
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn improved synthesis of poly(amidoamine)s for complexation with self-amplifying RNA and effective transfection†
Pratik
Gurnani
 *a,
Anna K.
Blakney
b,
Jonathan
Yeow
c,
Clément R.
Bouton
b,
Robin J.
Shattock
b,
Molly M.
Stevens
c and
Cameron
Alexander
*a
*a,
Anna K.
Blakney
b,
Jonathan
Yeow
c,
Clément R.
Bouton
b,
Robin J.
Shattock
b,
Molly M.
Stevens
c and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: pratik.gurnani@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk
bDepartment of Infectious Disease, Imperial College London, School of Medicine, St Mary's Hospital, Praed Street, London W2 1NY, UK
cDepartment of Materials and the Department of Bioengineering at Imperial College London, Prince Consort Rd, SW7 2AZ London, South Kensington, UK
First published on 19th August 2020
Abstract
Cationic polymers are widely used as materials to condense nucleic acids for gene-based therapies. These have been developed to mainly deliver DNA and RNA for cancer therapies but the ongoing COVID-19 pandemic has demonstrated an urgent need for new DNA and RNA vaccines. Given this, suitable manufacturing conditions for such cationic polymers which can protect the nucleic acid in the formulation and delivery stages but release the cargo in the correct cellular compartment effectively and safely are required. A number of polymers based on poly(amidoamine)s fit these criteria but their syntheses can be time-consuming, inefficient and poorly reproducible, precluding their adoption as manufacturable vaccine excipients. Here we report an improved synthesis of poly(cystamine bisacrylamide-co-4-amino-1-butanol), abbreviated as pABOL, via modifications in concentration, reaction time and reaction conditions. Optimisation of monomer contents and stoichiometries, solvents, diluents and temperature, combined with the application of microwaves, enabled the preparation of vaccine candidate pABOL materials in 4 h compared to 48 h reported for previous syntheses. These procedures were highly reproducible in multiple repeat syntheses. Transfection experiments with a model RNA showed that polymers of formulation with appropriate molar masses and mass distributions were as effective in model cell lines as polymers derived from the unoptimised syntheses which have been shown to have high efficacy as RNA vaccine formulation candidates.
Introduction
The application of polycations in the formulation of nucleic acid delivery systems has accelerated rapidly in recent years.1–4 Advances in gene editing technologies, and the possibilities for DNA- and RNA-based therapies and vaccines against cancers and infectious diseases, have all contributed to the focus on polycation-based gene delivery formulations.5–9 Within the large field of cationic polymers, materials based on oligo- or poly(amidoamine)s (pAMAMs) and poly(β-aminoesters) (pBAEs) have been extensively investigated.10–16 This is because these polymers are intrinsically biodegradable and can be synthesised from a range of commercially available bisacrylamide/bisacrylates (for pAMAMs/pBAEs respectively) and primary amines, thus enabling high chemical diversity and consequent broad utility across different nucleic acid delivery platforms.17 pAMAMs in particular have shown particular promise due to their limited mid-chain hydrolysis and higher water solubility due to their amide backbone, in comparison to esters in pBAEs. However, while the preparation of pAMAMs is relatively straight-forward for research investigations,18 in the case of rapid manufacture potentially required for high volume nucleic acid vaccine production, the current synthetic protocols are sub-optimal. In particular, there is a pressing need to understand if methods used for the preparation of pAMAMs can be tuned to produce materials with the right properties for delivery of mRNA or other nucleic acid vaccine candidates in a reproducible way, and in a manner which might allow for rapid and distributed manufacture.Accordingly, the aim of the study presented here was to investigate the synthetic parameters leading to reproducible and faster synthesis of a specific class of pAMAMs, whilst potentially making the procedure more amenable to microwave acceleration and continuous flow technologies for scale up and potential decentralised manufacture. The specific focus was to develop a more rapid synthesis of poly(cystamine bisacrylamide-co-4-amino-1-butanol), hereinafter referred to as pABOL, which has shown promise as a highly efficient delivery vector for self-amplifying RNA (saRNA) in candidate vaccine formulations.19 Pioneering work on this polymer,20 and related materials,21–24 initially established broad efficacy across a range of gene delivery applications, with the bioreducible disulfide linker greatly enhancing the expression of transgenes and minimising vector toxicity.25 In most reports, the synthesis of pABOL is conducted via the step-growth Michael-addition polymerisation between N,N′-cystaminebisacrylamide and 4-amino-1-butanol, either without solvent, or in high concentrations in apolar solvents. These methods typically allow the generation of polymers with molar masses of 103–104 g mol−1 and Đ values above 2, but reactions under these conditions require several days to reach high molar masses. The demand for sufficient cationic polymer for many millions of vaccine doses to be made rapidly available is incompatible with reactions which take long time periods. There is now a requirement to develop methods by which pABOL-type polymers can be produced to an appropriate specification for reproducible vaccine formulations in a minimum time frame. Accelerated polymerisations, may also be relevant in structure–property optimisation studies to screen structural (molar mass, branching etc.) and chemical (lipophilicity, basicity, charge density etc.). However, the current slow synthetic methods for this family of polymers significantly hinders structure optimisation, potentially useful for tailoring polymer vectors to an emerging nucleic acid vaccine candidate.
