
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceL- and rac-lactide polymerisation using scandium and aluminium permethylindenyl complexes†
Nichabhat
Diteepeng
 ,
Isobel A. P.
Wilson
,
Jean-Charles
Buffet
,
Zoë R.
Turner
and
Dermot
O'Hare
*
,
Isobel A. P.
Wilson
,
Jean-Charles
Buffet
,
Zoë R.
Turner
and
Dermot
O'Hare
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA Oxford, UK. E-mail: dermot.ohare@chem.ox.ac.uk
First published on 31st August 2020
Abstract
The synthesis and characterisation of constrained geometry scandium and aluminium permethylindenyl complexes Me2SB(RN,I*)ScCl(THF) (R = iPr (1), nBu (2) and Ph (3)), Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4), Me2SB(iPrN,I*)Sc(O-2,4-tBu-C6H3)(THF) (5), Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6), Me2SB(PhN,I*)Sc(O-2,6-iPr-C6H3)(THF) (7), Me2SB(tBuN,I*)AlCl(THF) (8), Me2SB(tBuN,I*)Al(O-2,6-Me-C6H3)(THF) (9) and Me2SB(tBuN,I*)Al(O-2,4-tBu-C6H3)(THF) (10) are reported. All complexes were characterised by NMR spectroscopy. Solid-state structures of 2–4, 6 and 8–10 were determined by X-ray crystallography. Ring-opening polymerisation of L- and rac-lactide using all complexes with the exception of 6 show first-order dependence on monomer concentration and produced polylactide with unimodal molecular weight distribution. First-order dependence on catalyst concentration was determined from L-lactide polymerisation using 4 and 9. Moderately heterotactic polylactide (Pr = 0.53–0.68) was achieved from rac-lactide polymerisation using 4, 5, 7 and 9. The effects of the metal centre (Sc and Al), the amido substituent (iPr, tBu, nBu and Ph) and the aryloxide initiating group (O-2,6-Me-C6H3, O-2,6-iPr-C6H3 and O-2,4-tBu-C6H3) on the catalytic activity are discussed.
Introduction
Polylactide (PLA) has diverse usage due to its biodegradability, biocompatibility and production from renewable feedstocks such as corn starch and sugar cane.1 Two stereogenic centres per lactide (LA) molecule result in L-LA (S,S-LA), D-LA (R,R-LA) and meso-LA (R,S-LA). A racemic mixture of L-LA and D-LA is referred to as rac-LA. Ring-opening polymerisation (ROP) of lactide initiated by single-site metal catalysts via a coordination–insertion mechanism can form well-controlled polymers in terms of molecular weight, molecular weight distribution and microstructure.2 Single-site initiators are based on Lewis acidic metal centre surrounded by ancillary ligand(s), and an initiating nucleophile which is commonly an alkoxide group.2aConstrained geometry complexes (CGCs) were originally developed in the academic literature by Bercaw et al. for scandium centres with a dicationic ligand and a dimethylsilyl ansa-bridge (SiMe2) linking a cyclopentadienyl ring and an amido ligand.3 Afterwards, Okuda et al. reported titanium and ferrocene CGCs containing a bridged amido-cyclopentadienyl {C5H4(tBu)−} ligand.4 Since then, several synthesis and applications of CGCs containing different substituted cyclopentadienyl, indenyl and fluorenyl groups, coordinating heteroatoms, ansa-linkages and metal centres were reported in the literature,5 particularly Group 4 CGCs for olefin polymerisations.6 The enhanced ability of Group 4 CGCs for ethylene polymerisation and (co)polymerisation of ethylene and α-olefins is ascribed to a smaller Cpcentroid–M–N bite angle than the typical Cpcentroid–M–Cpcentroid in metallocene systems and a reduced tendency to undergo chain transfer reactions, resulting in high molecular weight polymers.3a,7 More electron deficient metal centres (an amido moiety formerly donates two electrons less than a cyclopentadienyl-based ligand) also promote olefin insertion into the metal–carbon bond and increase reactivity.3a,7 Due to the higher thermal stability than related metallocenes, higher polymerisation temperatures are permitted by alkyl or dialkyl CGCs.8 The indenyl ligand (C9H7−, Ind, I) has been studied as an alternative to the cyclopentadienyl ligand (C5H5−, Cp).6h,8a,9 The indenyl ring slippage from η5 to η3-hapticity was observed when the formal number of metal electrons increased by two, resulting in a higher activity of ligand substitution reactions of electronically unsaturated complexes compared to their analogous Cp complexes.10 Permethylation of the indenyl ring has been proposed to increase steric congestion around the metal centre compared to the indenyl ligand, and afford kinetic stability to the metal–Ind* bond.11 Group 4 CGCs with variation of the amido moieties, ansa-bridges and permethylindenyl ligands have been developed by O'Hare and co-workers from Me2SB(tBuN,I*)TiCl2 (Chart 1a).12 These complexes are efficient for slurry-phase ethylene polymerisation and ethylene/1-hexene and ethylene/styrene (co)polymerisations. Another family of ansa-bridged permethylindenyl Group 4 metallocenes were used as catalysts for slurry-phase ethylene polymerisation and lactide polymerisation (Chart 1b).13 A bimodal molecular weight distribution was observed from poly(L-lactide) obtained from ROP of L-LA using an ansa-bridged permethylindenyl zirconium dichloride complex in the presence of benzyl alcohol.13b We recently reported constrained geometry scandium permethylindenyl aryloxide complexes, Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) and Me2SB(tBuN,I*)Sc(O-2,4-tBu-C6H3)(THF), as initiators for lactide polymerisation (Chart 1c).14 The single-site nature of scandium permethylindenyl CGCs leads to high molecular weight polylactide and unimodal molecular weight distribution (Mw/Mn < 1.2).
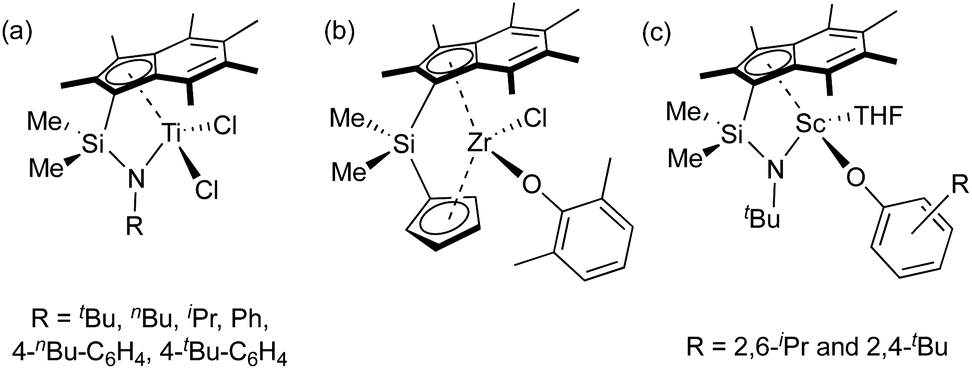 | ||
| Chart 1 Group 3 and 4 permethylindenyl complexes reported by O'Hare and co-workers.12–14 | ||
In this work, constrained geometry permethylindenyl complexes with variation of the metal centre (Sc and Al), an amido substituent (iPr, tBu, nBu and Ph) and an aryloxide initiating group (O-2,6-Me-C6H3, O-2,6-iPr-C6H3 and O-2,4-tBu-C6H3) were synthesised and studied as catalysts for polymerisation of L- and rac-lactide.
Results and discussion
Synthesis of constrained geometry scandium complexes
Reactions of Me2SB(RN,I*)Li2(THF)x (R = iPr,nBu and Ph) and ScCl3·3THF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio were carried out in benzene at room temperature (Scheme 1a). Me2SB(iPrN,I*)Sc(Cl)(THF) (1), Me2SB(nBuN,I*)Sc(Cl)(THF) (2) and Me2SB(PhN,I*)Sc(Cl)(THF) (3) were isolated as yellow solids in 35, 6 and 41% yield, respectively. A series of aryloxide complexes Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4), Me2SB(iPrN,I*)Sc(O-2,4-tBu-C6H3)(THF) (5), Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6) and Me2SB(PhN,I*)Sc(O-2,6-iPr-C6H3)(THF) (7) were synthesised from reactions between complexes 1–3 and appropriate potassium aryloxide salts (Scheme 1b). Complexes 4, 5 and 7 were isolated in 44, 34 and 41% yields, respectively. The 1H NMR spectra of 1–7 (see ESI†) show five singlets corresponding to the indenyl methyl protons at 1.50–3.00 ppm and two singlets corresponding to the silylmethyl groups between 0.50–1.20 ppm. Resonances of methylene protons of a THF molecule coordinated to the metal centre were also observed. The X-ray crystal structures of complexes 2–4 and 6 have been determined and are shown in Fig. 1. Selected bond lengths and angles are listed in Table 1.
:1 molar ratio were carried out in benzene at room temperature (Scheme 1a). Me2SB(iPrN,I*)Sc(Cl)(THF) (1), Me2SB(nBuN,I*)Sc(Cl)(THF) (2) and Me2SB(PhN,I*)Sc(Cl)(THF) (3) were isolated as yellow solids in 35, 6 and 41% yield, respectively. A series of aryloxide complexes Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4), Me2SB(iPrN,I*)Sc(O-2,4-tBu-C6H3)(THF) (5), Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6) and Me2SB(PhN,I*)Sc(O-2,6-iPr-C6H3)(THF) (7) were synthesised from reactions between complexes 1–3 and appropriate potassium aryloxide salts (Scheme 1b). Complexes 4, 5 and 7 were isolated in 44, 34 and 41% yields, respectively. The 1H NMR spectra of 1–7 (see ESI†) show five singlets corresponding to the indenyl methyl protons at 1.50–3.00 ppm and two singlets corresponding to the silylmethyl groups between 0.50–1.20 ppm. Resonances of methylene protons of a THF molecule coordinated to the metal centre were also observed. The X-ray crystal structures of complexes 2–4 and 6 have been determined and are shown in Fig. 1. Selected bond lengths and angles are listed in Table 1.
