
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-orthogonal triazolinedione (TAD) crosslinked protein nanocapsules affect protein adsorption and cell interaction†
Marie-Luise
Frey
a,
Johanna
Simon
ab,
Maximilian
Brückner
ab,
Volker
Mailänder
 ba,
Svenja
Morsbach
a and
Katharina
Landfester
*a
ba,
Svenja
Morsbach
a and
Katharina
Landfester
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de
bDepartment of Dermatology, University Medical Center of the Johannes Gutenberg-University Mainz, Langenbeckstrasse 1, 55131 Mainz, Germany
First published on 11th May 2020
Abstract
Albumin-based protein nanocarriers have been widely exploited as drug delivery systems, since they show excellent degradability, low toxicity, but at the same time provide high loading capacity and relevant uptake into cells. For the formation of protein nanocapsules, bio-orthogonal reactions are important so that the material to be encapsulated is not affected by the shell formation. We show that protein nanocapsules with narrow size distributions and low protein adsorption upon contact with blood serum can be synthesized by inverse miniemulsion and interfacial crosslinking of the protein using triazolinediones (TADs) as powerful dienophiles and enophiles, which smoothly perform electrophilic aromatic substitutions and Diels–Alder reactions under ambient conditions. Additionally, we compare the physicochemical and biological properties of TAD-crosslinked protein nanocapsules with conventional diisocyanate crosslinked nanocapsules. We discovered that the type of crosslinker reaction controls the cell uptake of protein nanocapsules towards cancer cells and the interaction with blood proteins since the crosslinkers react with different functional groups within the proteins leading to different surfaces of the protein nanocarriers.
Introduction
The delivery of drugs with suitable nanocarriers is one of the main tasks in modern nanomedicine.1,2 Choosing the suitable carrier material for this purpose is crucial and can be very challenging: The material is supposed to protect the drug from degradation during circulation in the body, but at the same time, it should also enable to release the cargo within a desired time frame.3 Recently, protein nanocapsules were developed as drug delivery systems, since they show excellent biodegradability and biocompatibility, while providing at the same time high loading capacity and encapsulation efficiency.4 Furthermore, certain proteins hold specific functions, which can be preserved after their processing into a nanocarrier. For example, nanocapsules consisting of ovalbumin, a protein derived from chicken egg white, can react as an adjuvant boosting the immune system.5Nanocapsules are a specific type of drug delivery vehicles which can be produced by an interfacial polymerization reaction in miniemulsion.6 For this purpose, water-soluble proteins are dissolved in the dispersed aqueous phase of an inverse miniemulsion while molecules functioning as crosslinkers are added to the continuous organic phase. They provide a covalent crosslinking at the droplet interface yielding nano-sized capsules with typical sizes of 50–500 nm. As already reported, proteins can easily be crosslinked in an addition reaction by using 2,4-toluene diisocyanate (TDI), which leads to a dense polymeric nanocapsule shell. However, TDI reacts unspecifically with different nucleophilic groups, like amine-, hydroxyl- or thiol-functionalities, which are mostly present in the side chains of the amino acids of the protein.4,7
In recent studies there have been several approaches to supply more specificity in the composition of the nanomaterial by using bioorthogonal chemistry.8,9 For example, Piradashvili et al. presented a method using a tetrazole–ene cycloaddition to synthesize protein nanocarriers.10 However, in the reported approach, UV irradiation for interfacial crosslinking is necessary, which may also provoke other photochemical reactions within the protein, destroying its native function.11 Strain-promoted azide–alkyne cycloadditions (SPAAC) become increasingly popular and provide high selectivity.12 However, the corresponding groups need to be introduced into the biomolecule before the click reaction, which may not inevitably occur in a bioorthogonal manner. Other bioorthogonal bioconjugation methods like e.g. hydrazone or oxime ligations are strongly dependent on the pH value and can be reversible in aqueous medium.8,13
Herein we report the synthesis of protein nanocapsules that were crosslinked with a 4-phenyl-triazolinedione (TAD) derivative, which specifically reacts with amino acids with delocalized π-electron systems.14–17 The ability of TAD to act as a strong dienophile in a Diels–Alder reaction has already been shown almost 60 years ago.18 Recent studies performed by Du Prez and coworkers again showed its versatility in current applications in polymer science.19–21 We show that particularly TAD's extraordinarily fast and specific reaction behavior leads to a remarkable responsiveness towards proteins in aqueous emulsion, forming nanocapsules with narrow size distribution, in spite of the reported sensitivity of TAD in regards to hydrolysis and reduction to the corresponding urazole derivative.22–25
TAD-crosslinked protein nanocapsules were synthesized with bovine serum albumin (BSA), human serum albumin (HSA) or ovalbumin (OVA) via an inverse miniemulsion procedure. Additionally, nanocapsules using only the amino acid tryptophan instead of full proteins were prepared. In all cases, the protein nanocapsules showed low plasma protein adsorption. The significant differences in the cell uptake of the TAD-crosslinked protein nanocapsules and TDI-crosslinked nanocapsules can be explained by a different protein conformation at the nanocapsule surfaces.