We report here a synthetic optimisation study, screening the effect of reaction parameters such as inert atmospheres, monomer concentration, reactant ratio and reaction temperature on the molecular weight distribution of pABOL. Through these improvements, we were able to minimise a radical branching side reaction, then accelerate the prevailing step-growth polymerisation with microwave assisted synthesis. The conditions established in this work were highly reproducible in comparison to unoptimized conditions. Finally, we highlight the importance of controlling the molecular weight distribution by evaluating the correlation of certain polymer physicochemical characteristics and their propensity to effectively deliver a model saRNA vaccine.
Experimental
Materials
All solvents were of analytic or HPLC grade and purchased from Sigma Aldrich or Fisher Scientific unless otherwise stated. All deuterated solvents were purchased from Sigma Aldrich. Reagents 4-aminobutanol (178330-5G), N,N′-bis(acryloyl)cystamine (A4929-5G) and triethylamine were purchased from Sigma-Aldrich. fLuc VEEV saRNA (9382 nt) was prepared as described previously.26Instrumentation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–550 g mol−1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Respectively, experimental molar mass (Mn,SEC) and dispersity (Đ) values of synthesized polymers were determined by conventional calibration using Cirrus GPC software.
500–550 g mol−1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Respectively, experimental molar mass (Mn,SEC) and dispersity (Đ) values of synthesized polymers were determined by conventional calibration using Cirrus GPC software.
Polymer synthesis
Formulation of saRNA polyplexes
Stock solutions of pABOLs and saRNA were prepared first by directly dissolving these materials in molecular grade water and stored at 4 °C for up to 3 months. The concentrations of the stock solutions are 0.24 μg μL−1 (fLuc VEEV saRNA), and 5.00 μg μL−1 (pABOLs) respectively.In vitro transfection of saRNA polyplexes
Transfections were performed in HEK293T.17 cells (ATCC, USA) that were maintained in culture in complete Dulbecco's Modified Eagle's Medium (cDMEM) (Gibco, ThermoFisher, UK) containing 10% (v/v) fetal calf serum (FCS), 5 mg mL−1L-glutamine and 5 mg mL−1 penicillin and streptomycin (ThermoFisher, UK). Cells were plated at a density of 50000 cells per well in a 96 well plate 24 h prior to transfection. At the time of transfection, the media was completely removed and replaced by 50 μL of transfection media (DMEM with 5 mg mL−1L-glutamine). 100 μL of the polyplex solution was added to each well and incubated for 4 h. Then, the media was replaced with cDMEM and the cells were allowed to culture for 24 hours, at which time 50 μL of media was removed and 50 μL of ONE-Glo™ D-luciferin substrate (Promega, UK) was added and mixed well by pipetting. The total volume was transferred to a white 96-well plate (Costar, UK) and analyzed on a FLUOstar Omega plate reader (BMG LABTECH, UK).