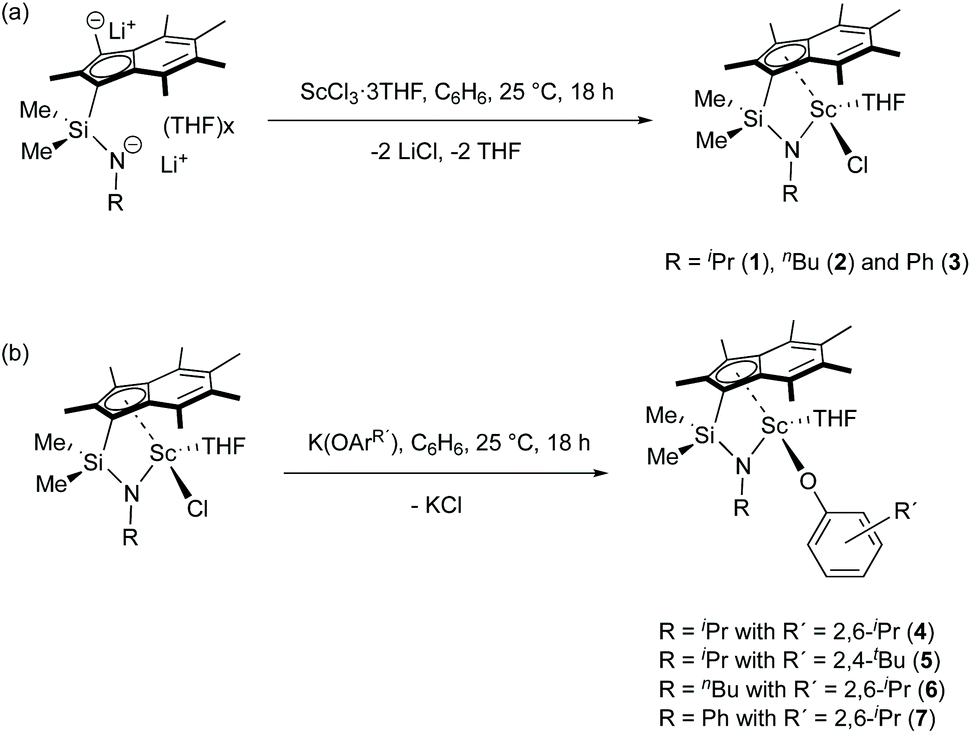 | ||
| Scheme 1 (a) Synthesis of Me2SB(RN,I*)ScCl(THF) (1–3) and (b) Me2SB(RN,I*)Sc(OArR′)(THF) (4–7). | ||
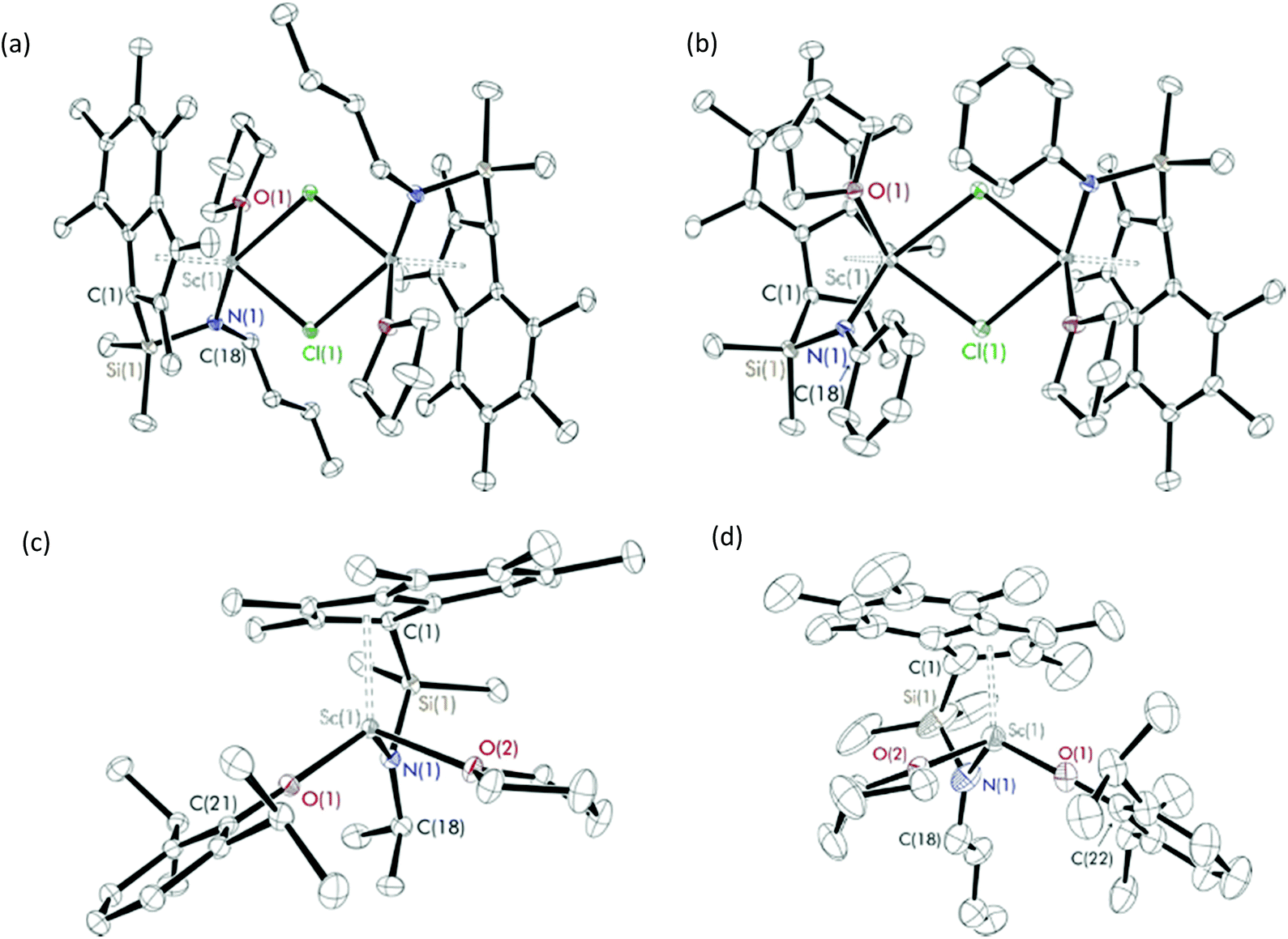 | ||
| Fig. 1 Solid-state structures of (a) Me2SB(nBuN,I*)Sc(Cl)(THF) (2), (b) Me2SB(PhN,I*)Sc(Cl)(THF) (3), (c) Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4) and (d) Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6). Ellipsoids are drawn at the 30% probability level and H atoms omitted for clarity. | ||
| Complex | 2 | 3 | 4 | 6 | Refa |
|---|---|---|---|---|---|
| a Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF).14 b Sc(1)–O(1)–C(21) for 4 and Sc(1)–O(1)–C(22) for 6. | |||||
| Sc(1)–I*cent | 2.1836(1) | 2.1845(1) | 2.1704(1) | 2.1735(1) | 2.1718(1) |
| Sc(1)–Cl(1) | 2.5700(5) | 2.5732(5) | — | — | — |
| Sc(1)–Cl(1′) | 2.6273(5) | 2.6055(5) | — | — | — |
| Sc(1)–O(1) | 2.2257(12) | 2.2174(12) | 1.9344(10) | 1.9298(1) | 1.9450(9) |
| Sc(1)–O(2) | — | — | 2.1705(10) | 2.1820(1) | 2.1686(9) |
| Sc(1)–N(1) | 2.0412(14) | 2.0909(14) | 2.0458(12) | 2.0265(1) | 2.0593(11) |
| I*cent–Sc(1)–N(1) | 102.77 | 102.50 | 103.82(1) | 103.63 | 103.99(1) |
| Sc(1)–Cl(1)–Sc(1) | 105.173(15) | 102.264(15) | — | — | — |
| Sc(1)–O(1)–COArb | — | — | 176.91(9) | 169.94(1) | 175.63(9) |
Single crystals suitable of X-ray diffraction studies of 2 and 3 were grown at room temperature of saturated benzene solution and pentane solution, respectively, and found to crystallise in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and C2/c. The solid-state structures of 2 and 3 are dimeric, consisting of two chloride-bridged scandium centres. Each scandium centre has a distorted square pyramidal geometry, evidenced by the τ5 values of 0.22 and 0.02 for 2 and 3,15 respectively, with η5-coordination with the C9Me6 ring. The oxygen of the THF ligand and nitrogen of the amido group also coordinate to the metal centre. The Sc(1)–I*cent bond length of 3 (2.1845(1) Å) is slightly longer than 2 (2.1836(1) Å) due to the increased steric bulk of the phenyl group on the amido ligand compared with the n-butyl group. The two C9Me6 rings on 2 have a trans arrangement while those on 3 have a cis arrangement. Therefore, the plane containing scandium and chlorine atoms of 2 is planar while that of 3 is puckered with an interplanar angle of 24.3° (Fig. S61†) in order to reduce steric repulsion between the C9Me6 rings.
and C2/c. The solid-state structures of 2 and 3 are dimeric, consisting of two chloride-bridged scandium centres. Each scandium centre has a distorted square pyramidal geometry, evidenced by the τ5 values of 0.22 and 0.02 for 2 and 3,15 respectively, with η5-coordination with the C9Me6 ring. The oxygen of the THF ligand and nitrogen of the amido group also coordinate to the metal centre. The Sc(1)–I*cent bond length of 3 (2.1845(1) Å) is slightly longer than 2 (2.1836(1) Å) due to the increased steric bulk of the phenyl group on the amido ligand compared with the n-butyl group. The two C9Me6 rings on 2 have a trans arrangement while those on 3 have a cis arrangement. Therefore, the plane containing scandium and chlorine atoms of 2 is planar while that of 3 is puckered with an interplanar angle of 24.3° (Fig. S61†) in order to reduce steric repulsion between the C9Me6 rings.
The average Sc–Cl bond lengths of 2 and 3 (2.5987 and 2.5894 Å) are comparable to those observed from reported complexes.16 Compared to 3, an analogous Cp-based scandium CGC [Me2SB(PhN,C5Me4)Sc(μ-Cl)(THF)]2 reported by Hou et al. has comparable Sc–Cl (2.545 Å), Sc–Cpcent (2.171 Å), Sc–N (2.142 Å) and Sc–O (2.224 Å) bond lengths.16e The crystal structure of [Cp2Sc(μ-Cl)]2 was reported with a Sc–Cl distance of 2.575 Å.16a Another THF-free complex [Sc(N2NC3,Me)Sc(μ-Cl)]2 where N2NC3,Me = MeN{(CH2)3NSiMe3}21a was reported with a Sc–Cl bond length of 2.5685 Å.16c [Sc(C8H8)(μ-Cl)(THF)]216g and [Sc(C8H6(1,4-SiMe3)2)(μ-Cl)]2(THF)16b were reported with Sc–Cl bond distances of 2.5972 and 2.5155 Å, respectively. A scandium chloride complex containing C5Me4SiMe2CH2Ch2Ph ligand has a tetrameric structure with the average Sc–Cl bond length of 2.5243 Å.16f The crystal structures of 2 and 3 also show similar Sc(1)–I*cent bond lengths to those of cyclopentadienyl based scandium chloride complexes (2.12–2.18 Å).16a,d,f In contrast to the dimeric structure of 2 and 3, their titanium analogues were described as monomeric with no THF ligand coordinated to the metal centre.12a The Ti–Cpcent (2.03 Å) and Ti–N (1.89–1.94 Å) bond lengths of Me2SB(RN,I*)TiCl2 (R = tBu, iPr and 4-tBu-C6H4) are smaller than those of 2 and 3.
Solid-state structures of Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4) and Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6) are monomeric with a distorted tetrahedral geometry at the scandium centre, indicated by the τ4 parameters of 0.89 and 0.86 for 4 and 6,17 respectively. The bond distances of Sc(1)–O(1) and Sc(1)–N(1) of 4 (1.9344(10) and 2.0458(12) Å) are slightly longer than those of 6 (1.9298(1) and 2.0265(1) Å). The bond lengths of Sc(1)–I*cent, Sc(1)–O(1), Sc(1)–O(2) and Sc(1)–N(1) of 4 and 6 are comparable to those previously reported from Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF)14 (Table 1). As a consequence of the less sterically demanding nBu group on the amido ligand of 6 compared to the iPr group on 4 or tBu group on Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF), the aryloxide group is more oriented towards the amido ligand on 6 than 4 or Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF). Hence, the Sc(1))–O(1))–COAr angle of 169.94(1) in 6 is considerably smaller than that of 176.91(9)° in 4 and 175.63(9)° in Me2SB(tBuN,I*)Sc(O-2,6-iPr-C6H3)(THF).
