
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of surface charge on the formulation of elongated PEG-b-PDLLA nanoparticles†
Roxane
Ridolfo
a,
David S.
Williams
*b and
Jan C. M.
van Hest
 *a
*a
aBio-Organic Chemistry, Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513 (STO 3.41), 5600 MB Eindhoven, The Netherlands. E-mail: j.c.m.v.hest@tue.nl
bDepartment of Chemistry, College of Science, Swansea University, Swansea, UK. E-mail: d.s.williams@swansea.ac.uk
First published on 31st March 2020
Abstract
Polymeric vesicles (polymersomes) are an important class of nanoparticles for various biomedical applications. Their interaction with biological systems is strongly dependent on their topological features such as size, shape and surface charge. We have recently developed a versatile method that enables the formation of tubes or bowl-shaped vesicles (stomatocytes) out of spherical vesicles composed of the biodegradable block copolymer poly(ethylene glycol)-poly(D,L-lactic acid) (PEG-b-PDLLA), via a dialysis process. Applying this method for particles with different surface charge is however a far from trivial task, as the shape change process is affected by spontaneous membrane curvature, which is strongly influenced by the presence of charged polymer end groups. Here we describe an optimized procedure to attain an effective shape transformation toward tubes of PEG-b-PDLLA polymersomes containing PEG blocks with amine (A) or carboxylic acid (CA) end groups. The salt concentration employed during the dialysis process turns out to be key, with the CA-polymersomes requiring substantially lower concentrations than unmodified or A-ones. The ability to control now reliably shape and surface charge in these polymer vesicles, allows a future systematic analysis of the effect of these topological parameters on the biological response of the nanoparticles.
Introduction
Nanoparticle morphology is recognised to be a key determinant of performance in the field of nanomedicine.1–6 Contrary to other particle features such as size and surface charge, shape only recently received attention as an important factor determining the behaviour of particles in a biological context. High-aspect ratio nanoparticles have been shown to have improved biophysical properties with regard to for example flow characteristics (influencing circulation and distribution) and interactions with cells/tissues.6–8An important class of nanoparticles that have been used in a diverse range of applications including drug delivery,9 radiotherapy,10 cancer cell targeting,11 and nanoreactors12 are polymeric vesicles, or polymersomes.13,14 The compartmentalized nature of polymersomes facilitates encapsulation of various types of cargo within their inner lumen, protecting them from unwanted degradation whilst facilitating their delivery to cells through passive or active uptake (using homing motifs such as peptides or antibodies).4,15 The shape effect of course also holds for polymersomes, and this has inspired researchers to develop effective methods for the construction of non-spherical vesicular structures.
For this purpose different processes have been reported such as polymerisation-induced self-assembly (PISA),16–18 film re-hydration,19–21 or co-solvent/evaporation.7 The PRINT technology22 and moulding23,24 have also proven to be very useful to create nanoparticles with different shapes, albeit that these techniques predominantly have been used for solid systems. In our group we have constructed non-spherical polymersomes by employing a dialysis procedure; block copolymers were dissolved in an organic solvent, after which water was added to induce polymer assembly. The organic solvent was subsequently removed via dialysis. By careful control over the latter step a shape change could be realized. We were able to demonstrate this both for non-degradable block copolymers such as poly(ethylene glycol)-poly(styrene)25,26 and, more recently, biodegradable block copolymers.1,27 In this way, we have generated tubular polymersomes (nanotubes) and bowl-shaped vesicles known as stomatocytes comprising poly(ethylene glycol)-poly(D,L-lactide) (PEG-b-PDLLA) block copolymers.
Surface charge is another important feature of nanoparticles that can have a major effect on their performance as drug delivery systems. Particle charge has been shown to influence phagocytosis, cellular uptake and biodistribution.28–30 Reports on various polymeric nanoparticles indicate better cellular uptake and biodistribution for positively charged particles.31–33 Negatively charged particles were usually less toxic34,35 and more prone to permeate tissues for targeted delivery.36,37 Internalization pathways have also been linked to surface charge, with positively charged particles tending towards internalization via clathrin receptors, while negatively charged counterparts are prone for caveolae mediated uptake.33 With this in mind, methods should be available in which both shape and surface charge can be effectively engineered to tailor the performance of nanoparticles in vivo.
Various strategies exist for the incorporation of charge onto polymeric nanoparticles including synthetic approaches and post-modification.30,33,35,38–42 Herein, we present a facile approach whereby the surface charge of non-spherical polymersomes was modified by blending uncharged methoxy-terminated PEG-b-PDLLA block copolymers with either amino (A) or carboxyl (CA) terminated variants. Assembly was conducted using the solvent switch methodology, where water was slowly added to a solution of polymer in organic solvent. By systematically varying the salt concentration during the subsequent dialysis procedure, we were able to transform the differently functionalized spherical polymersomes into nanotubes and, under certain conditions, stomatocytes. Having attained a robust route to the fabrication of functionalized polymersomes (either spherical or tubular) with different surface groups, their toxicity was tested against human retinal (ARPE-19) cells – where they yielded no evidence of harmful effects to cells.