Results and discussion
Previous reports of the synthesis of pABOL have utilised protocols where N,N′-bis(acryloyl)cystamine (CBA), 4-aminobutanol (ABOL) and triethylamine in a methanol/water are mixed and heated with stirring at 45 °C under a nitrogen atmosphere for up to 14 days.19 We thus commenced our studies by following this synthesis (Fig. 1) and observed a clear viscous solution at the end of two weeks heating at 45 °C (Table 1, entry 1). NMR spectra (Fig. 1B) and DMF-SEC revealed successful polymerisation, with an Mw,SEC = 45 kg mol−1 and a relatively broad molecular weight distribution (Đ = 3.62). The origins of this high Đ appeared to be due to an additional high molecular weight peak in the chromatogram, which was postulated to be caused by chain branching through a side reaction of radical polymerisation (Fig. 1C). | ||
| Fig. 1 (A) Synthetic scheme for step-growth Michael-addition polymerisation of N,N′-cystaminebisacrylamide and 4-amino-1-butanol to produce pABOL, (B) 1H NMR spectrum of pABOL; (C) size exclusion chromatogram of product obtained via ‘conventional’ synthesis. | ||
| Description | Entry | [ABOL] (M) | [CBA] (M) | [ABOL]/[CBA] | Atmosphere | T (°C) |
|---|---|---|---|---|---|---|
| Original | 1 | 3.2 | 3.2 | 1 | Argon | 45 |
| Dilution | 2 | 0.60 | 0.60 | 1 | Argon | 45 |
| Dilution + air | 3 | 0.60 | 0.60 | 1 | Air | 45 |
| Temperature | 4 | 0.60 | 0.60 | 1 | Air | 60 |
| 5 | 0.60 | 0.60 | 1 | Air | 25 | |
| [M] | 6 | 3.2 | 3.2 | 1 | Air | 60 |
| 7 | 0.8 | 0.8 | 1 | Air | 60 | |
| 8 | 0.45 | 0.45 | 1 | Air | 60 | |
| 9 | 0.24 | 0.24 | 1 | Air | 60 | |
| Monomer ratio | 10 | 0.60 | 0.60 | 1 | Air | 60 |
| 11 | 0.606 | 0.60 | 1.01 | Air | 60 | |
| 12 | 0.615 | 0.60 | 1.025 | Air | 60 | |
| 13 | 0.63 | 0.60 | 1.05 | Air | 60 | |
| 14 | 0.66 | 0.60 | 1.1 | Air | 60 | |
| 15 | 0.72 | 0.60 | 1.2 | Air | 60 | |
| Repeatability | 16 | 0.60 | 0.60 | 1 | Air | 60 |
| 17 | 3.2 | 3.2 | 1 | Argon | 45 | |
| Microwave | 18 | 0.60 | 0.60 | 1 | Air | 100 |
These results suggest that the high reaction concentration (3.2 M) and inert conditions both favour this side reaction. We thus conducted a systematic study, varying the reaction conditions to mitigate this side reaction. Previous data indicated that pABOL of Mw ∼ 8 kg mol−1 yields high saRNA transfection in animal models. Therefore, throughout the optimisation study we targeted polymers around this molecular weight.
Systematic optimisation of polymerisation
Immediately comparing the argon deoxygenated conditions at lower dilution (Table 1, entry 2), we observed a steady increase in molar mass without the appearance of a bimodal high molar mass peak. A high molar mass shoulder however, was observed after 10 d in the SEC chromatograms, indicating some radical polymerisation was still occurring at diluted deoxygenated conditions (Fig. 2A). This was further evidenced by the minimal increase in Mn,SEC whilst Mw,SEC continued to climb after this time point. Noticeably, the diluted conditions appear to retard the side reaction, however, do not completely inhibit it. Comparatively at the 10 d time point under air (Table 1, entry 3), no further increase in molecular weight was observed, likely due to the radical scavenging mechanism of oxygen (Fig. 2B). This is further exemplified in the Đ values, with samples taken from polymerisations performed under air maintaining Đ < 1.6, while Đ for polymers prepared under inert atmosphere rose to ∼4.5 over the 2 weeks reaction (Fig. 2C).