Synthesis of constrained geometry aluminium complexes
Me2SB(tBuN,I*)Al(Cl)(THF) (8) was prepared in 49% yield via the salt elimination reaction of Me2SB(tBuN,I*)Li2(THF)x and AlCl3·THF in benzene at room temperature (Scheme 2a). The aryloxide complexes, Me2SB(tBuN,I*)Al(O-2,6-Me-C6H3)(THF) (9) and Me2SB(tBuN,I*)Al(O-2,4-tBu-C6H3)(THF) (10), were synthesised by reactions of 8 and K(O-2,6-Me-C6H3) or K(O-2,4-tBu-C6H3) in benzene at room temperature (Scheme 2b), and were isolated in 41 and 24% yield, respectively. Reaction of 8 with K(O-2,6-iPr-C6H3) to form Me2SB(tBuN,I*)Al(O-2,6-iPr-C6H3)(THF) was carried out. However, several attempts to isolate clean product of Me2SB(tBuN,I*)Al(O-2,6-iPr-C6H3)(THF) were unsuccessful. The 1H NMR spectra of 8–10 (Fig. S18, S20 and S22†) show two sets of resonances corresponding to a mixture of two isomers.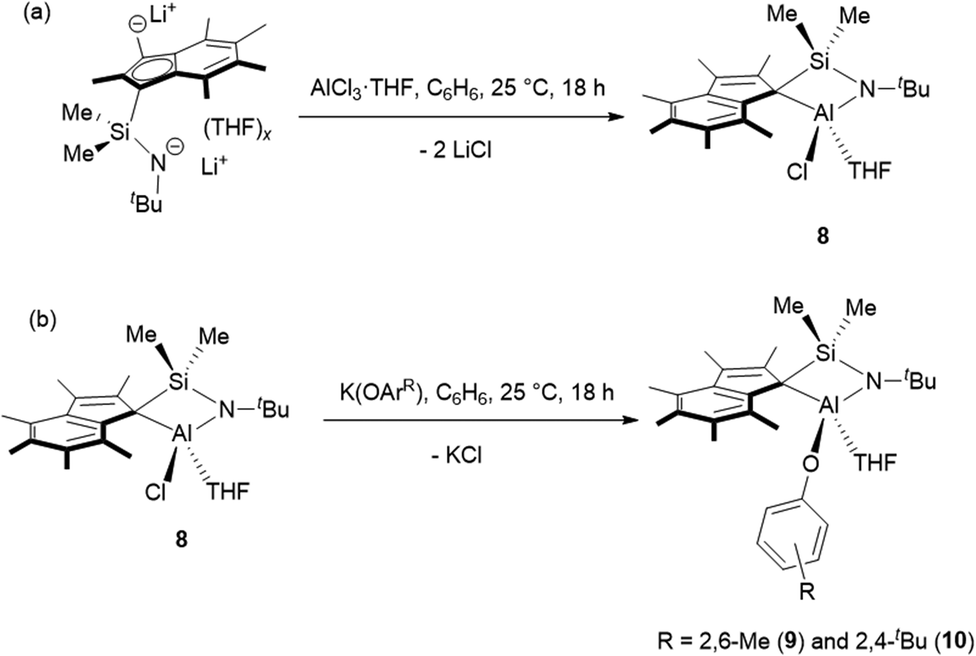 | ||
| Scheme 2 (a) Synthesis of Me2SB(tBuN,I*)Al(Cl)(THF) (8), (b) Me2SB(tBuN,I*)Al(O-2,6-Me-C6H3)(THF) (9) and Me2SB(tBuN,I*)Al(O-2,4-tBu-C6H3)(THF) (10). | ||
Diffraction-quality crystals were grown from a concentrated benzene solution of 8 and 9 and a pentane solution of 10 at room temperature. X-ray crystal structures of 8 and 9 (Fig. 2) were obtained for one isomer, while for complex 10 (Fig. 3), both isomers were obtained (Fig. S22†). In contrast to the scandium constrained geometry complexes (1–7), 8–10 display σ-instead of π-bonding interactions between the metal centre and C9Me6 ring due to the absence of accepting d-orbitals on the aluminium centre. The hapticity of one between the C9Me6 ring and the metal centre is consistent with the known Group 1318 and 1519 cyclopentadienyl constrained geometry complexes reported in literature. Cowley et al. synthesised and crystallographically characterised complexes Me2SB(tBuN,C5Me4)M(CH3)(THF) (M = Al and Ga).18b The C5Me4 ring possesses a localised diene structure, and the σ-attachment occurs at the metal centre at an α position with respect to the SiMe2 group affording a five-membered ring M–C–C–Si–N ring. The solid-state structures of 8–10 show σ-bonds between the aluminium centre and the carbon on the C9Me6 ring adjacent to SiMe2 group, and the nitrogen of the tBuN moiety linked between the metal centre and the SiMe2 group. The four-membered ring of Al–C–Si–N is perpendicular to the C9Me6 ring. Distorted tetrahedral geometry at the aluminium centre was observed, confirmed by the τ4 values of 0.81, 0.76 and 0.72 for 8, 9 and 10, respectively.17 The presence of the four-membered ring species was reported by Rieger et al. for the solid-state structure of aluminium constrained geometry cyclopentadienyl complex containing the lutidinyl moiety.18c
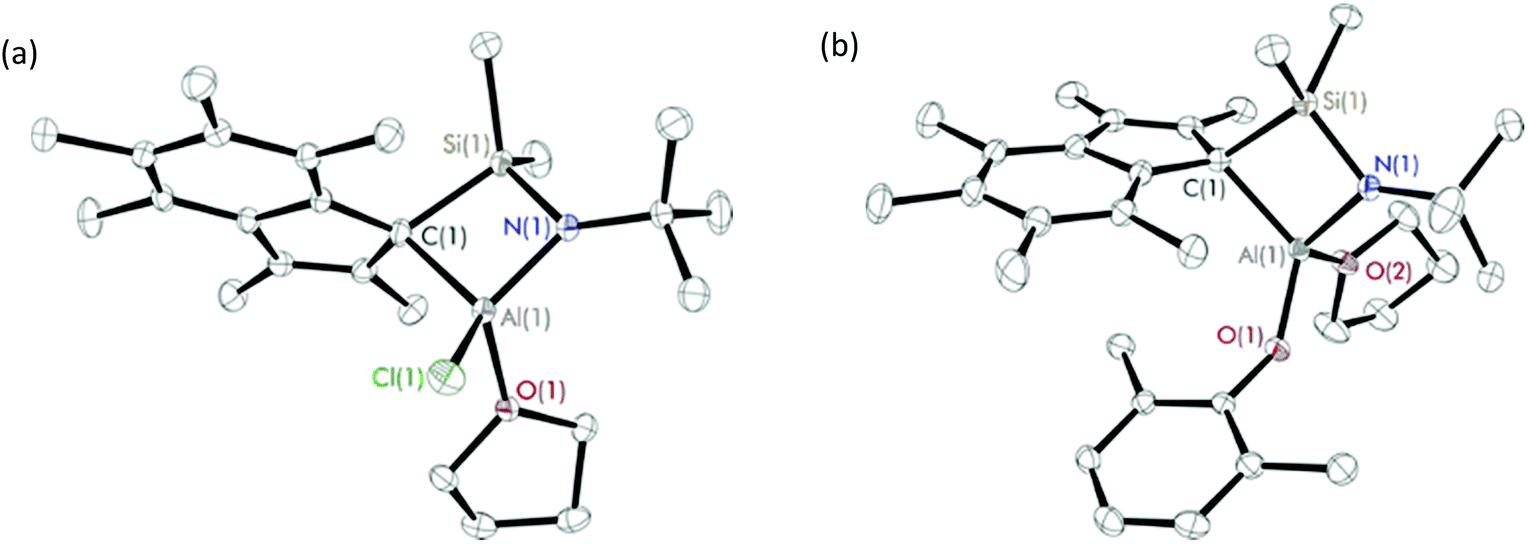 | ||
| Fig. 2 Solid-state structures of (a) Me2SB(tBuN,I*)Al(Cl)(THF) (8) and (b) Me2SB(tBuN,I*)Al(O-2,6-Me-C6H3)(THF) (9). Ellipsoids are drawn at the 30% probability level and H atoms omitted for clarity. | ||
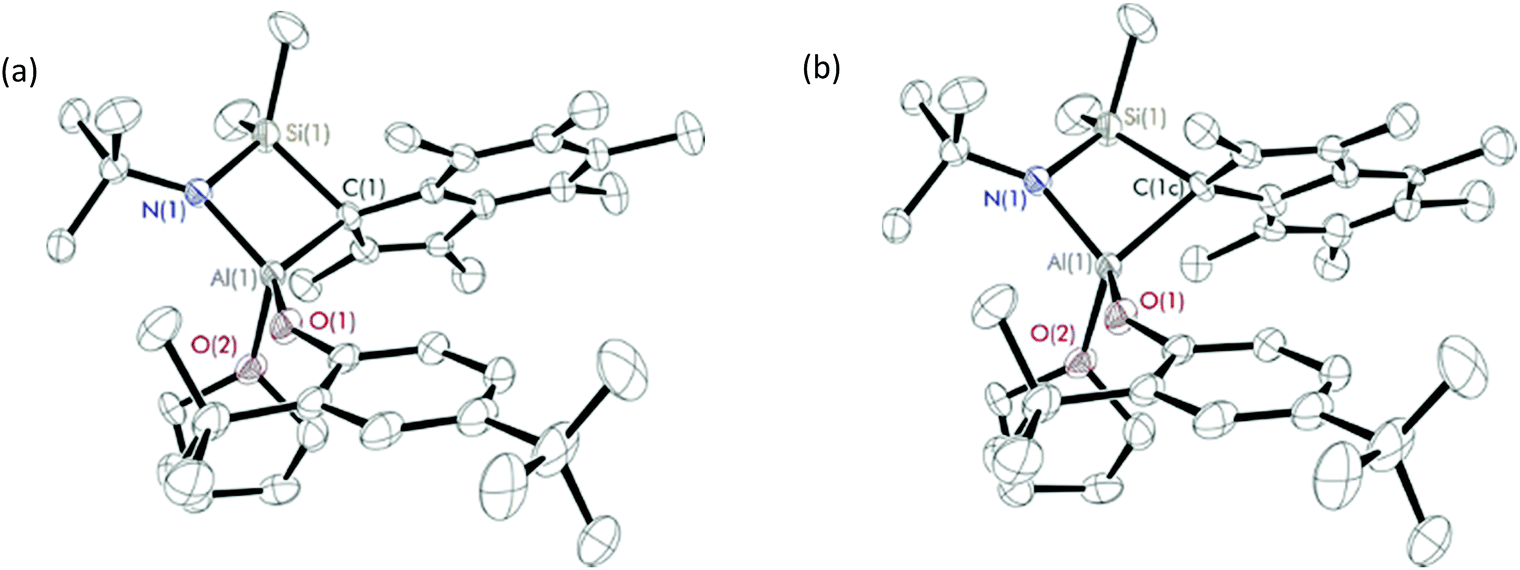 | ||
| Fig. 3 Solid-state structures of isomer 1 (a) and 2 (b) of Me2SB(tBuN,I*)Al(O-2,4-tBu-C6H3)(THF) (10). Ellipsoids are drawn at the 30% probability level and H atoms omitted for clarity. | ||
In contrast to the analogous dimeric scandium complexes (2 and 3), 8 was obtained as a monomer with smaller M(1)–Cl(1) bond length of 2.1375(7) Å than those in 2 and 3 (2.5700–2.5732 Å). A similar trend was observed for the aryloxide complexes 9 and 10 with the shorter M(1)–O(1), M(1)–O(2) and M(1)–N(1) distances comparing to those of 4 and 6. The C(1)–Al(1)–O(1) and Al(1)–O(1)–COAr angles of 9 (124.96(8) and 143.72(14)°) are considerably smaller than those of 10 (133.04(13) and 147.2(2)°) as a result of the less sterically-hindered aryloxide group on 9.