Experimental section
Materials
All chemicals were used without further purification. Bovine serum albumin (BSA), human serum albumin (HSA) and ovalbumin (OVA) were purchased from Sigma Aldrich, as well as 2,4-toluene diisocyanate (TDI, Mw = 174.16 g mol−1), 4,4′-methylenebis(phenylisocyanate) (Mw = 250.25 g mol−1), polyvinylalcohol (PVA – Mowiol 4–88), L-tryptophan (Mw = 204.23 g mol−1, HPLC grade), dry toluene, DMSO and sodium dodecyl sulfate (SDS).Ethyl carbazate (Mw = 104.11 g mol−1), dichloromethane (HPLC grade) and NaCl were purchased from VWR. Tetramethylammoniumhydroxid (TMAOH – 1.0 M, aqueous solution) was purchased from Alfa Aesar. The block copolymer poly((ethylene-co-butylene)-b-(ethylene oxide) P(E/B)-b-EO) used as surfactant was synthesized according to previously published procedures from Schlaad et al.26
Human blood serum
Human blood serum was collected at the Transfusion Center of the University Clinic of Mainz (Germany) from ten healthy donors after physical examination. All experiments were performed in accordance with the Guidelines of the University Medical Center Mainz and in accordance with the Declaration of Helsinki, and Experiments were approved by the ethics committee “Landesärztekammer Rheinland-Pfalz” (837.439.12 (8540-F)) which is the institutional ethics committee for the University Medical Center Mainz. Informed consents were obtained from human participants of this study.All serum batches were pooled and stored at −20 °C.
Methods
For ultrasonication a Branson Sonifier W-450-Digital was used with a 1/2′ tip. For ultrasonication in a sonication bath a Bandelin Sonorex RK52H from Bandelin electronic Berlin was used.Scanning electron microscopy (SEM) was performed by placing a drop of diluted nanocapsule dispersion onto a silica wafer and drying under ambient conditions. SEM measurements were operated on a 1530 Gemini LEO (Zeiss) field emission microscope.
Transmission electron microscopy (TEM) was performed by placing a drop of diluted nanocapsule dispersion onto a 300-mesh carbon-coated copper grid and drying under ambient conditions. TEM measurements were operated on a Jeol 1400 transmission electron microscope.
Average sizes and size distributions were measured by dynamic light scattering (DLS) at 20 °C using an instrument from ALV GmbH consisting of a goniometer and an ALV-5000 multiple-tau full digital correlator with 320 channels. As a light source a helium–neon laser (JDS Uniphase with a single mode intensity of 25 mW operating at a laser wavelength of 632.8 nm) was used. Measurements were performed in quartz light scattering cuvettes (inner diameter 18 mm, Hellma, Müllheim) and dispersions were filtered dust-free prior to the measurements.
Zeta potential measurements were performed by diluting the nanocapsule dispersions in potassium chloride solution (10−3 M, 25 °C, pH 6.8) with a Malvern Zeta sizer (Malvern Instruments, UK).
Fluorescence intensity measurements were operated in 96-well plates on the Infinite M1000 plate reader from Tecan, Switzerland.