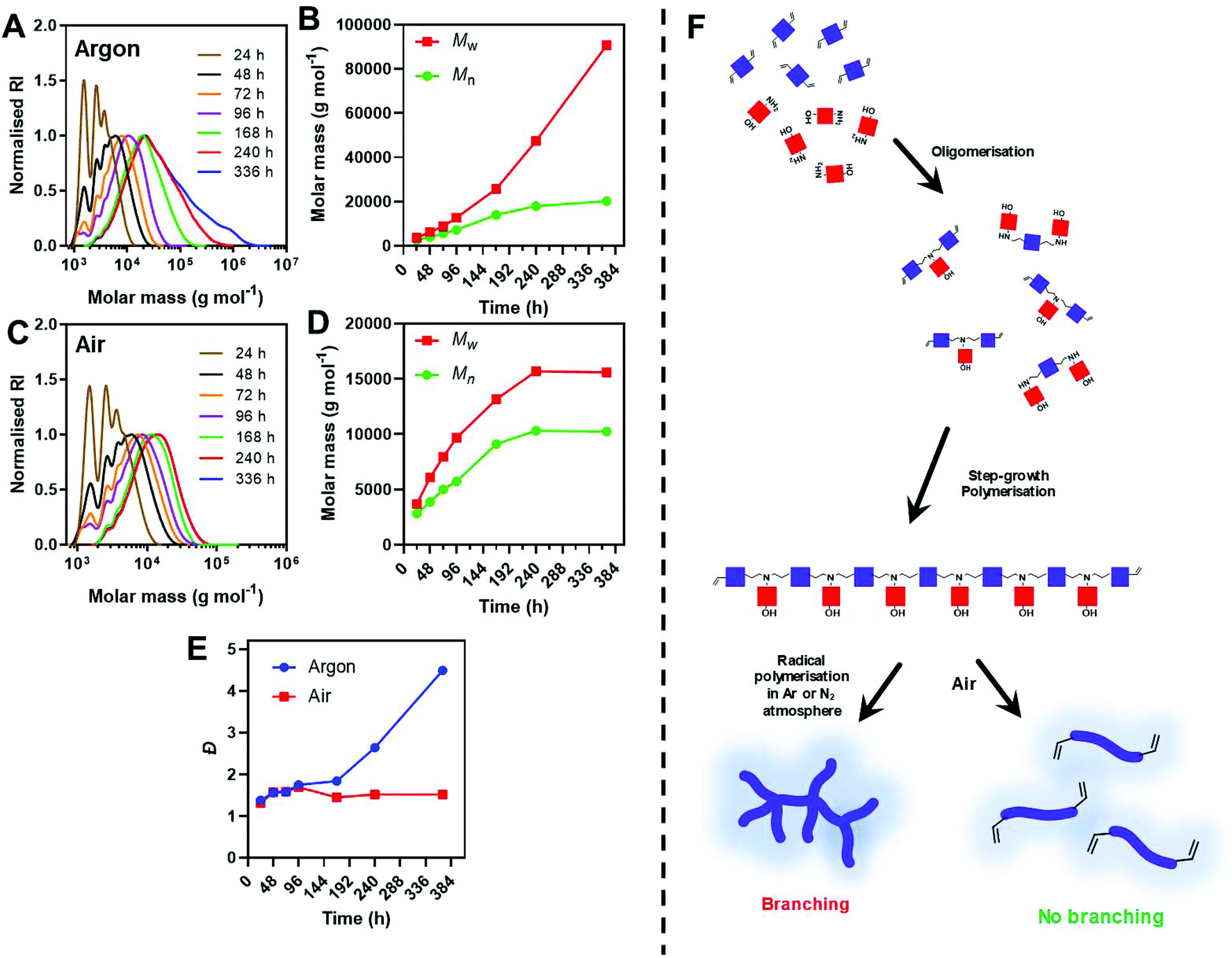 | ||
| Fig. 2 (A and C) DMF-SEC chromatograms and (B and D) molecular weight evolution of pABOL at [M] = 0.60 M, T = 45 °C polymerisation in an argon or air atmosphere. (E) Evolution of dispersity over time for pABOL. (F) Schematic representation of step-growth polymerisation and subsequent radical polymerisation leading to branching under inert conditions and inhibition of radical polymerisation under air. | ||
We anticipate that the steadily increasing high molar mass shoulder was a result of successive branching events, otherwise inhibited under an air atmosphere (Fig. 2D). The diluted conditions significantly slowed the rate of polymerisations taking 96 h to reach Mw,SEC > 8 kg mol−1, whilst concentrated conditions (5-fold higher concentration), reached these molar masses within 48 h.