X-ray crystal structures of 10 show two components of the complex in which the C9Me6 ring featuring in two different positions (Fig. 3, Table S6†). The C9Me6 ring in isomer 1 and isomer 2 are labelled as C(1)–C(9) and C(1c)–C(9c), respectively. The direction of the six-membered ring on the C9Me6 ring of isomer 1 is at the front side of the five-membered ring, while that of isomer 2 is at the backside of the five-membered ring. Bond lengths and angles of the two isomers of 10 are shown in Table 2. The significant differences between the C(1)–Al(1)–O(1) and C(1)–Al(1)–O(2) angles in isomer 1 and those in isomer 2 were observed, and reflect a different conformation of the C9Me6 ring found in the solid state structures of 10. The ratio of these two components from the crystal structure of 10 (59:41) is consistent with those from the solution 1H NMR spectrum (55:45) (Fig. S22†). Two isomers found in the 1H NMR spectra of 8 and 9 (Fig. S18 and S20†) are also proposed to be attributed to the different C9Me6 ring position.
| Complex | 8 | 9 | 10 (Isomer 1) | 10 (Isomer 2) |
|---|---|---|---|---|
| Al(1)–Cl(1) | 2.1375(7) | — | — | — |
| Al(1)–O(1) | 1.8600(13) | 1.7150(15) | 1.712(2) | 1.712(2) |
| Al(1)–O(2) | — | 1.8917(15) | 1.880(2) | 1.880(2) |
| Al(1)–N(1) | 1.8047(15) | 1.8221(18) | 1.815(2) | 1.815(2) |
| Al(1)–C(1) | 2.0257(18) | 2.027(2) | 2.051(4) | 2.032(8) |
| C(1)–Al(1)–O(1) | 110.78(7) | 124.96(8) | 133.04(13) | 111.5(2) |
| C(1)–Al(1)–O(2) | — | 115.06(8) | 101.68(15) | 129.1(3) |
| C(1)–Al(1)–N(1) | 89.06(7) | 88.36(8) | 89.13(14) | 86.8(3) |
| Si(1)–C(1)–Al(1) | 82.08(7) | 82.67(8) | 81.13(16) | 80.4(3) |
| C(1)–Si(1)–N(1) | 94.17(7) | 94.34(8) | 94.87(14) | 90.0(2) |
| Al(1)–O(1)–COAr | — | 143.72(14) | 147.2(2) | 147.2(2) |
Polymerisation of L- and rac-lactide using scandium complexes
Me2SB(iPrN,I*)Sc(Cl)(THF) (1), Me2SB(nBuN,I*)Sc(Cl)(THF) (2) and Me2SB(PhN,I*)Sc(Cl)(THF) (3) were tested as initiators for the polymerisation of L-lactide in the presence of benzyl alcohol. In situ protonolysis is commonly used for lactide polymerisation catalysed by scandium alkyl,20 amide21 or chloride13b,14 complexes. It is hypothesised that benzyl alcohol reacts in situ with the chloride ligand of 1–3 to form the benzyloxide group which initiates the polymerisation via coordination–insertion mechanism. Under the same conditions, the polymerisation rate follows the order of 1 > 2 > 3 with kobs values of 1.21, 0.89 and 0.57 h−1, respectively (Table 3, entries 1–3). This indicates the effect of increasing nucleophilicity of the amido substituent on polymerisation activity (iPr > nBu > Ph). The introduction of the electron donating substituent on the amido ligand can increase the Lewis acidity of the metal centre, which is favourable for scandium–alkoxide bond cleavage. Kinetic measurements show a first-order dependence on L-lactide concentration (Fig. 4). No initiation period was observed with high monomer conversion reached within 2–4.5 h. Polymer molecular weights determined by GPC are in a fair agreement with those calculated for one chain per metal centre, and narrow Mw/Mn values (1.17) were observed. The presence of polylactide terminated with OCH2Ph end-group was observed in the 1H NMR spectrum (Fig. S51†). End-group analysis by MALDI-ToF mass spectrometry (Fig. S56†) also shows peaks corresponding to polylactide with OCH2Ph and OH end-groups and peak envelopes separated by Δ(m/z) of 72.0 Da indicating intermolecular transesterification.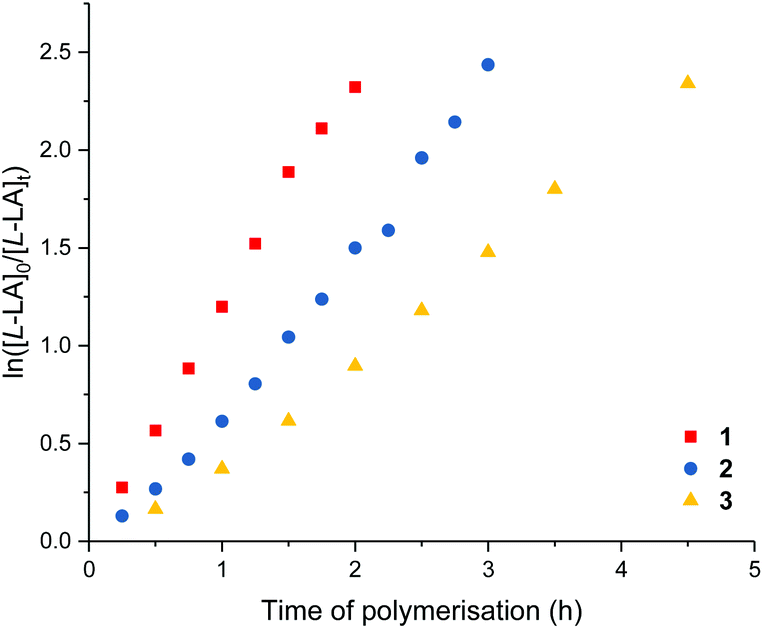 | ||
| Fig. 4 Plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation. Red squares: ROP of Llactide using 1, kobs = 1.21 ± 0.03 h−1, R2 = 0.995. Blue circles: ROP of L-lactide using 2, kobs = 0.89 ± 0.02 h−1, R2 = 0.994. Yellow triangles: ROP of L-lactide using 3, kobs = 0.57 ± 0.02 h−1, R2 = 0.992. Conditions: [L-LA]0:[Sc]0:[BnOH]0 = 400:1:1, [L-LA]0 = 0.5 M, 7.0 mL toluene at 70 °C. | ||
| Entry | Complex | LA | [LA]0:[Sc]0 |
T (°C) | t (h) | Conv.b (%) | k obs (h−1) | M n(GPC)c (g mol−1) | M n(Calcd)d (g mol−1) | M w/Mn | P r |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Conditions for ROP using 1–3: [LA]0:[Sc]0:[BnOH]0 = 400:1:1, [LA]0 = 0.5 M, 7.0 mL toluene and conditions for ROP using 4, 5 and 7: [LA]0:[Sc]0 as stated, [LA]0 = 0.5 M, 7.0 mL toluene.
b Measured by 1H NMR spectroscopic analyses.
c Determined by GPC in THF against PS standards using the appropriate Mark–Houwink corrections.22
d Calculated Mn for PLA synthesised by using 1–3 = conv.(%) × 400 × 144.1 + 108.1, calculated Mn for PLA synthesised with 4 and 7 = conv. (%) × [LA]0:[Sc]0 × 144.1 + 178.1 and calculated Mn for PLA synthesised by using 5 = conv. (%) × [LA]0:[Sc]0 × 144.1 + 206.2.
