymer, 1997, 38(9), 2291–2294 CrossRef CAS.
- Y. Zhang, Z. Ke, X. Zhu, H. Bi and K. Nie, Eur. Polym. J., 2002, 38(2), 333–337 CrossRef CAS.
- L. Pasquato, M. Modena, L. Cotarca, P. Delogu and S. Mantovani, J. Org. Chem., 2000, 65, 8224–8228 CrossRef CAS PubMed.
- L. Cotarca, T. Geller and J. Répási, Org. Process Res. Dev., 2017, 21(9), 1439–1446 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00989j |
| This journal is © The Royal Society of Chemistry 2020 |