Research and discussion
Using either monomethoxy, carboxyl- or NHBoc-terminated poly(ethylene glycol) macroinitiators of narrow polydispersity, block copolymers (BCPs) were synthesized through the organocatalyzed ring-opening polymerization (ROP) of D,L-lactide monomers using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a catalyst (S1).27 This yielded BCPs with a well-defined (45 repeats, 6.5 kDa), hydrophobic PDLLA block (after reaction for 2 h at room temperature) with a hydrophilic PEG block of either 1 or 2 kDa for uncharged and charged BCPs, respectively (Fig. 1A and B). This difference in length was chosen to ensure that the incorporated charged groups were accessible at the particle surface. All BCPs had polydispersity values of approximately 1.1, which highlights the excellent control that was achieved using this method (Fig. 1A/S2–S3). Subsequent deprotection of the PEG-terminal amine was performed using trifluoroacetic acid (TFA) in DCM to yield the primary amine, with no detrimental effect on the reaction or polydispersity. The use of acid-terminated PEG did not appear to interfere with the polymerisation. The composition of all BCPs was assessed using 1H NMR spectroscopy, which confirmed that precise polymerization control was achieved (Fig. 1A/S4–S6). In this way, a range of well-defined polymers were prepared as a first step in formulating biocompatible polymersomes that would be able to display either negative or positive charge at neutral pH through blending with the non-charged variant.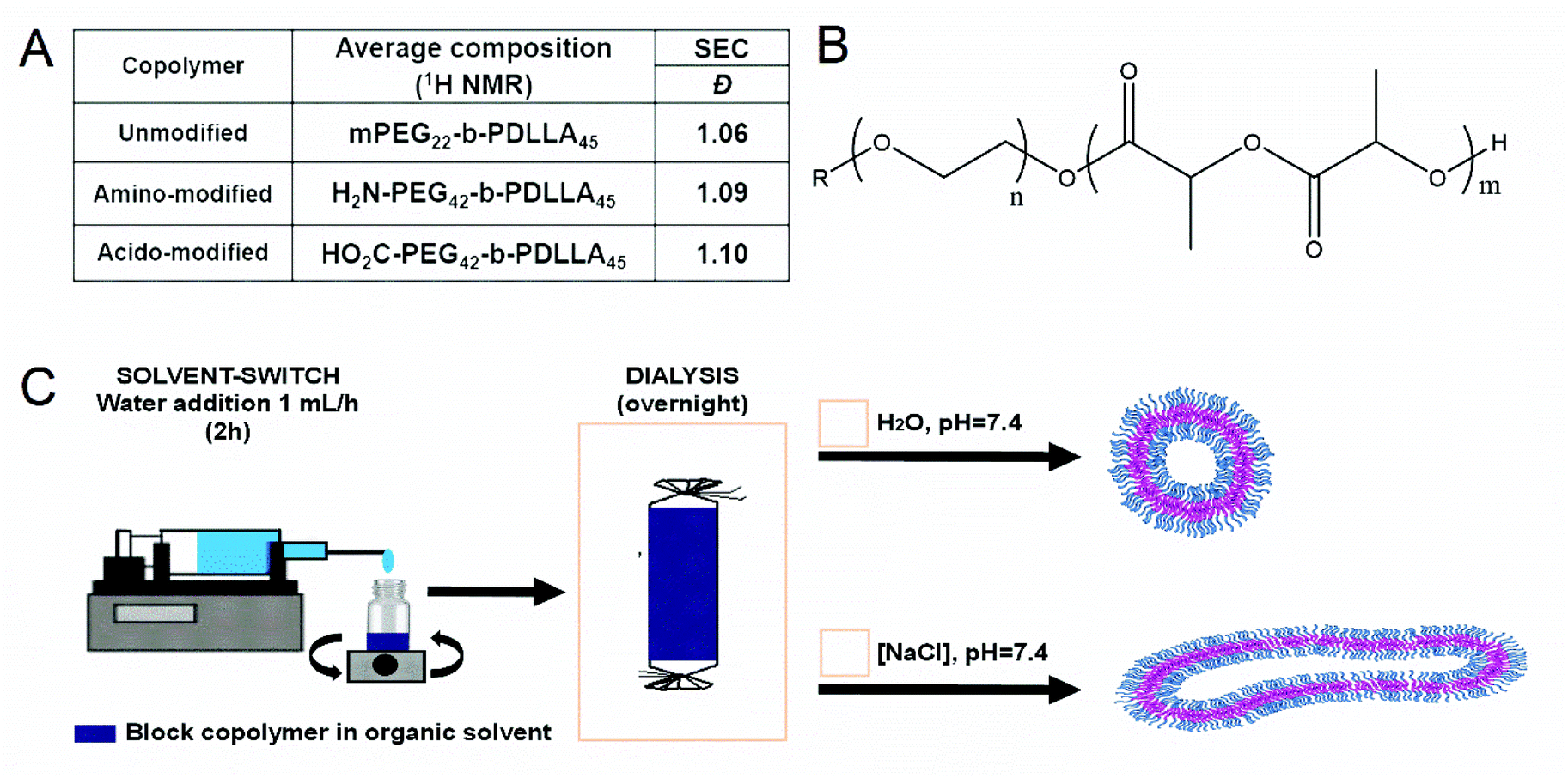 | ||
| Fig. 1 (A) Characterisation of synthesized poly(ethylene glycol)-block-poly(D,L-lactide) (PEG-b-PDLLA) copolymers; (B) molecular structure of PEG-b-PDLLA; (C) formulation scheme for the preparation of PEG-b-PDLLA vesicles. | ||
Self-assembly of BCPs was performed using the solvent switch method, by dissolving the polymer in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF:dioxane prior to dropwise addition of water up to 50 vol% over 2 h (Fig. 1C).27 The formation of polymersomal nanostructures was indicated by the increased turbidity of the solution, without the formation of unwanted aggregates. A- or CA-modified polymersomes were formed using the same process, with the introduction of 10 wt% (with respect to total polymer content) of either A or CA terminated BCPs, respectively. From cryo-TEM measurements, polymersomal morphologies were evident in all three samples, with some influence of charge upon the size (Fig. 2A–C). Unmodified polymersomes, comprising neutral PEG-b-PDLLA, attained sizes of approximately 500 nm which was also the case for the A-modified samples; however, there was a significant difference in size for the CA-doped samples which were clearly smaller (ca. 200–300 nm). This was attributed to the difference in solvent interactions between A- and CA-containing polymers that would influence the kinetics of polymersome formation, thereby altering the average size of the nanostructures. A uniform membrane thickness of 15–20 nm was observed for all formulations.