 | ||
| Fig. 3 (A and B) DMF-SEC chromatograms and (C) molecular weight evolution of pABOL at [M] = 0.60 M and air atmosphere at 25 °C, 45 °C and 60 °C under conventional heating at 100 °C under using microwave assisted heating. | ||
With these conditions providing low viscosity solutions and exhibiting minimal side reactions we sought to accelerate further the synthetic procedure utilising microwave irradiation. Microwave accelerated synthesis has two major advantages: firstly, microwaves homogenously heat solvents, leading to reliable heat gradients; secondly most microwave synthesis procedures occur under high pressure conditions, increasing the boiling point of solvent.29,30 These effects both synergise and enable synthesis at significantly higher temperatures and faster reaction rates than conventional heating. Furthermore, microwave accelerated synthesis is highly amenable to continuous flow reactors as they offer fast, but energy efficient synthetic routes to scalable manufacture, important in rapid response supply chains.31
To evaluate pABOL synthesis using microwave assisted heating, we utilised a Biotage microwave synthesiser set to 200 W, heating to 100 °C and monitored the molecular weight evolution over 24 h (Table 1, entry 18). Under these conditions, we observed a significantly faster increase in molecular weight, reaching ∼8 kDa in ∼4 h rather than 48 h, a 12-fold acceleration compared to conventional heating at 60 °C (Fig. 3B and C). Chromatograms were comparable under microwave conditions and conventional, with similar Đ regardless of heating method.
 | ||
| Fig. 4 (A) DMF-SEC chromatograms and (B) molecular weight evolution of ABOL + CBA polymerisations at 60 °C and an air atmosphere at [M] = 3.2, 0.8, 0.45 and 0.24 M. (C) DMF-SEC chromatograms and (D) Mw,SEC of final polymers after 48 h reaction between CBA and ABOL at different ABOL/CBA ratios. Reactions were performed at 60 °C and an air atmosphere at [M] = 0.60 M. | ||
To achieve high molar masses for step-growth polymerisations it is imperative to have a near equimolar ratio between the A and B components. However, if the ratios are not perfectly equimolar (i.e. 1–10% out), it is possible to tune the molecular weight and end-group of the final polymers. We prepared 5 polymerisations at an equimolar ratio and added 1%, 2.5%, 5%, 10% and 20% (Table 1, entries 11–15) excess ABOL and heated the tubes for 48 h at 60 °C under an air atmosphere. With 