|
|||||||||||
| 1 | 1 | L- | 400:1 |
70 | 2 | 90 | 1.21 ± 0.03 | 42290 | 51984 |
1.17 | 0.00 |
| 2 | 2 | L- | 400:1 |
70 | 3 | 91 | 0.89 ± 0.02 | 44920 |
52560 |
1.17 | 0.00 |
| 3 | 3 | L- | 400:1 |
70 | 4.5 | 90 | 0.57 ± 0.02 | 39810 |
51984 |
1.16 | 0.00 |
| 4 | 4 | L- | 600:1 |
70 | 2.25 | 87 | 1.18 ± 0.05 | 69700 |
99319 |
1.16 | 0.00 |
| 5 | 4 | L- | 800:1 |
70 | 3 | 86 | 0.81 ± 0.01 | 103670 |
150618 |
1.14 | 0.00 |
| 6 | 4 | L- | 1000:1 |
70 | 3.5 | 85 | 0.66 ± 0.02 | 91500 |
122663 |
1.13 | 0.00 |
| 7 | 4 | L- | 1200:1 |
70 | 5 | 87 | 0.46 ± 0.01 | 103670 |
150618 |
1.14 | 0.00 |
| 8 | 4 | L- | 1000:1 |
60 | 8 | 91 | 0.34 ± 0.01 | 105980 |
130309 |
1.09 | 0.00 |
| 9 | 4 | L- | 1000:1 |
80 | 2.5 | 92 | 1.35 ± 0.04 | 85090 |
132750 |
1.18 | 0.00 |
| 10 | 4 | L- | 1000:1 |
100 | 1.25 | 90 | 2.68 ± 0.09 | 75280 |
129868 |
1.19 | 0.00 |
| 11 | 4 | rac- | 1000:1 |
70 | 2.5 | 93 | 1.27 ± 0.04 | 83100 |
134191 |
1.21 | 0.59 |
| 12 | 5 | L- | 1000:1 |
70 | 0.5 | 91 | 6.32 ± 0.37 | 77560 |
131337 |
1.18 | 0.00 |
| 13 | 5 | rac- | 1000:1 |
70 | 0.5 | 91 | 7.40 ± 0.50 | 64540 |
131337 |
1.23 | 0.68 |
| 14 | 7 | L- | 1000:1 |
70 | 4 | 84 | 0.48 ± 0.01 | 69570 |
121222 |
1.15 | 0.00 |
| 15 | 7 | rac- | 1000:1 |
70 | 3.5 | 86 | 0.58 ± 0.01 | 64820 |
124104 |
1.17 | 0.63 |
Complexes 1–3 exhibit superior performance compared to reported scandium monoamide or monoalkyl complexes.20c,21a Carpentier et al. reported scandium alkyl complex supported by phenoxy-aminopyridinate ligand for polymerisation of rac-LA with iPrOH as co-initiator ([rac-LA]0:[Sc]0:[iPrOH]0 = 500:1:1).20c Only 4% conversion was achieved after 1.5 h in toluene at 60 °C. Okuda et al. used bis(phenolato)scandium amide complex with iPrOH to polymerise 83% of 300 equivalents of rac-lactide after 72 h.21a
Me2SB(iPrN,I*)Sc(O-2,6-iPr-C6H3)(THF) (4), Me2SB(iPrN,I*)Sc(O-2,4-tBu-C6H3)(THF) (5) and Me2SB(PhN,I*)Sc(O-2,6-iPr-C6H3)(THF) (7) were used as initiators for ring-opening polymerisation of L- and rac-lactide (Table 3, entries 4–15). First-order dependence on monomer concentration was observed in all cases, evidenced by linear plots of ln([L-LA]0/[L-LA]t) vs. time (see ESI†). Under the same conditions, complexes 4 and 5 exhibited greater polymerisation rate than 7 suggesting the effect of the electron donating ability of amido substituent on polymerisation activity (iPr > Ph). Despite bearing the same amido substituent (iPrN), polymerisations using 5 are significantly greater than those using 4 which could be attributed to the 2,4-substitution pattern of the aryloxide ligand of 5, relative to the 2,6-substitution of 4, resulting in reduced steric crowding around the metal centre and an increased rate of lactide insertion into the metal–aryloxide bond in the initiation step.23 For 4, 5 and 7, the polymerisation rate of rac-lactide is faster than those of L-lactide, suggesting a preference for racemic linkages. The polymer tacticity measured by homonuclear decoupled 1H{1H} NMR spectroscopy showed that 4, 5 and 7 produced slightly heterotactic polylactide with Pr values of 0.59–0.68, suggesting the initiators favour racemic enchainment with chain-end control, where the next monomer to insert has an opposing stereocentre from the last monomer. This suggests that the substituent on the amido group has marginal influence on the stereoselectivity. Isotactic pure poly(L-lactide) was formed without epimerisation during polymerisation of L-lactide with 4, 5 and 7, confirmed by a single resonance in the methine region of the 1H{1H} NMR spectra (see ESI†). Catalytic studies of Me2SB(nBuN,I*)Sc(O-2,6-iPr-C6H3)(THF) (6) were not performed as adequate quantities could not be obtained in suitable yield.
Detailed kinetic studies were performed using 4. Polymerisation of L-lactide with 4 using different catalyst loading was carried out at 70 °C in toluene. Concentration of L-lactide was maintained at 0.5 M while that of 4 was varied giving the monomer to catalyst ratio of 600, 800, 1000 and 1200. The polymerisation data are summarised in Table 3 (entries 4–7). First-order dependence on L-lactide was observed from all conditions evidenced by linear plots of ln([L-LA]0/[L-LA]t) vs. time with an induction period of 0.5 h (Fig. 5). The gradient of 0.89 is indicative of first-order dependence on the concentration of 4 (Fig. 6). The propagation rate constant (kp) of 1120 ± 29 M−1 h−1 was calculated from plot of kobsvs. [4]0 (Fig. 7). The overall rate law was determined as −d[L-LA]/dt = kp[L-LA][4].
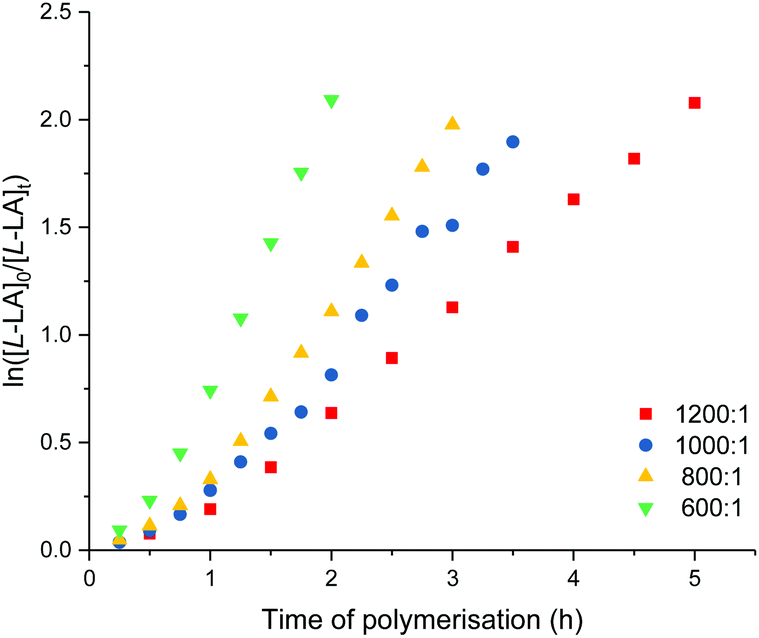 | ||
| Fig. 5 Plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation. ROP of L-lactide using 4. [L-LA]0/[Sc]0 = 1200, red square: kobs = 0.46 ± 0.01 h−1, R2 = 0.996. [L-LA]0/[Sc]0 = 1000, blue circle: kobs = 0.66 ± 0.02 h−1, R2 = 0.989. [L-LA]0/[Sc]0 = 800, yellow triangle: kobs = 0.81 ± 0.01 h−1, R2 = 0.996. [L-LA]0/[Sc]0 = 600, green down triangle: kobs = 1.18 ± 0.05 h−1, R2 = 0.985. Conditions: [L-LA]0 = 0.5 M, 7.0 mL toluene at 70 °C. | ||
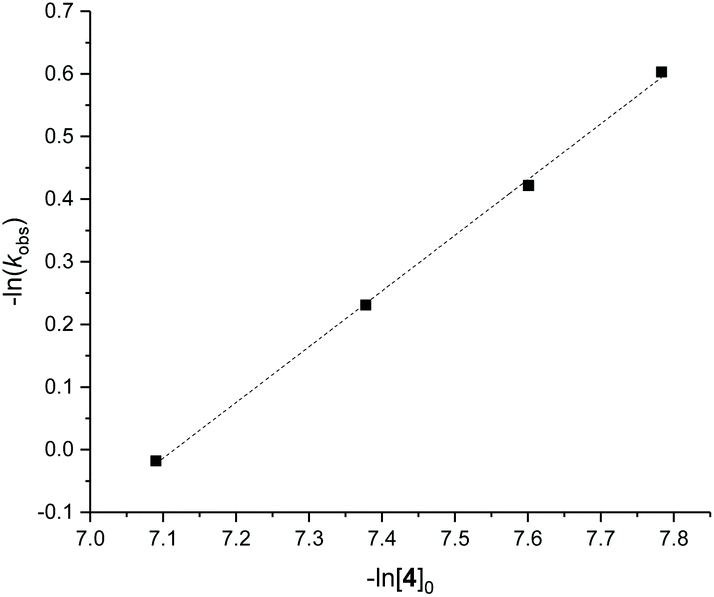 | ||
| Fig. 6 Plot of −ln(kobs) vs. −ln([4]0) for ROP of L-LA using 4 shows that the order of reaction with respect to [4]0 is equal to 0.89 ± 0.02. R2 = 0.999. Conditions: [L-LA]0 = 0.5 M, 7.0 mL toluene at 70 °C. | ||
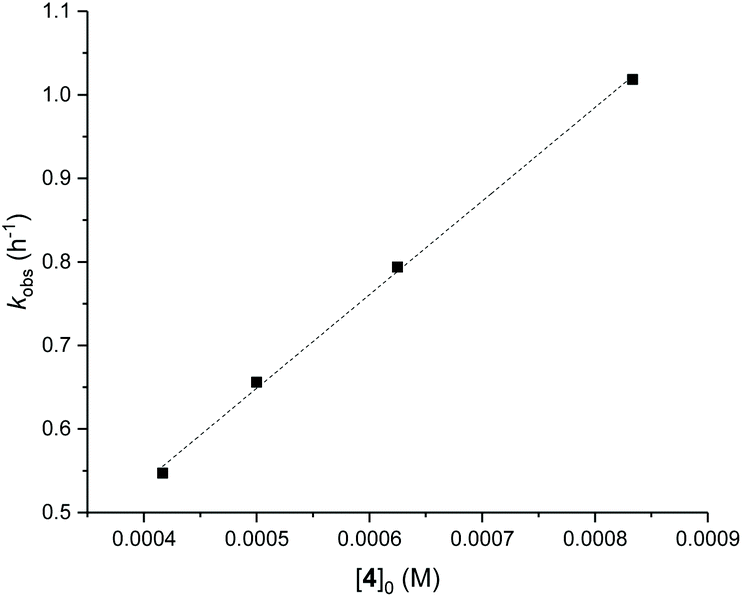 | ||
| Fig. 7 Plot of kobsvs. [4]0 for ROP of L-LA using 4, kp = 1120 ± 29 M−1 h−1. R2 = 0.998. Conditions: [L-LA]0 = 0.5 M, 7.0 mL toluene at 70 °C. | ||
The 1H NMR spectra of oligomers synthesised by 4 and 5 (Fig. S52 and S53†) show signals corresponding to O-2,6-iPr-C6H3 and O-2,4-tBu-C6H3 end-groups, suggesting that the ROP of L-lactide proceeds via a coordination–insertion mechanism (Scheme S1†). The presence of polylactide with iPrNH and OH end-groups was observed from MALDI-ToF mass spectra (Fig. S57 and S58†), suggesting the role of the amido ligand as an initiator. Peaks corresponding to cyclic polylactide and a repeating unit of Δ(m/z) = 72.0 Da between peak envelopes were also observed, indicating an occurrence of intra- and intermolecular transesterification reactions, respectively. Therefore, the considerably lower than calculated Mn(GPC) values could be attributed to double-site initiator from the amido and aryloxide ligands.