:1 mixture of THF:dioxane prior to dropwise addition of water up to 50 vol% over 2 h (Fig. 1C).27 The formation of polymersomal nanostructures was indicated by the increased turbidity of the solution, without the formation of unwanted aggregates. A- or CA-modified polymersomes were formed using the same process, with the introduction of 10 wt% (with respect to total polymer content) of either A or CA terminated BCPs, respectively. From cryo-TEM measurements, polymersomal morphologies were evident in all three samples, with some influence of charge upon the size (Fig. 2A–C). Unmodified polymersomes, comprising neutral PEG-b-PDLLA, attained sizes of approximately 500 nm which was also the case for the A-modified samples; however, there was a significant difference in size for the CA-doped samples which were clearly smaller (ca. 200–300 nm). This was attributed to the difference in solvent interactions between A- and CA-containing polymers that would influence the kinetics of polymersome formation, thereby altering the average size of the nanostructures. A uniform membrane thickness of 15–20 nm was observed for all formulations.
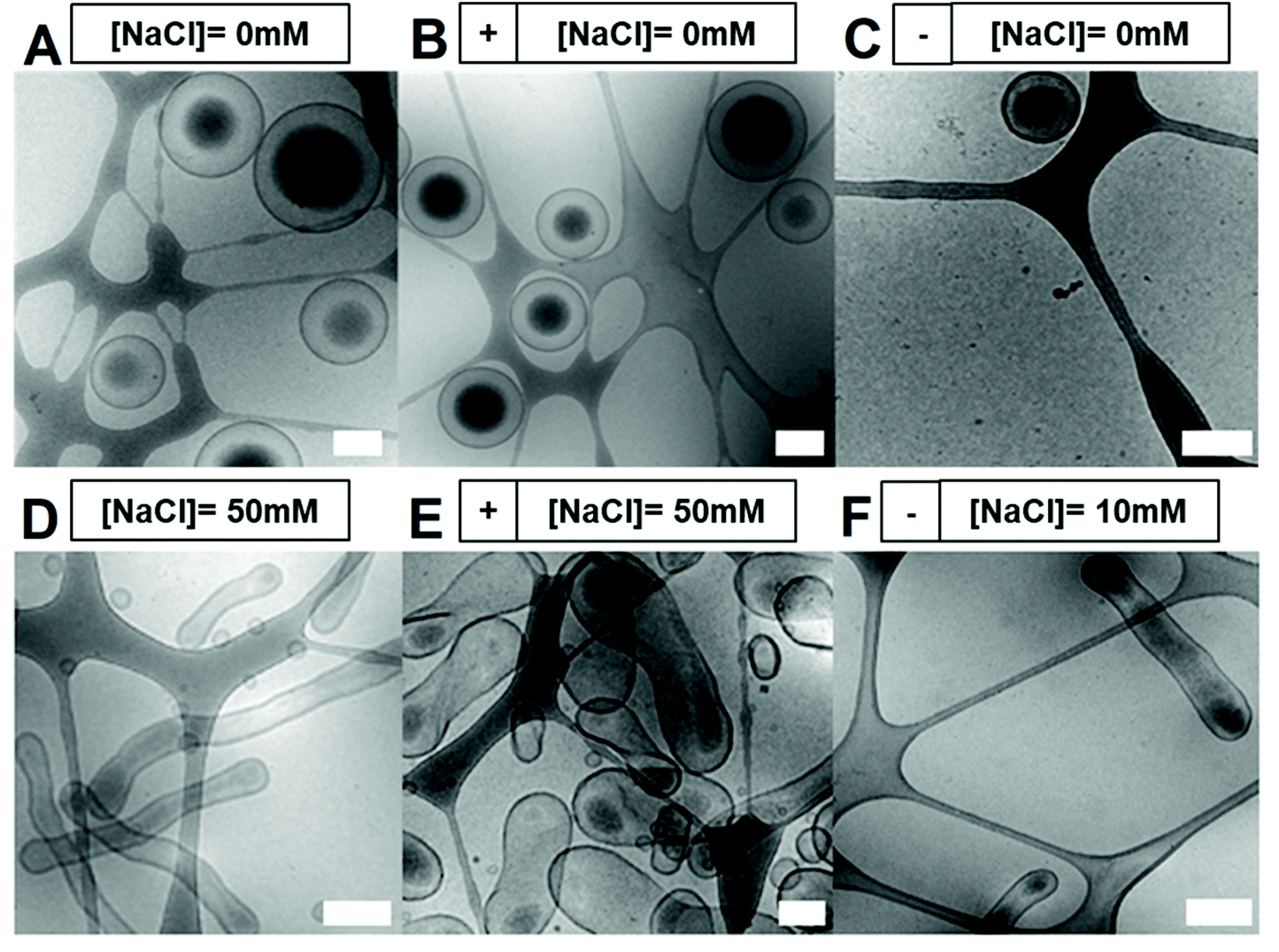 | ||
| Fig. 2 Cryo-TEM images of polymersomes with or without shape transformation. No shape transformation was observed when dialysing polymersomes against water (A = PEG-b-PDLLA/B = 10 wt% NH2-PEG-b-PDLLA/C = 10 wt% CO2H-PEG-b-PDLLA). Shape transformation was observed when dialysing polymersomes against 50 mM NaCl (D = PEG-b-PDLLA/E = 10 wt% NH2-PEG-b-PDLLA) or 10 mM NaCl (F = 10 wt% CO2H-PEG-b-PDLLA). All scale bars = 0.2 μm. | ||
Inducing shape transformations of PDLLA-based block copolymers from a spherical to elongated tubular morphology was achieved using osmotically induced deflation, as described previously.27 However, the introduction of charged polymers affected the shape transformation process markedly, showing different morphological changes under the same dialysis conditions (overnight dialysis against water for spheres, or salted water for elongated shapes). Unmodified and CA-modified particles formed well-defined elongated tubes, with an average size of 100 nm (width) × 1 μm (length), whereas A-modified charged variants were not as elongated and more closely