1% extra ABOL we observed no difference in final Mw, compared to the purely equimolar polymerisation, however with higher amounts of excess ABOL a decreasing trend in Mw with increasing excess ABOL was evident (Fig. 4C and D). At 20% excess ABOL, no polymerisation or only oligomerisation was observed. This data also provides important information on the weighing/stoichiometric error allowed (max. 1%) to avoid molecular weight discrepancy.
700 ± 5500 g mol−1 and Đ = 3.51 ± 1.07 (Fig. 5A–C). The variation in Mw and Đ for the ‘old’ conditions is most likely a consequence of the bimodal distribution, absent in the ‘new’ conditions. Overall, the new conditions presented here generated polymers with more reproducible properties.
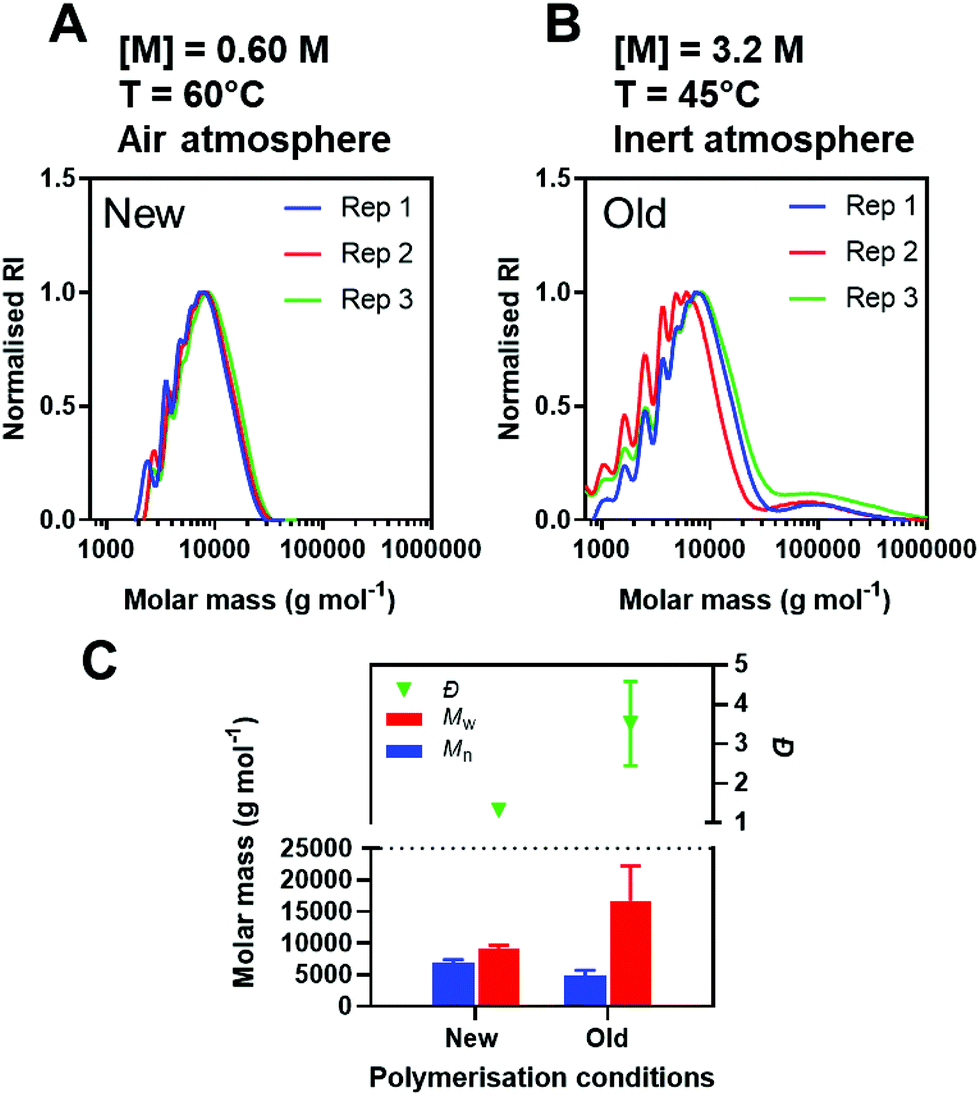 | ||
| Fig. 5 (A and B) DMF-SEC chromatograms and (C) Mw of final polymers after 48 h reaction between CBA and ABOL performed in triplicate. Reactions were performed at 60 °C, [M] = 0.60 M and an air atmosphere compared to 45 °C, [M] = 3.2 M and an argon atmosphere. | ||
551 nt.33 saRNA constructs have shown great promise in vaccine applications, as their manufacture can be conducted in vitro in a pathogen agnostic manner but can achieve similar efficacy and protective immunity at 1000-fold lower doses than conventional mRNA.34–36
 | ||
| Fig. 6 (A) Transfection of polyplexes with saRNA encoding luciferase at N/P = 45 in HEK 293T.17 cells 24 h after treatment. (B) GPC chromatograms of polymers used in transfection studies. | ||
There were marked differences in saRNA expression delivered by the various polyplexes derived from the synthesised polymers, dependent on molecular weight distribution (Fig. 6A). Three polymers (P2, P3 and P6) displayed lower luciferase expression (∼101 RLU), whilst the other three exhibited up to 5 orders of magnitude higher luminescence. Comparing the molecular weight distributions of these two groups (Table S8†), it was clear that polymers with bimodal distributions and thus high Đ severely reduces saRNA transfection. Overall, these results indicate transfection efficiency is significantly enhanced using polymers with monomodal distributions, highlighting the need to minimise radical induced branching as shown in this study. The key question for future development of these materials is therefore what the best compromise between speed and reliability of synthetic method is, while retaining effective transfection in a vaccine setting in vivo.