HeLa cells (obtained from ATCC® CCL-2™) were expanded after arrival, a master cell bank was frozen down and cell stocks were stored in liquid nitrogen. Cells were used for 3 months for experiments, then the old batch was discarded. A new vial was thawed. Old and new batch from the master cell bank of HeLa cells were tested to be Mycoplasma free using Microsart® AMP Mycoplasma Test Kit (Sartorius, Germany) according to the manufacturer's instruction. By these measures we ensured that all experiments were as close as possible to the original vial obtained from ATCC and that the cell cultures were free from mycoplasma. HeLa cells were cultured in Dulbecco Modified Eagle Medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, 100 mg mL−1 streptomycin and 2 mM glutamine (all Thermo Fisher, Germany). Cells were split about two times a week at 80–90% confluency. For cell uptake analysis, cells were detached with 2.5% trypsin (Gibco, Germany) and seeded out in 24-well plates (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) in cell culture medium with 10% FBS. After overnight incubation, the medium was changed to serum-free medium.
000 cells per well) in cell culture medium with 10% FBS. After overnight incubation, the medium was changed to serum-free medium.
For flow cytometry analysis, cells were detached and measurements were performed on an Attune NxT flow cytometer (Thermo Fisher, USA).
Confocal laser scanning microscopy (cLSM) was conducted on the LSM SP5 STED Leica Laser Scanning Confocal Microscope (Leica, Germany), composed of an inverse fluorescence microscope DMI 6000CS equipped with a multi-laser combination using a HCX PL APO CS 63 × 1.4 oil objective.
Synthesis of MDI-TAD
This procedure was adopted from a previously published procedure from Du Prez and coworkers.23The yellow solid was filtered off and the filtrate was washed with 200 mL dichloromethane. The dichloromethane was removed under reduced pressure and the product was dried and stored at 4 °C.
Yield: 750.5 mg (2.071 mmol, 76%) pink solid. 1H-NMR (250 MHz, d6-DMSO): δ (ppm) = 7.53–7.29 (m, 9H), 4.10 (s, 2H).
Nanocapsule synthesis
The nanocapsules were prepared adopting to previously published procedures from Piradashvili et al. using an inverse miniemulsion process.450 mg of protein or L-tryptophan was dissolved in 500 μL of demineralized water and 8.0 mg of NaCl was added to the mixture. In the case of L-tryptophan the pH was adjusted to pH 11 with NaOH. In the case of TAD-TAD nanocapsules just NaCl was added into the aqueous phase. 35.0 mg of P((E/B)-b-EO) were dissolved in 7.5 g of toluene and the mixture was added at once to the stirring aqueous solution. This mixture was pre-emulsified by vigorously stirring for approx. 30 min. Afterwards, the pre-emulsion was subjected to ultrasound for 3 min (70%, 20 s pulse, 10 s pause) and stirred shortly after at 700 rpm at room temperature. A solution of 10.0 mg P((E/B)-b-EO) in 5.0 g of toluene was prepared and 88 mg of MDI-TAD were added to the solution. To improve the solubility of MDI-TAD in the mixture, it was homogenized in an ultrasound bath at 40 °C for 5 min. The MDI-TAD solution was then added dropwise over a period of one minute to the stirring miniemulsion and the mixture stirred for another 24 h at room temperature.
Transfer of TAD nanocapsules into aqueous medium
5 mL of nanocapsule dispersion were split in three Eppendorf tubes (1.667 mL each) and centrifuged at 4000 rpm for 30 min. The supernatant was replaced against the same volume of toluene and the pellet was redispersed in the fresh solvent. This process was repeated one more time. The first supernatant was distributed into fresh Eppendorf tubes and also centrifuged once more. The pellets generated from the centrifugation steps were concentrated together with 500 μL of fresh toluene and redispersed in 5 mL of aqueous SDS solution (0.1 wt%) by shaking in a sonication bath while the nanocapsule dispersion was added in 100 μL steps. The sonication process was performed for 5 min and afterwards the dispersion was placed in a cooling bath and additionally sonified (Branson Digital Sonifier W-450-D) for 30 s, 10%, 10 s pulse, 10 s pause. Afterwards the dispersion was placed on a stirring plate and the vial was stirred open over night at 1000 rpm to evaporate the solvent.Tryptophan-TAD nanocapsules with encapsulated magnetite-nanoparticles
