
Monodisperse oligo(δ-valerolactones) and oligo(ε-caprolactones) with docosyl (C22) end-groups†

Abstract
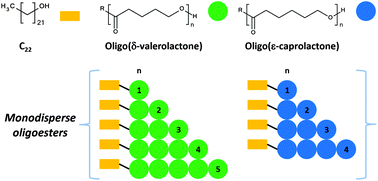
Linear aliphatic oligoesters derived from δ-valerolactone (VL) and ε-caprolactone (CL) were synthesised by ring-opening polymerisation (ROP) using 1-docosanol (C22H45OH, C22OH) and aluminum isopropoxide [Al(OiPr)3] as initiator and catalyst, respectively. Monodisperse species such as monomers, dimers, trimers, and tetramers with terminal groups α-hydroxyl-ω-docosyl (C22)-(C22VL and C22CL) were isolated by flash column chromatography (FCC). Characterisation by MALDI-TOF and 1H NMR spectroscopy corroborated the chemical nature of the samples. This work represents the first time in the literature that monodisperse species derived from VL are isolated. The thermal properties of C22VL and C22VL showed a pattern suggesting that the crystallinity was proportional to the length of the chain. The presence of C22 groups promoted the nucleation of the crystalline domain in the monomers (C22VL1 and C22CL1), increasing their respective melting temperatures (Tm) relative to those of their non-monodisperse oligomers (C22PVL or C22PCL). Finally, monodisperse species and non-monodisperse oligomers were analysed by POM.
 Please wait while we load your content...
Please wait while we load your content...