Conclusions
In conclusion, we have optimised the A2 + B2 step-growth polymerisation between CBA and ABOL, yielding conditions that provide low viscosity solutions and minimal side reactions. Our findings suggest that at high concentrations the radical branching side reaction is relatively favourable, with dilution this is partially retarded but is completely inhibited under an air atmosphere. The optimised conditions are now highly reproducible and the timeframe to achieve the 8 kg mol−1 target can be accelerated using a microwave synthesiser. These conditions and accelerated timeframe make this synthesis more amenable to scale up, particularly in continuous flow. In terms of the future rapid manufacture of vaccines, the processes to produce these materials are robust, and show small batch-batch variability even with low durations of synthesis. Furthermore, the use of microwaves and simple conditions will facilitate the transfer to flow chemistries, further enhancing the suitability of this method for rapid and scalable manufacture of vaccine excipients. These microwave conditions, utilising standardized equipment, may also provide a reliable method for reproducible decentralized batch synthesis, useful in disease outbreak scenarios where traditional supply chains are constrained. Given the chemical versatility of pAMAM cationic polymer platform, through variation of the two components, we anticipate the findings will extend to synthesis processes with other amines and bisacrylamides. To date we do not yet know the optimal set of physicochemical characteristics which produce the best saRNA vaccine properties in vivo, and the correlation between in vitro and in vivo efficacy remains challenging. Nevertheless, the data presented here provide indicators towards more rapid and scalable synthesis of promising nucleic acid vaccine formulation components.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research is funded by the Department of Health and Social Care using UK Aid funding and is managed by the Engineering and Physical Sciences Research Council (EPSRC, grant number: EP/R013764/1). The views expressed in this publication are those of the author(s) and not necessarily those of the Department of Health and Social Care. We thank Dr Yunqing Zhu for useful discussions on pABOL synthesis. This research was also funded through the Royal Society through a Wolfson Research Merit Award [WM150086]. We also thank Douglas Crackett and Paul Cooling for expert technical assistance and Carol Turrill for outstanding administrative support.References
- A. C. Anselmo, Y. Gokarn and S. Mitragotri, Nat. Rev. Drug Discovery, 2019, 18, 19–40 CrossRef CAS PubMed.
- R. Kedmi, N. Veiga, S. Ramishetti, M. Goldsmith, D. Rosenblum, N. Dammes, I. Hazan-Halevy, L. Nahary, S. Leviatan-Ben-Arye, M. Harlev, M. Behlke, I. Benhar, J. Lieberman and D. Peer, Nat. Nanotechnol., 2018, 13, 214–219 CrossRef CAS PubMed.
- I. Lostale-Seijo and J. Montenegro, Nat. Rev. Chem., 2018, 2, 258–277 CrossRef.
- K. Muhammad, J. Zhao, I. Ullah, J. T. Guo, X. K. Ren and Y. K. Feng, Biomater. Sci., 2020, 8, 64–83 RSC.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- R. Mout, M. Ray, Y. W. Lee, F. Scaletti and V. M. Rotello, Bioconjugate Chem., 2017, 28, 880–884 CrossRef CAS PubMed.
- J. C. Kaczmarek, P. S. Kowalski and D. G. Anderson, Genome Med., 2017, 9, 60–76 CrossRef PubMed.
- A. K. Blakney, Y. Abdouni, G. Yilmaz, R. Liu, P. F. McKay, C. R. Bouton, R. J. Shattock and C. R. Becer, Biomacromolecules, 2020, 21, 2482–2492 CrossRef CAS PubMed.
- F. Saviano, T. Lovato, A. Russo, G. Russo, C. R. Bouton, R. J. Shattock, C. Alexander, F. Quaglia, A. K. Blakney, P. Gurnani and C. Conte, J. Mater. Chem. B, 2020, 8, 4940–4949 RSC.
- A. Akinc, D. G. Anderson, D. M. Lynn and R. Langer, Bioconjugate Chem., 2003, 14, 979–988 CrossRef CAS PubMed.
- J. C. Meredith, J. Mater. Chem., 2009, 19, 34–45 RSC.
- R. A. Cordeiro, A. Serra, J. F. J. Coelho and H. Faneca, J. Controlled Release, 2019, 310, 155–187 CrossRef CAS PubMed.
- E. Ben-Akiva, S. E. Witte, R. A. Meyer, K. R. Rhodes and J. J. Green, Biomater. Sci., 2019, 7, 14–30 RSC.
- Y. Rui, D. R. Wilson, J. Choi, M. Varanasi, K. Sanders, J. Karlsson, M. Lim and J. J. Green, Sci. Adv., 2019, 5, eaay3255 CrossRef CAS PubMed.
- S. Z. Duan, D. S. Cao, X. D. Li, H. F. Zhu, M. Lan, Z. Z. Tan, Z. Y. Song, R. Y. Zhu, L. C. Yin and Y. B. Chen, Biomater. Sci., 2020, 8, 290–301 RSC.