50 mg of L-tryptophan were dissolved in 350 μL of demineralized water and 50 μL of conc. NaOH-solution (pH 11) and 8.0 mg of NaCl as well as 100 μL of an aqueous magnetite-nanoparticle solution (5 wt%) was added to the mixture. 35.0 mg of P((E/B)-b-EO) were dissolved in 7.5 g of toluene and the mixture was added at once to the stirring aqueous solution. This mixture was pre-emulsified by vigorous stirring for about 30 min. Afterwards, the pre-emulsion was subjected to ultrasound for 3 min (70%, 20 s pulse, 10 s pause) and stirred shortly after at 700 rpm at room temperature. A solution of 10.0 mg P((E/B)-b-EO) in 5.0 g of toluene was prepared and 88 mg of MDI-TAD were added to the solution. To improve the solubility of MDI-TAD in the mixture, it was homogenized in an ultrasound bath at 40 °C for 5 min. The MDI-TAD solution was then added dropwise over a period of one minute to the stirring miniemulsion and the mixture stirred for another 24 h at room temperature.Transfer of magnetite (Fe3O4) nanoparticles into aqueous medium
This procedure was adopted from a previously published procedure from Bannwarth et al.2750 mg of magnetite nanoparticles (stabilized with oleic acid) were placed in a screwcap vial and 5 mL of a TMAOH solution (1 M) was added. The mixture was placed in an ultrasound bath for one hour. Afterwards, the nanoparticles were centrifuged at 20000 rpm for 40 min and the supernatant was removed. The pellet was resuspended in a small amount of a 0.1 wt% TMAOH solution and afterwards the whole dispersion was added to 50 mL of a 0.1 wt% TMAOH solution while placing the whole solution into an ultrasound bath. 50 mg of carboxymethylated PVA (CPVA) were dissolved in 50 mL of hot water and slowly added to the redispersed nanoparticles. The whole mixture was homogenized in an ultrasound bath for 5 min and stirred overnight. On the next day, the nanoparticles were centrifuged at 14000 rpm for 30 min, the supernatant was removed and the pellet was redispersed in 1 mL of demineralized water.
Cellular uptake and protein corona analysis
For cellular uptake experiments, the nanocapsules were washed several times by centrifugation to remove excess SDS. The SDS concentration in the dispersion was checked by a colorimetric assay.28 Then protein nanocapsules (75 μg mL−1) were added to cells in serum-free cell culture medium (−HS) or cell culture medium containing 10% human serum (+HS) for 2 h or 24 h at 37 °C.Purification of protein corona
Nanocapsules (0.05 m2) were incubated with 1 mL of human serum at 37 °C for 1 h. Protein coated nanocapsules were centrifuged (20000g, 30 min, 4 °C), the supernatant was removed and the pellet was washed with 1 mL PBS solution. To fully remove all loosely bound proteins, this step was repeated three times. Finally, the strongly attached corona proteins were desorbed from the nanocapsules’ surface with an aqueous solution containing 2 wt% SDS supplemented with 62.5 mM Tris-HCl. The sample was incubated for 5 min at 95 °C. In a last step, the dispersion was centrifuged and the supernatant containing the corona proteins was isolated. The protein concentration was analyzed using a Pierce 660 nm Protein Assay (Thermo Fisher, Germany) according to manufacturer's instructions.
SDS-PAGE of the protein corona
Corona proteins (2 μg in 26 μL) were incubated with sample buffer (10 μL) and reducing agent (4 μL) for 10 min at 70 °C and applied onto a 10% NuPAGE Bis-Tris Gel. The SDS-PAGE was run at 120 mV for 1 h. The protein bands were visualized by the SilverQuest Silver Staining Kit according to the manufacturer's instructions. All components were obtained from Thermo Fisher, Germany.In solution digestion
In a first step, SDS was removed via Pierce detergent removal columns (Thermo Fisher, Germany) according to the manufacturer's instruction. Tryptic in solution digestion was carried out according to former reports.29,30 Proteins were precipitated using a ProteoExtract protein precipitation kit (Merck Millipore, Germany) for at least 2 h. After centrifugation, the protein pellet was resuspended in RapiGest SF (Waters, USA) dissolved in 50 mM ammonium bicarbonate (Sigma Aldrich, Germany). The samples were incubated at 80 °C for 15 min. For reduction, dithiothreitol (5 mM, Sigma Aldrich, Germany) was added to the solution for 45 min at 56 °C and for alkylation iodoacetoamide (15 mM, Sigma Aldrich, Germany) was added for 1 h in the dark. Finally, a protein:trypsin ratio of 50:1 was used for tryptic digestion (incubation time 16 h at 37 °C). The digestion was stopped with hydrochloric acid (Sigma Aldrich, Germany) and the resulting peptide sample was centrifuged (14000g, 15 min, 4 °C) to remove degradation products of RapiGest SF.