Complexes 4, 5 and 7 show better activity for L-lactide polymerisation with high monomer loading (600–1200) compared to other metallocene catalysts in the literature.13b,24 Zirconocene bis(ester enolate) complex (Ph2C(Cp,Flu)Zr[OC(OiPr)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe2]2) reported by Chen et al. polymerised 200 equivalents of L-lactide (toluene, 80 °C) up to 85% within 5 h.24a Other zirconocene complexes (rac-C2H4(Ind)2Zr[OC(OiPr)CMe2]2 and Cp2Zr[OC(OiPr)CMe2]2) from the same research group were reported to be poorly active under the same conditions.24a A series of Cp and Ind-based group 4 complexes reported by O'Hare et al. were found to be active in L-lactide polymerisation.24b (Ind)2ZrMe(OtBu) was the fastest catalyst with the kobs values of 0.317 and 0.293 h−1 for polymerisation of L- and rac-lactide (50 equivalents) at 100 °C in chloroform-d1, respectively. Me2SB(Cp,I*)ZrCl(O-2,6-Me-C6H3) presented a second-order dependence on L-lactide concentration (kobs = 3.23 M−1 h−1) for the polymerisation with [L-LA]0:[Zr]0:[BnOH]0 ratio of 50:1:2 in chloroform-d at 80 °C.13b Okuda et al. reported the yttrocene complex Li[(Me2Si(Cp,NC2H4OMe))2Y].24cL-Lactide polymerisation in toluene at 75 °C with [L-LA]0:[Y]0 ratio of 127 gave polymer after 2 h with Mn value double that expected and Mw/Mn of 1.44. Cui et al. reported rac-lactide polymerisation using scandium aryloxide complex supported by a pentadentate (N2O3) salen-type ligand in THF at room temperature (71% conversion, 2 h).25 Scandium alkoxide complexes containing a phosphasalen ligand were found to be inactive for rac-lactide polymerisation attributed to the formation of an unreactive single-lactide insertion product.26
CMe2]2) reported by Chen et al. polymerised 200 equivalents of L-lactide (toluene, 80 °C) up to 85% within 5 h.24a Other zirconocene complexes (rac-C2H4(Ind)2Zr[OC(OiPr)CMe2]2 and Cp2Zr[OC(OiPr)CMe2]2) from the same research group were reported to be poorly active under the same conditions.24a A series of Cp and Ind-based group 4 complexes reported by O'Hare et al. were found to be active in L-lactide polymerisation.24b (Ind)2ZrMe(OtBu) was the fastest catalyst with the kobs values of 0.317 and 0.293 h−1 for polymerisation of L- and rac-lactide (50 equivalents) at 100 °C in chloroform-d1, respectively. Me2SB(Cp,I*)ZrCl(O-2,6-Me-C6H3) presented a second-order dependence on L-lactide concentration (kobs = 3.23 M−1 h−1) for the polymerisation with [L-LA]0:[Zr]0:[BnOH]0 ratio of 50:1:2 in chloroform-d at 80 °C.13b Okuda et al. reported the yttrocene complex Li[(Me2Si(Cp,NC2H4OMe))2Y].24cL-Lactide polymerisation in toluene at 75 °C with [L-LA]0:[Y]0 ratio of 127 gave polymer after 2 h with Mn value double that expected and Mw/Mn of 1.44. Cui et al. reported rac-lactide polymerisation using scandium aryloxide complex supported by a pentadentate (N2O3) salen-type ligand in THF at room temperature (71% conversion, 2 h).25 Scandium alkoxide complexes containing a phosphasalen ligand were found to be inactive for rac-lactide polymerisation attributed to the formation of an unreactive single-lactide insertion product.26
The effect of temperature on L-lactide polymerisation activity using 4 was studied with polymerisation temperature varied from 60–100 °C (Table 3, and Fig. 8). The enthalpy of activation (ΔH‡) of 53 kJ mol−1 and entropy of activation (ΔS‡) of −95 J mol−1 K−1 were calculated from an Eyring plot of ln(kobs/T) vs. 1/T (Fig. S76†). These values are comparable to those reported, and suggest the ordered transition state in a coordination–insertion mechanism.14,27 As expected, the polymerisation activity increased at higher temperatures. Mw/Mn values and discrepancy between Mn(GPC) values and those calculated were observed to increase with increased temperature, attributed to transesterification reactions.
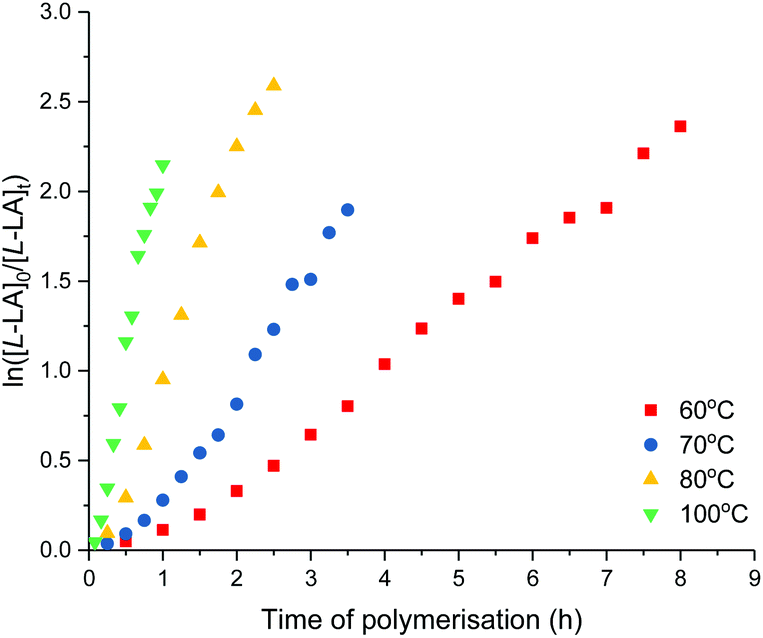 | ||
| Fig. 8 Plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation. ROP of Llactide using 4. 60 °C, red square: kobs = 0.34 ± 0.01 h−1, R2 = 0.995. 70 °C, blue circle: kobs = 0.66 ± 0.02 h−1, R2 = 0.989. 80 °C, yellow triangle: kobs = 1.35 ± 0.04 h−1, R2 = 0.996. 100 °C, green down triangle: kobs = 2.68 ± 0.09 h−1, R2 = 0.989. Conditions: [L-LA]0 = 0.5 M, [L-LA]0:[4]0 = 1000, 7.0 mL toluene. | ||
Polymerisation of L- and rac-lactide using aluminium complexes
Me2SB(tBuN,I*)Al(Cl)(THF) (8) was found to be less active than the analogous scandium chloride complexes (1–3) for L-lactide polymerisation in the presence of benzyl alcohol even at higher polymerisation temperature (100 °C) and lower ratio of [L-LA]0:[Al]0:[BnOH]0 (100:1:1). L-Lactide conversion reached 55% after 7.5 h which first-order dependence on L-lactide concentration was observed (kobs = 0.11 h−1, Fig. S84†). The polymerisations were quenched after 23 h with 80% conversion. The experimental Mn value (17 600 g mol−1) is higher than that calculated for one chain per metal centre (11636 g mol−1) with moderate Mw/Mn value of 1.37. OCH2Ph terminated polylactide was observed from the 1H NMR (Fig. S54†) and MALDI-ToF mass spectra (Fig. S59†).
Me2SB(tBuN,I*)Al(O-2,6-Me-C6H3)(THF) (9) and Me2SB(tBuN,I*)Al(O-2,4-tBu-C6H3)(THF) (10) show comparable activity for polymerisation of L-lactide at 100 °C in toluene with more than 80% conversion reached after 9 h (Table 4, entries 3 and 10). Kinetic studies show the first-order dependency on L-lactide concentration, supported by linear plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation (Fig. S85 and S100†) with kobs values of 0.24 and 0.19 h−1 for the polymerisation with 9 and 10, respectively. Isotactic poly(L-lactide) was produced with an absence of epimerisation occurring during polymerisation as evidenced by a singlet in the methine region of the homonuclear decoupled 1H{1H} NMR spectrum (Fig. S42 and S50†).Polymerisation of L-lactide using 9 were also carried out at 70, 80 and 90 °C with [L-LA]0:[Al]0 = 100:1 (Table 4 and Fig. 9). The enthalpy of activation (ΔH‡) of 71 kJ mol−1 and entropy of activation (ΔS‡) of −69 J mol−1 K−1 were calculated from an Eyring plot of ln(kobs/T) vs. 1/T (Fig. S90†). Rate of L-lactide polymerisation using 9 at 70 °C is comparable to those using hemi-salen aluminium alkyl complexes with iPrOH (kobs = 0.04–0.06 h−1) under the same conditions reported by Pang et al.28
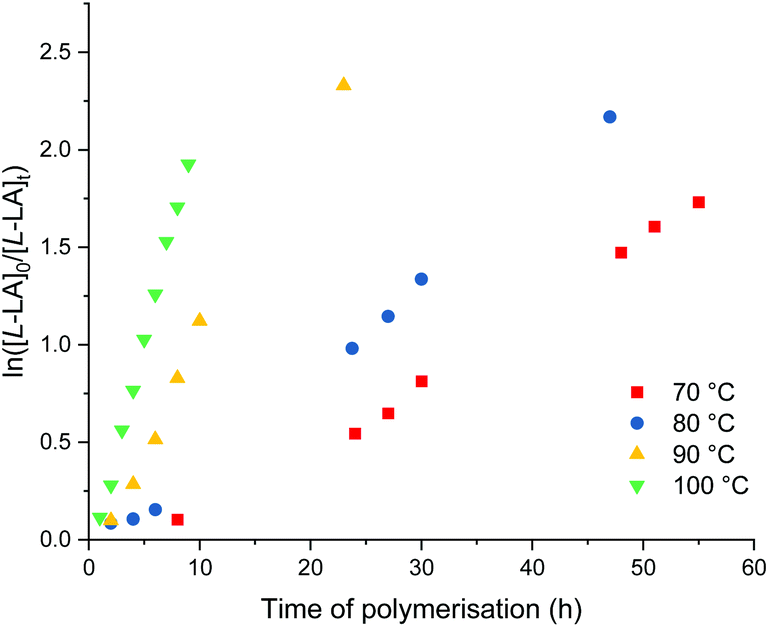 | ||
| Fig. 9 Plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation. ROP of L-lactide using 9. 70 °C, Red square: kobs = 0.03 ± 0.01 h−1, R2 = 0.993. 80 °C, Blue circle: kobs = 0.04 ± 0.01 h−1, R2 = 0.993. 90 °C, Yellow triangle: kobs = 0.11 ± 0.01 h−1, R2 = 0.989. 100 °C, green down triangle: kobs = 0.24 ± 0.01 h−1, R2 = 0.995. Conditions: [L-LA]0 = 0.5 M, [L-LA]0:[9]0 = 100:1, 4.0 mL toluene. | ||
| Entry | complex | LA | [LA]0:[Al]0 |
T (°C) | t (h) | Conv.b (%) | k obs (h−1) | M n(GPC)c (g mol−1) | M n(calcd)d (g mol−1) | M w/Mn |
|---|---|---|---|---|---|---|---|---|---|---|
| a Conditions: [LA]0 = 0.5 M, 4.0 mL toluene. b Measured by 1H NMR spectroscopic analyses. c Determined by GPC in THF against PS standards using the appropriate Mark–Houwink corrections.22 d Calculated Mn for PLA synthesised by using 9 = conv. (%) × 100 × 144.1 + 122.2 and calculated Mn for PLA synthesised by using 10 = conv. (%) × 100 × 144.1 + 206.2. | ||||||||||
| 1 | 9 | L- | 100 | 70 | 55 | 82 | 0.04 ± 0.01 | 11600 |
11938 |
1.44 |
| 2 | 9 | L- | 100 | 80 | 47 | 89 | 0.05 ± 0.01 | 15290 |
12947 |
1.41 |
| 3 | 9 | L- | 100 | 90 | 23 | 90 | 0.11 ± 0.01 | 14520 |
13091 |
1.37 |
| 4 | 9 | L- | 100 | 100 | 9 | 86 | 0.24 ± 0.01 | 12420 |
12515 |
1.35 |
| 5 | 9 | L- | 200 | 100 | 10 | 81 | 0.20 ± 0.01 | 21230 |
23466 |
1.27 |
| 6 | 9 | L- | 300 | 100 | 11 | 79 | 0.17 ± 0.01 | 25310 |
34274 |
1.33 |
| 7 | 9 | L- | 500 | 100 | 24 | 85 | 0.08 ± 0.01 | 46040 |
61364 |
1.33 |
| 8 | 9 | L- | 700 | 100 | 24 | 77 | 0.06 ± 0.01 | 53110 |
77792 |
1.31 |
| 9 | 9 | L- | 1000 | 100 | 27 | 76 | 0.05 ± 0.01 | 74390 |
109638 |
1.16 |
| 10 | 9 | rac- | 100 | 100 | 8 | 87 | 0.30 ± 0.01 | 12070 |
12659 |
1.29 |
| 11 | 10 | L- | 100 | 100 | 10 | 82 | 0.19 ± 0.01 | 13310 |
12022 |
1.37 |
The polymerisation of rac-lactide using 9 at 100 °C shows the first-order dependence on rac-lactide concentration (Fig. S99†) with a similar rate to L-lactide (kobs = 0.30 and 0.24 h−1 for rac- and L-lactide, respectively). The polymer tacticity studied by 1H{1H} NMR spectroscopy showed slight heterotactic polylactide with Pr values of 0.53–0.57 (Fig. S48 and S49†), suggesting chain-end controlled rac-lactide polymerisation using 9 where the stereocentre in the last unit on the propagating chain favours the racemic-enchainment. Although isoselectivity in rac-lactide polymerisation has been generally obtained from using aluminium catalysts,29 some known aluminium complexes were reported to produce heterotactic polylactides.29i,30 Gibson et al. prepared aluminium methyl complex supported by tetradentate phenoxy-amine ligand.30b Heterotactic polylactide (Pr = 0.57) was produced after 280 h with [rac-LA]0:[Al]0 = 50 in toluene at 70 °C. Aluminium methyl complexes supported by asymmetric [ONNO′]-type Salan ligand reported by Hormnirun et al. polymerised 100 equivalents of rac-LA with benzyl alcohol in toluene at 70 °C (more than 80% conversion after 300 h).30e Heterotactic polylactides were formed with Pr values of 0.64–0.74.