resembled peanut shaped morphologies as opposed to tubes (Fig. 2D/E). During the shape-change process, an internal volume reduction of ca. 50% occurred, leading to an irreversible change in morphology owing to the semi-permeability characteristics of the membrane. We hypothesized that addition of charged groups at the surface of the polymersomes would induce distinct (lateral) electrostatic interactions, influencing the membrane flexibility, its spontaneous curvature and, as a result, the shape change pathway. Spontaneous membrane curvature (influenced by the packing of polymer chains in the membrane, their interactions with each other and surrounding solvent) is a controlling factor in the shape transformation of polymersomes into non-spherical morphologies.1,43
As the ionic strength of the dialysis medium will have an effect on the charge repulsion in the polymersome membrane, we set out to systematically vary the salt concentration during the dialysis process upon the shape transformations of the A- and CA-modified polymersome variants (Fig. 3). The use of 10 mM sodium chloride solution resulted in small bi-layered spheres for A-doped assemblies, but elongated tubes for CA-doped particles. Raising the salt concentration of the solution to 25 mM NaCl resulted in the formation of large peanut-like shapes for assemblies with amine surface, often shorter than the ones obtained with 50 mM NaCl. However, when the same concentration of salt was used for the CA-modified polymersomes, this yielded elongated tubes together with a small population of bowl-shaped structures (stomatocytes). At 100 mM NaCl, mainly stomatocytes were formed from the A-doped polymersomes. Stomatocytes were also seen for the polymersomes with CA surface, in equal amounts with the elongated tubes. These findings highlight the complex nature of polymersomal shape transformations, where oblate and prolate morphologies can be formed through controlled osmotic deflation and are significantly influenced by salt concentration, composition and the molecular characteristics of the bilayer.43 Based on these results, 10 mM and 50 mM NaCl were chosen for dialysis of CA- or A-modified and unmodified polymersomes, respectively, in order to controllably obtain tubular morphologies.