- K. L. Kozielski, A. Ruiz-Valls, S. Y. Tzeng, H. Guerrero-Cazares, Y. Rui, Y. X. Li, H. J. Vaughan, M. Gionet-Gonzales, C. Vantucci, J. Kim, P. Schiapparelli, R. Al-Kharboosh, A. Quinones-Hinojosa and J. J. Green, Biomaterials, 2019, 209, 79–87 CrossRef CAS PubMed.
- A. Akinc, D. M. Lynn, D. G. Anderson and R. Langer, J. Am. Chem. Soc., 2003, 125, 5316–5323 CrossRef CAS PubMed.
- T. Lovato, V. Taresco, A. Alazzo, C. Sansone, S. Stolnik, C. Alexander and C. Conte, J. Mater. Chem. B, 2018, 6, 6550–6558 RSC.
- A. K. Blakney, Y. Zhu, P. F. McKay, C. R. Bouton, J. Yeow, J. Tang, K. Hu, K. Samnuan, C. L. Grigsby, R. J. Shattock and M. M. Stevens, ACS Nano, 2020, 14, 5711–5727 CrossRef CAS PubMed.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2006, 116, 130–137 CrossRef CAS PubMed.
- Z. Zhong, Y. Song, J. F. J. Engbersen, M. C. Lok, W. E. Hennink and J. Feijen, J. Controlled Release, 2005, 109, 317–329 CrossRef CAS PubMed.
- D. M. Lynn, D. G. Anderson, D. Putnam and R. Langer, J. Am. Chem. Soc., 2001, 123, 8155–8156 CrossRef CAS PubMed.
- D. M. Lynn and R. Langer, J. Am. Chem. Soc., 2000, 122, 10761–10768 CrossRef CAS.
- Y. Liu, D. Wu, Y. Ma, G. Tang, S. Wang, C. He, T. Chung and S. Goh, Chem. Commun., 2003, 2630–2631 RSC.
- C. Lin, C.-J. Blaauboer, M. M. Timoneda, M. C. Lok, M. van Steenbergen, W. E. Hennink, Z. Zhong, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2008, 126, 166–174 CrossRef CAS PubMed.
- P. Gurnani, A. K. Blakney, R. Terracciano, J. E. Petch, A. J. Blok, C. R. Bouton, P. F. McKay, R. J. Shattock and C. Alexander, Biomacromolecules, 2020, 21, 3242–3253 CrossRef CAS PubMed.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- J. Yeow, R. Chapman, A. J. Gormley and C. Boyer, Chem. Soc. Rev., 2018, 47, 4357–4387 RSC.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- K. Kempe, C. R. Becer and U. S. Schubert, Macromolecules, 2011, 44, 5825–5842 CrossRef CAS.
- T. N. Glasnov and C. O. Kappe, Macromol. Rapid Commun., 2007, 28, 395–410 CrossRef CAS.
- J. K. Stille, J. Chem. Educ., 1981, 58, 862 CrossRef CAS.
- P. F. McKay, K. Hu, A. K. Blakney, K. Samnuan, J. C. Brown, R. Penn, J. Zhou, C. R. Bouton, P. Rogers, K. Polra, P. J. C. Lin, C. Barbosa, Y. K. Tam, W. S. Barclay and R. J. Shattock, Nat. Commun., 2020, 11, 3523 CrossRef PubMed.
- C. Zhang, G. Maruggi, H. Shan and J. Li, Front. Immunol., 2019, 10, 594 CrossRef CAS PubMed.
- A. B. Vogel, L. Lambert, E. Kinnear, D. Busse, S. Erbar, K. C. Reuter, L. Wicke, M. Perkovic, T. Beissert, H. Haas, S. T. Reece, U. Sahin and J. S. Tregoning, Mol. Ther., 2018, 26, 446–455 CrossRef CAS PubMed.
- A. J. Geall, A. Verma, G. R. Otten, C. A. Shaw, A. Hekele, K. Banerjee, Y. Cu, C. W. Beard, L. A. Brito, T. Krucker, D. T. O'Hagan, M. Singh, P. W. Mason, N. M. Valiante, P. R. Dormitzer, S. W. Barnett, R. Rappuoli, J. B. Ulmer and C. W. Mandl, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14604–14609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00912a |
| This journal is © The Royal Society of Chemistry 2020 |