All polymerisations produced polymers with monomodal molecular weight distribution and moderate Mw/Mn values (1.29–1.44, Table 4). Experimental Mn values are also consistent with those calculated for one chain per metal centre, suggesting a well-controlled and living manner of polymerisation can be attained under harsh experimental conditions including high temperature and long polymerisation time. The polymerisation of L-lactide with 9 using various monomer to catalyst ratios was carried out at 100 °C in toluene to determine the kinetic order dependence on catalyst concentrations. The concentration of L-lactide remains at 0.5 M, while the concentration of 9 was varied, providing the ratio of [L-LA]0:[9]0 = 200, 300, 500, 700 and 1000. The polymerisation data are summarised in Table 4 (entries 5–9). Plots of first-order dependence on L-lactide concentration are shown in Fig. 10. The gradient of 0.79 from the plot of −ln(kobs) vs. −ln[9]0 is indicative of the first-order dependence on catalyst concentration (Fig. S97†).
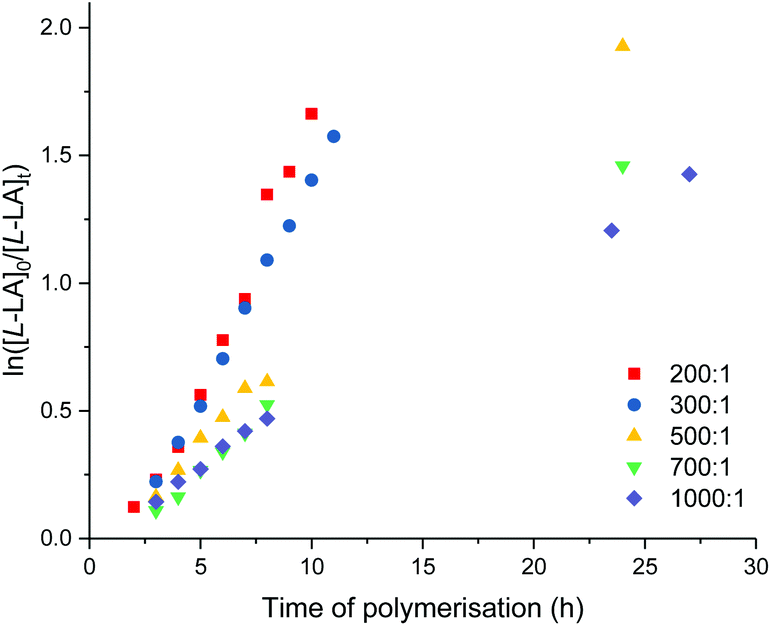 | ||
| Fig. 10 Plots of ln([L-LA]0/[L-LA]t) vs. time of polymerisation. ROP of L-lactide using 9. [L-LA]0/[9]0 = 200, red square: kobs = 0.20 ± 0.01 h−1, R2 = 0.980. [L-LA]0/[9]0 = 300, blue circle: kobs = 0.17 ± 0.01 h−1, R2 = 0.998. [L-LA]0/[9]0 = 500, yellow triangle: kobs = 0.08 ± 0.01 h−1, R2 = 0.997. [L-LA]0/[9]0 = 700, green down triangle: kobs = 0.06 ± 0.01 h−1, R2 = 0.993. [L-LA]0/[9]0 = 1000, purple diamond: kobs = 0.05 ± 0.01 h−1, R2 = 0.996. Conditions: [L-LA]0 = 0.5 M, 4.0 mL toluene at 100 °C. | ||
The propagation rate constant (kp) of 70 ± 11 M−1 h−1 was calculated from the plot between kobsvs. [9]0 (Fig. S98†). The overall rate law was determined as −d[L-LA]/dt = kp[L-LA][9]. At a [L-LA]0:[Al]0 ratio of 200 and 300, Mn(GPC) values are similar to those calculated. However, polylactide obtained from high monomer loading ([L-LA]0:[Al]0 = 500, 700 and 1000) show molecular weights lower than those predicted with moderate Mw/Mn values (1.16–1.33). The MALDI-ToF mass spectrum of polymer synthesised by 9 (Fig. S60†) shows peaks corresponding to polylactide with O-2,6-Me-C6H3 and OH end-groups. Other peaks are assigned to polylactide terminated with tBuNH and OH end-groups. Double-site initiator from the amido and aryloxide ligands, which was previously observed from ROP initiated by 4, results in the mismatch between the Mn(GPC) values and those calculated for one polymer chain per metal centre.
Conclusions
A series of new scandium (1–7) and aluminium (8–10) constrained geometry permethylindenyl complexes were reported. Scandium complexes (1–5 and 7) are highly active catalysts for lactide polymerisation whereas aluminium complexes (8–10) show moderate activity. First-order dependence on lactide concentration was observed in all polymerisations. First-order dependence on catalyst concentration was measured from polymerisation of L-lactide using 4 and 9 with kp values of 1120 ± 29 and 70 ± 11 M−1 h−1, respectively. Polymers with Mn(GPC) values lower than those calculated for one polymer chain per metal centre were observed, attributed to the double-site nature of scandium and aluminium complexes using these amido and aryloxide ligands.Complexes with a more electron donating substituent on the amido ligand (iPr > nBu > Ph) show greater polymerisation activity as observed from L-lactide polymerisation using 1–3 with one equivalent of benzyl alcohol and L-and rac-lactide polymerisation using 4, 5 and 7. The effect of the less sterically demanding aryloxide substituent was observed with the scandium system as 5 (O-2,4–tBu-C6H3) shows higher activity than 4 (O-2,6-iPr-C6H3). Complexes 1–5 and 7–10 produced isotactic poly(L-lactide) without epimerisation occurring during polymerisation. Moderate heterotactically enriched polylactide (Pr = 0.53–0.68) was obtained from polymerisation of rac-lactide using 4, 5, 7 and 9, suggesting minor influence of the metal centre, the amido substituent and the aryloxide group on the stereoselectivity.
Experimental section
General polymerisation procedure
To a stock solution of 1–3 and 8 (31.25 μmol) in toluene (5.00 mL), benzyl alcohol (31.25 μmol) was added. L-Lactide (2.50 mmol) was added into an ampoule and dissolved in 4.0 mL of toluene. The catalyst stock solution (1.0 mL) was added to the solution of lactide in the ampoule, corresponding to an initial lactide concentration of 0.5 M and a [L-LA]0:[Sc]0:[BnOH]0 ratio of 400:1:1. The polymerisation ampoule was then stirred at in the preheated oil bath at desired temperature.
A stock solution of 4, 5, 7, 9, 10 (17.50 μmol) in benzene (2.50 mL) was prepared. The stock solution of catalyst (3.50 μmol, 0.50 mL) was added into a toluene solution of lactide (0.50 g, 3.50 mmol, 6.50 mL) in the ampoule, corresponding to an initial lactide concentration of 0.5 M and a monomer-to-catalyst ratio of 1000:1. The polymerisation ampoule was then stirred at in the preheated oil bath at desired temperature.
Aliquots (ca. 0.1 mL) were taken at appropriate time intervals and quenched with THF (ca. 0.3 mL). The volatiles were evaporated to give PLA. The monomer to polymer% conversion was determined using 1H NMR spectroscopy and measured by integration of the CHMe resonances of the unreacted monomer and PLA. After the chosen time, the reaction was quenched with THF. The polymer was isolated by addition of pentane to a concentrated solution of PLA to yield a precipitate which was washed with pentane and dried under vacuum at 30 °C.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. D., J.-C. B. and Z. R. T. would like to thank SCG Chemicals Co., Ltd (Thailand) for financial support and for a SCG Research Fellowship (Z. R. T.). Chemical Crystallography (University of Oxford) is thanked for the use of the diffractometers.Notes and references
- (a) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS; (b) X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS; (c) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS; (d) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC.
- (a) W. E. Piers, P. J. Shapiro, E. E. Bunel and J. E. Bercaw, Synlett, 1990, 1990, 74–84 CrossRef; (b) P. J. Shapiro, E. Bunel, W. P. Schaefer and J. E. Bercaw, Organometallics, 1990, 9, 867–869 CrossRef CAS; (c) P. J. Shapiro, W. P. Schaefer, J. A. Labinger, J. E. Bercaw and W. D. Cotter, J. Am. Chem. Soc., 1994, 116, 4623–4640 CrossRef CAS.
- J. Okuda, Chem. Ber., 1990, 123, 1649–1651 CrossRef CAS.
- (a) J. Okuda, Dalton Trans., 2003, 2367–2378 RSC; (b) J. Cano and K. Kunz, J. Organomet. Chem., 2007, 692, 4411–4423 CrossRef CAS; (c) H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691–2720 CrossRef CAS.