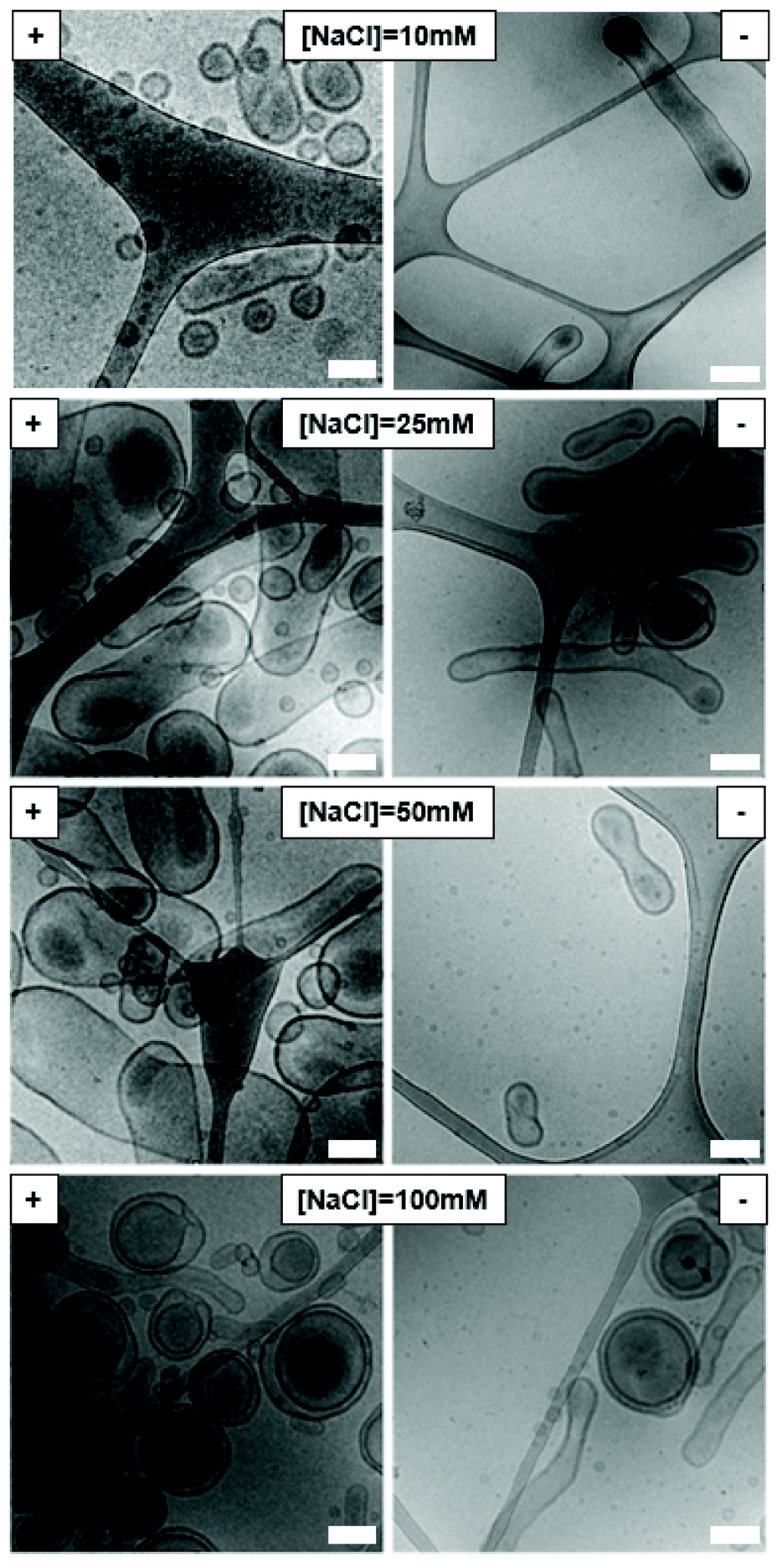 | ||
| Fig. 3 Cryo-TEM images of modified polymersomes after shape transformation using different salt concentrations. (−) 10 wt% CO2H-PEG-b-PDLLA and (+) 10 wt% NH2-PEG-b-PDLLA based polymersomes. All scale bars = 0.2 μm. | ||
Further physical characterisation of polymersomes and tubes was performed using DLS (Fig. 4). The average particle size was, in agreement with the cryo-TEM images, in the region of 400–500 nm for unmodified and A-modified polymersomes, whereas their CA-modified counterparts were smaller at around 200–300 nm. It should be noted that DLS is not best suited for non-spherical particles, as it is based on the Stokes–Einstein relationship, and this consideration should thus be taken into account during the analysis of the tubes.44 All particles possessed a negative zeta-potential due to the negative character of poly(ethylene glycol)-rich membranes in solution, which was diminished in case of the amine-containing polymersomes.45–47 No significant difference in zeta-potential was observed between unmodified and CA-modified polymersomes. Although the zeta-potential values did not yield major differences between the different polymersomal formulations in terms of surface charge, the presence of carboxylic acid and primary amine groups at the surface of polymersomes could still induce different interactions in a biological context.