- (a) Y.-X. Chen, P.-F. Fu, C. L. Stern and T. J. Marks, Organometallics, 1997, 16, 5958–5963 CrossRef CAS; (b) Y.-X. Chen and T. J. Marks, Organometallics, 1997, 16, 3649–3657 CrossRef CAS; (c) A. L. McKnight, M. A. Masood, R. M. Waymouth and D. A. Straus, Organometallics, 1997, 16, 2879–2885 CrossRef CAS; (d) M. Kamigaito, T. K. Lal and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4649–4660 CrossRef CAS; (e) E. Y. X. Chen, W. J. Kruper, G. Roof and D. R. Wilson, J. Am. Chem. Soc., 2001, 123, 745–746 CrossRef CAS; (f) J. M. Santos, M. R. Ribeiro, M. F. Portela, H. Cramail, A. Deffieux, A. Antiñolo, A. Otero and S. Prashar, Macromol. Chem. Phys., 2002, 203, 139–145 CrossRef CAS; (g) Y. Zhang, Y. Mu, C. Lü, G. Li, J. Xu, Y. Zhang, D. Zhu and S. Feng, Organometallics, 2004, 23, 540–546 CrossRef CAS; (h) H. Wang, H.-S. Chan, J. Okuda and Z. Xie, Organometallics, 2005, 24, 3118–3124 CrossRef CAS; (i) J. Li, W. Gao, Q. Wu, H. Li and Y. Mu, J. Organomet. Chem., 2011, 696, 2499–2506 CrossRef CAS.
- D. W. Carpenetti, L. Kloppenburg, J. T. Kupec and J. L. Petersen, Organometallics, 1996, 15, 1572–1581 CrossRef CAS.
- (a) D. B. Millward, A. P. Cole and R. M. Waymouth, Organometallics, 2000, 19, 1870–1878 CrossRef CAS; (b) P.-J. Sinnema, L. van der Veen, A. L. Spek, N. Veldman and J. H. Teuben, Organometallics, 1997, 16, 4245–4247 CrossRef CAS.
- (a) F. Amor and J. Okuda, J. Organomet. Chem., 1996, 520, 245–248 CrossRef CAS; (b) J. Klosin, W. J. Kruper, P. N. Nickias, G. R. Roof, P. De Waele and K. A. Abboud, Organometallics, 2001, 20, 2663–2665 CrossRef CAS; (c) L. Li, M. V. Metz, H. Li, M.-C. Chen, T. J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12725–12741 CrossRef CAS; (d) S. K. Noh, J. Lee and D.-h. Lee, J. Organomet. Chem., 2003, 667, 53–60 CrossRef CAS; (e) S. K. Noh, M. Lee, D. H. Kum, K. Kim, W. S. Lyoo and D.-H. Lee, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1712–1723 CrossRef CAS; (f) J. Wang, H. Li, N. Guo, L. Li, C. L. Stern and T. J. Marks, Organometallics, 2004, 23, 5112–5114 CrossRef CAS.
- (a) M. E. Rerek and F. Basolo, J. Am. Chem. Soc., 1984, 106, 5908–5912 CrossRef CAS; (b) J. M. O'Connor and C. P. Casey, Chem. Rev., 1987, 87, 307–318 CrossRef; (c) M. J. Calhorda, C. C. Romão and L. F. Veiros, Chem. – Eur. J., 2002, 8, 868–875 CrossRef CAS.
- T. K. Miyamoto, M. Tsutsui and L.-B. Chen, Chem. Lett., 1981, 10, 729–730 CrossRef.
- (a) T. J. Williams, J.-C. Buffet, Z. R. Turner and D. O'Hare, Catal. Sci. Technol., 2018, 8, 5454–5461 RSC; (b) T. J. Williams, A. D. H. Smith, J.-C. Buffet, Z. R. Turner and D. O'Hare, Mol. Catal., 2020, 486, 110872 CrossRef CAS.
- (a) J. V. Lamb, J.-C. Buffet, Z. R. Turner and D. O'Hare, Polym. Chem., 2019, 10, 1386–1398 RSC; (b) J. V. Lamb, J.-C. Buffet, J. E. Matley, C. M. R. Wright, Z. R. Turner and D. O'Hare, Dalton Trans., 2019, 48, 2510–2520 RSC; (c) J. V. Lamb, J. C. Abell, J. E. McLaren, J.-C. Buffet, Z. R. Turner and D. O'Hare, Mol. Catal., 2020, 484, 110735 CrossRef CAS; (d) J. V. Lamb, J.-C. Buffet, Z. R. Turner and D. O'Hare, Macromolecules, 2020, 53, 929–935 CrossRef CAS.
- N. Diteepeng, J.-C. Buffet, Z. R. Turner and D. O’Hare, Dalton Trans., 2019, 48, 16099–16107 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) J. L. Atwood and K. D. Smith, J. Chem. Soc., Dalton Trans., 1973, 2487–2490 RSC; (b) N. C. Burton, F. G. N. Cloke, P. B. Hitchcock, H. C. de Lemos and A. A. Sameh, J. Chem. Soc., Chem. Commun., 1989, 1462–1464 RSC; (c) B. D. Ward, S. R. Dubberley, A. Maisse-François, L. H. Gade and P. Mountford, J. Chem. Soc., Dalton Trans., 2002, 4649–4657 RSC; (d) K. A. Tupper and T. D. Tilley, J. Organomet. Chem., 2005, 690, 1689–1698 CrossRef CAS; (e) B. Wang, M. Nishiura, J. Cheng and Z. Hou, Dalton Trans., 2014, 43, 14215–14218 RSC; (f) A. Fridrichová, A. Růžička, M. Lamač and M. Horáček, Inorg. Chem. Commun., 2017, 76, 62–66 CrossRef; (g) Z. Zhou, J. Greenough, Z. Wei and M. A. Petrukhina, Acta Crystallogr., Sect. C: Struct. Chem., 2017, 73, 420–423 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) P. Jutzi, J. Dahlhaus, B. Neumann and H.-G. Stammler, Organometallics, 1996, 15, 747–752 CrossRef CAS; (b) J. M. Pietryga, J. D. Gorden, C. L. B. Macdonald, A. Voigt, R. J. Wiacek and A. H. Cowley, J. Am. Chem. Soc., 2001, 123, 7713–7714 CrossRef CAS; (c) M. Weger, P. Pahl, F. Schmidt, B. S. Soller, P. J. Altmann, A. Pöthig, G. Gemmecker, W. Eisenreich and B. Rieger, Macromolecules, 2019, 52, 7073–7080 CrossRef CAS.
- R. J. Wiacek, C. L. B. Macdonald, J. N. Jones, J. M. Pietryga and A. H. Cowley, Chem. Commun., 2003, 430–431 RSC.
- (a) M. Mazzeo, M. Lamberti, I. D'Auria, S. Milione, J. C. Peters and C. Pellecchia, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1374–1382 CrossRef CAS; (b) V. Rad'Kov, T. Roisnel, A. Trifonov, J. F. Carpentier and E. Kirillov, Eur. J. Inorg. Chem., 2014, 4168–4178 CrossRef; (c) J. El Haj Hassan, V. Radkov, V. Dorcet, J. F. Carpentier and E. Kirillov, J. Organomet. Chem., 2016, 823, 34–39 CrossRef CAS; (d) H. Xie, C. Wu, D. Cui and Y. Wang, J. Organomet. Chem., 2018, 875, 5–10 CrossRef CAS.
- (a) H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef; (b) M. Mazzeo, R. Tramontano, M. Lamberti, A. Pilone, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 9338–9351 RSC; (c) Y. Chapurina, J. Klitzke, O. d. L. Casagrande Jr., M. Awada, V. Dorcet, E. Kirillov and J.-F. Carpentier, Dalton Trans., 2014, 43, 14322–14333 RSC.
- J. R. Dorgan, J. Janzen, D. M. Knauss, S. B. Hait, B. R. Limoges and M. H. Hutchinson, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3100–3111 CrossRef CAS.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS.
- (a) Y. Ning, Y. Zhang, A. Rodriguez-Delgado and E. Y. X. Chen, Organometallics, 2008, 27, 5632–5640 CrossRef CAS; (b) J.-C. Buffet, G. R. Harris, J. J. Coward, T. A. Q. Arnold, Z. R. Turner and D. O'Hare, J. Organomet. Chem., 2016, 801, 87–95 CrossRef CAS; (c) K. Beckerle, K. C. Hultzsch and J. Okuda, Macromol. Chem. Phys., 1999, 200, 1702–1707 CrossRef CAS.
- Y. Cui, W. Gu, Y. Wang, B. Zhao, Y. Yao and Q. Shen, Catal. Sci. Technol., 2015, 5, 3302–3312 RSC.
- C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2015, 54, 2204–2212 CrossRef CAS.
- (a) M. H. Chisholm and E. E. Delbridge, New J. Chem., 2003, 27, 1167–1176 RSC; (b) A. F. Douglas, B. O. Patrick and P. Mehrkhodavandi, Angew. Chem., Int. Ed., 2008, 47, 2290–2293 CrossRef CAS; (c) I. Peckermann, A. Kapelski, T. P. Spaniol and J. Okuda, Inorg. Chem., 2009, 48, 5526–5534 CrossRef CAS; (d) J. Börner, I. dos Santos Vieira, A. Pawlis, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2011, 17, 4507–4512 CrossRef; (e) H. Sun, J. S. Ritch and P. G. Hayes, Dalton Trans., 2012, 41, 3701–3713 RSC; (f) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS.
- B. Gao, R. Duan, X. Pang, X. Li, Z. Qu, Z. Tang, X. Zhuang and X. Chen, Organometallics, 2013, 32, 5435–5444 CrossRef CAS.
- (a) N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS; (b) M. Wisniewski, A. L. Borgne and N. Spassky, Macromol. Chem. Phys., 1997, 198, 1227–1238 CrossRef CAS; (c) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS; (d) C. P. Radano, G. L. Baker and M. R. Smith, J. Am. Chem. Soc., 2000, 122, 1552–1553 CrossRef CAS; (e) Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS; (f) N. Nomura, R. Ishii, M. Akakura and K. Aoi, J. Am. Chem. Soc., 2002, 124, 5938–5939 CrossRef CAS; (g) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS; (h) Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS; (i) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS; (j) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2010, 29, 491–500 CrossRef CAS.
- (a) H. Ma, G. Melillo, L. Oliva, T. P. Spaniol, U. Englert and J. Okuda, Dalton Trans., 2005, 721–727 RSC; (b) Z. Tang and V. C. Gibson, Eur. Polym. J., 2007, 43, 150–155 CrossRef CAS; (c) F. Hild, P. Haquette, L. Brelot and S. Dagorne, Dalton Trans., 2010, 39, 533–540 RSC; (d) E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469–11473 RSC; (e) P. Sumrit and P. Hormnirun, Macromol. Chem. Phys., 2013, 214, 1845–1851 CrossRef CAS; (f) E. D. Cross, L. E. N. Allan, A. Decken and M. P. Shaver, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1137–1146 CrossRef CAS; (g) K. Press, I. Goldberg and M. Kol, Angew. Chem., 2015, 127, 15071–15074 CrossRef; (h) S. Gesslbauer, R. Savela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 9, 7912–7920 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complex syntheses and characterisations, NMR spectroscopy, MALDI-ToF mass spectrometry, X-ray crystallography and polymerisation data. CCDC 2014306–2014312. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0py00980f |
| This journal is © The Royal Society of Chemistry 2020 |