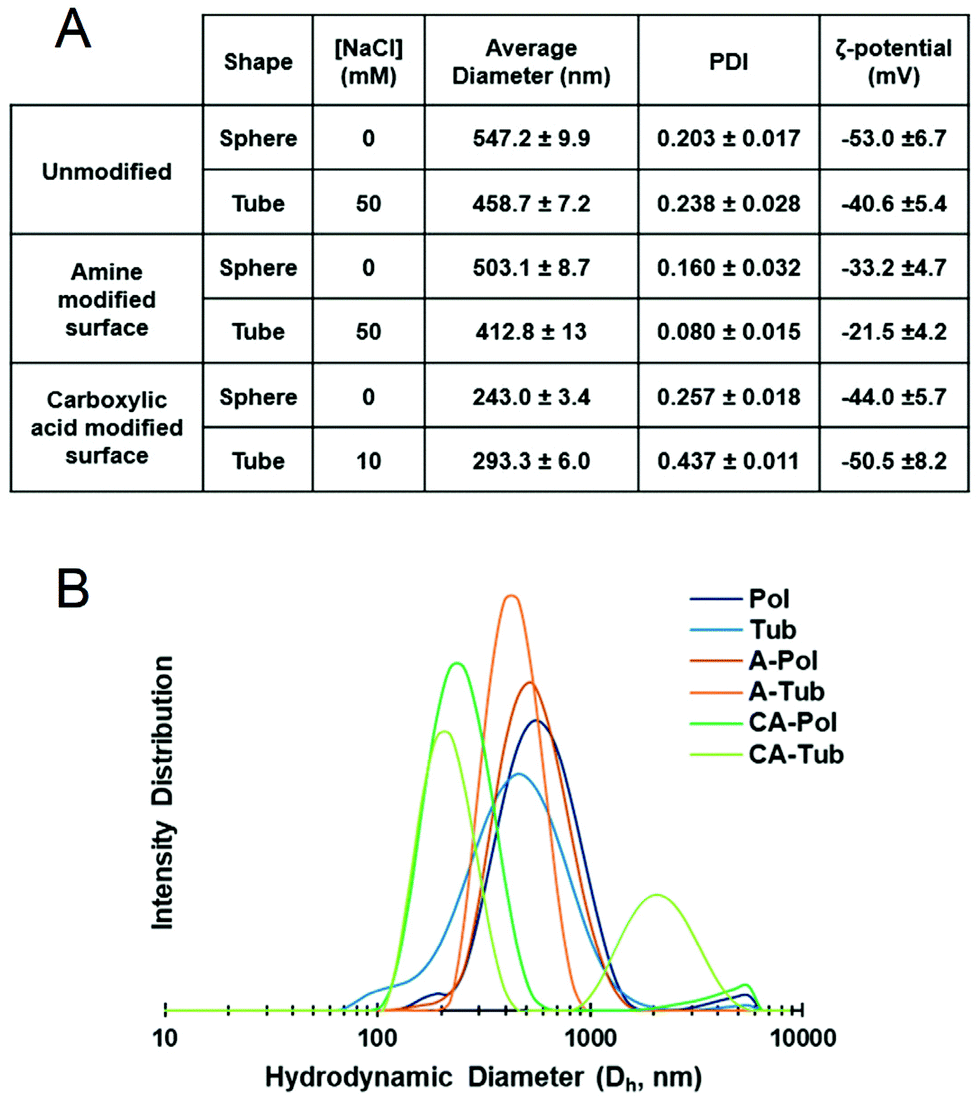 | ||
| Fig. 4 Physical data for polymersomal formulations. (A) Tabulated results of dynamic light scattering (DLS) measurements and (B) intensity distribution data (n = 3 for each sample, standard deviation reported as ±x in A). | ||
To evaluate the suitability of the differently shaped and charged particles for application in biology, we investigated their cytotoxicity. In keeping with our assertions regarding the biodegradable, and therefore biocompatible nature of PEG-b-PDLLA block copolymers, there was no evident toxicity towards human retinal (ARPE-19) cells at concentrations up to 1.25 mg mL−1 (with 24 h incubation), which was very high compared to other polymeric systems as presented in the literature where studies are usually limited to 0.5 mg mL−1 due to onset of toxicity (Fig. 5).48–51 Taken together, these results highlight the suitability of this platform for further studies in biomedical research to investigate the effect of both charge and shape on biological performance.
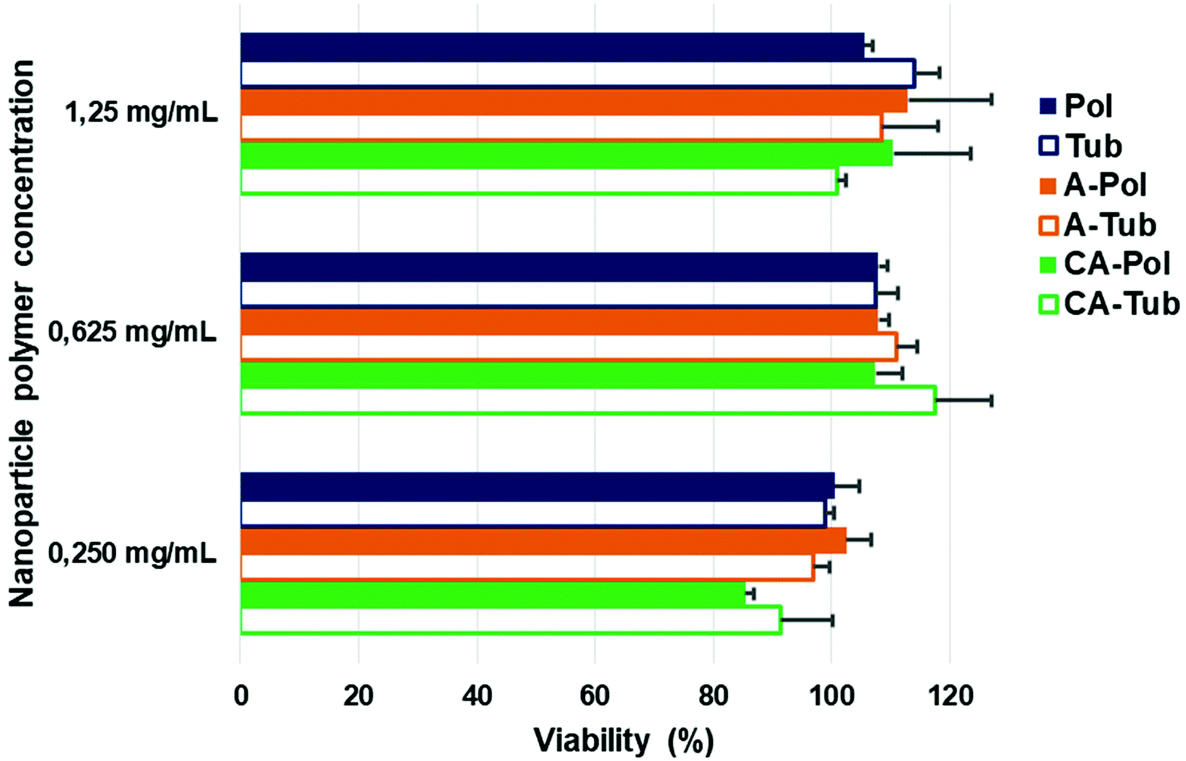 | ||
| Fig. 5 Toxicity studies of spherical and tubular polymersomes towards ARPE-19 cells after 24 h incubation (viability determined using AlamarBlue assays). | ||
Conclusion
In this work, we have explored the influence of formulation factors, such as salt concentration, upon the assembly of PEG-b-PDLLA copolymers bearing 10 wt% of either carboxylic acid or amine-modified derivatives. Using optimised conditions we have generated tubular and spherical polymersomes using copolymer blends and characterised their morphology using both cryo-TEM and DLS. Furthermore, neither spherical nor tubular polymersomes (with different surface characteristics) showed any toxicity to cells up to concentrations of 1.25 mg mL−1. This platform constitutes a robust technology that can be readily tuned in terms of morphology and composition to suit future applications in, for example, therapeutic drug delivery.Abbreviations
| A | Amine group |
| ARPE-19 | Retinal pigmented epithelium cell line 19 |
| BCPs | Block copolymers |
| CA | Carboxylic acid group |
| CryoTEM | Cryogenic transmission electron microscopy |
| DLS | Dynamic light scattering |
| PEG-b-PDLLA | Poly(ethylene glycol)-block-poly(D,L-lactide) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Imke Welzen-Pijpers and Alexander Mason for CryoTEM measurements. We thank the European Union's Horizon 2020 research and innovation programme Marie Sklodowska-Curie Innovative Training Networks Nanomed (no. 676137) for funding. We thank the Ser Cymru II programme for support of DSW; this project received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 663830.References
- I. A. B. Pijpers, L. K. E. A. Abdelmohsen, D. S. Williams and J. C. M. Van Hest, ACS Macro Lett., 2017, 6, 1217–1222 CrossRef CAS PubMed.
- A. Banerjee, J. Qi, R. Gogoi, J. Wong and S. Mitragotri, J. Controlled Release, 2016, 238, 176–185 CrossRef CAS PubMed.
- M. Beck-Broichsitter, J. Nicolas and P. Couvreur, Eur. J. Pharm. Biopharm., 2015, 97, 304–317 CrossRef CAS PubMed.
- Y. Wei, X. Gu, Y. Sun, F. Meng, G. Storm and Z. Zhong, J. Controlled Release, 2020, 319, 407–415 CrossRef CAS PubMed.
- Y. Altay, S. Cao, H. Che, L. K. E. A. Abdelmohsen and J. C. M. Van Hest, Biomacromolecules, 2019, 20, 4053–4064 CrossRef CAS PubMed.
- D. S. Williams, I. A. B. Pijpers, R. Ridolfo and J. C. M. van Hest, J. Controlled Release, 2017, 259, 29–39 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- N. P. Truong, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- V. Balasubramanian, B. Herranz-Blanco, P. V. Almeida, J. Hirvonen and H. A. Santos, Prog. Polym. Sci., 2016, 60, 51–85 CrossRef CAS.
- S. J. Roobol, T. A. Hartjes, J. A. Slotman, R. M. de Kruijff, G. Torrelo, T. E. Abraham, F. Bruchertseifer, A. Morgenstern, R. Kanaar, D. C. van Gent, A. B. Houtsmuller, A. G. Denkova, M. E. van Royen and J. Essers, Nanotheranostics, 2020, 4, 14–25 CrossRef PubMed.
- S. Pakizehkar, N. Ranji, A. N. Sohi and M. Sadeghizadeh, Polym. Adv. Technol., 2020, 31, 160–177 CrossRef CAS.
- H. Che, J. Zhu, S. Song, A. F. Mason, S. Cao, I. A. B. Pijpers, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Angew. Chem., 2019, 131, 13247–13252 CrossRef.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. Van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- M. Gai, J. Simon, I. Lieberwirth, V. Mailänder, S. Morsbach and K. Landfester, Polym. Chem., 2020, 11, 527–540 RSC.
- S. Varlas, R. Keogh, Y. Xie, S. L. Horswell, J. C. Foster and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 20234–20248 CrossRef CAS PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- E. Hinde, K. Thammasiraphop, H. T. T. Duong, J. Yeow, B. Karagoz, C. Boyer, J. J. Gooding and K. Gaus, Nat. Nanotechnol., 2017, 12, 81–89 CrossRef CAS PubMed.
- P. J. Photos, L. Bacakova, B. Discher, F. S. Bates and D. E. Discher, J. Controlled Release, 2003, 90, 323–334 CrossRef CAS PubMed.
- Y. Geng and D. E. Discher, J. Am. Chem. Soc., 2005, 127, 12780–12781 CrossRef CAS PubMed.
- L. Luo, F. Xu, H. Peng, Y. Luo, X. Tian, G. Battaglia, H. Zhang, Q. Gong, Z. Gu and K. Luo, J. Controlled Release, 2020, 318, 124–135 CrossRef CAS PubMed.
- J. L. Perry, K. P. Herlihy, M. E. Napier and J. M. Desimone, Acc. Chem. Res., 2011, 44, 990–998 CrossRef CAS PubMed.
- W. Wang, H. Guo, J. Gao and J. Yu, Chin. J. Mater. Res., 1998, 12, 628–631 CAS.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS PubMed.
- H. Che, L. N. J. de Windt, J. Zhu, I. A. B. Pijpers, A. F. Mason, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Chem. Commun., 2020, 1–4 Search PubMed.
- K. T. Kim, J. Zhu, S. A. Meeuwissen, J. J. L. M. Cornelissen, D. J. Pochan, R. J. M. Nolte and J. C. M. Van Hest, J. Am. Chem. Soc., 2010, 132, 12522–12524 CrossRef CAS PubMed.
- L. K. E. A. Abdelmohsen, D. S. Williams, J. Pille, S. G. Ozel, R. S. M. Rikken, D. A. Wilson and J. C. M. Van Hest, J. Am. Chem. Soc., 2016, 138, 9353–9356 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- X. Xing, W. Ma, X. Zhao, J. Wang, L. Yao, X. Jiang and Z. Wu, Langmuir, 2018, 34, 12583–12589 CrossRef CAS PubMed.
- X. J. Du, J. L. Wang, S. Iqbal, H. J. Li, Z. T. Cao, Y. C. Wang, J. Z. Du and J. Wang, Biomater. Sci., 2018, 6, 642–650 RSC.
- N. Hasan, J. Cao, J. Lee, S. P. Hlaing, M. A. Oshi, M. Naeem, M. H. Ki, B. L. Lee, Y. Jung and J. W. Yoo, Pharmaceutics, 2019, 11, 1–17 Search PubMed.
- S. Jeon, J. Clavadetscher, D. K. Lee, S. V. Chankeshwara, M. Bradley and W. S. Cho, Nanomaterials, 2018, 8, 1028 CrossRef PubMed.
- K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435–3446 CrossRef CAS PubMed.
- A. M. El Badawy, R. G. Silva, B. Morris, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2011, 45, 283–287 CrossRef CAS PubMed.
- A. K. Kohli and H. O. Alpar, Int. J. Pharm., 2004, 275, 13–17 CrossRef CAS PubMed.
- H. Koo, H. Moon, H. Han, J. H. Na, M. S. Huh, J. H. Park, S. J. Woo, K. H. Park, I. Chan Kwon, K. Kim and H. Kim, Biomaterials, 2012, 33, 3495–3493 Search PubMed.
- H. K. Na, H. Kim, J. G. Son, J. H. Lee, J. K. Kim, J. Park and T. G. Lee, Appl. Surf. Sci., 2019, 483, 1069–1080 CrossRef CAS.
- S. Matsusaka, Adv. Powder Technol., 2019, 30, 2851–2858 CrossRef CAS.
- Y. N. Lin, L. Su, J. Smolen, R. Li, Y. Song, H. Wang, M. Dong and K. L. Wooley, Mater. Chem. Front., 2018, 2, 2230–2238 RSC.
- M. Shen, H. Cai, X. Wang, X. Cao, K. Li, S. H. Wang, R. Guo, L. Zheng, G. Zhang and X. Shi, Nanotechnology, 2012, 23, 1–10 CrossRef PubMed.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914–922 CrossRef CAS PubMed.
- R. S. M. Rikken, H. Engelkamp, R. J. M. Nolte, J. C. Maan, J. C. M. Van Hest, D. A. Wilson and P. C. M. Christianen, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409–427 CrossRef CAS PubMed.
- S. Liufu, H. Xiao and Y. Li, Powder Technol., 2004, 145, 20–24 CrossRef CAS.
- Y. Song, A. Feng, Z. Liu and D. Li, Electrophoresis, 2019, 1–8 Search PubMed.
- A. Vila, H. Gill, O. McCallion and M. J. Alonso, J. Controlled Release, 2004, 98, 231–244 CrossRef CAS PubMed.
- Y. Liu, C. Xu, X. Fan, X. J. Loh, Y. L. Wu and Z. Li, Mater. Sci. Eng., C, 2020, 108, 1–9 Search PubMed.
- S. C. Kim, D. W. Kim, Y. H. Shim, J. S. Bang, H. S. Oh, S. W. Kim and M. H. Seo, J. Controlled Release, 2001, 72, 191–202 CrossRef CAS PubMed.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jérôme, J. Marchand-Brynaert, O. Feron and V. Préat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS PubMed.
- P. Nguyen-Tri, T.-O. Do and T. A. Nguyen, Smart Nanocontainers, ELSEVIER, 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00280a |
| This journal is © The Royal Society of Chemistry 2020 |