Liquid chromatography-mass spectrometry (LC-MS)
Peptide samples were diluted with 0.1% formic acid and 50 fmol of Hi3 EColi Standard (Waters, USA) was added for absolute protein quantification. LC-MS measurements were performed with a nanoACQUITY UPLC system coupled to a Synapt G2-Si mass spectrometer (Waters, USA). A nanoLockSpray source was used for electrospray ionization (ESI) in positive ion mode and the mass spectrometer was operated in resolution mode performing data-independent experiments (MSE). Data was processed with MassLynx 4.1 and all proteins were identified with Progenesis QI. A reviewed human data base was downloaded from Uniprot.The absolute amount of each protein was calculated based on the TOP3/Hi3 approach.31 A list of all identified proteins and the absolute amount in fmol or relative amount in % is given in a separated excel document.
Visualization of intracellular localization by cLSM
To visualize the cellular uptake behavior of the protein nanocapsules in HeLa cells, confocal laser scanning microscopy (cLSM) images were taken. Nanocapsules containing the dye SR101 were excited with a 561 nm laser and detected with an emission filter at 569–624 nm. The cell membrane was stained with CellMask DeepRed (stock solution: 5 mg mL−1, Thermo Fisher), excited with a 633 nm laser and detected with an emission filter at 680–695 nm. For the visualization of intracellular localization, 5 × 104 cells in 200 μL DMEM with 10% FBS were seeded out into 8-well μ-slides with a glass bottom (ibidi). After an overnight incubation at 37 °C and 5% CO2 in the incubator, the cells were treated with the respective sample in medium with 10% FBS. After the 2 h and 24 h incubation with a nanocapsule concentration of 75 μg mL−1, the wells were washed with PBS to remove unbound nanocapsules. Afterwards, the cells were directly imaged. Cell membrane staining was conducted shortly before the cLSM analysis for 5 min in the dark using CellMask Deep Red (Dilution of stock solution: 1:1000 in PBS).
Cell viability assay
HeLa cells were seeded out into 96-well plates (Greiner Bio-One, Austria) with a cell number of 10 × 103 cells per well in 100 μL DMEM with 10% FBS. After overnight incubation at 37 °C and 5% CO2, the medium was removed. The respective protein nanocapsules were prepared in 10% FBS supplemented DMEM and added in a volume of 100 μL to the cells (75 μg mL−1 final concentration). Water (Ampuwa®) was used as positive control. All samples were handled in triplicates. HeLa cells were incubated at 37 °C and 5% CO2 for 2 and 24 h before conducting the viability assay. A CellTiter-Glo® Luminescent Cell Viability Assay (Promega, Germany) was performed according to the manufacturer's instructions. The luminescence signal was measured with an Infinite M1000 plate reader (Tecan, Switzerland).Results and discussion
Protein nanocapsules were synthesized by an interfacial bio-orthogonal crosslinking reaction in inversion miniemulsion adding MDI-TAD as difunctional TAD-derivative to the organic continuous phase (see Fig. 1). The aqueous droplet phase either consists of the protein BSA (bovine serum albumin) or the amino acid tryptophan (Trp). As control reaction, also no further reactant was added allowing TAD-TAD reactions (see Fig. 1A). Upon reaction, the intensive pink color of the MDI-TAD molecules vanished within a few minutes and the emulsion turned slightly yellow, indicating conversion of MDI-TAD. After reaction, the sizes of the formed nanocapsules as revealed by dynamic light scattering (DLS) are about 120 nm in diameter for Trp-TAD and 240 nm for BSA-TAD, with a narrow size distribution for Trp-TAD nanocapsules (PDI of 0.100, see Table 1 and Fig. S1†). The morphology analyzed via scanning electron microscopy (SEM) showed a well-defined morphology for Trp-TAD nanocapsules as well as for BSA-TAD nanocapsules (Fig. 1B, middle and right panel). Pure TAD crosslinking lead to a more undefined and film-like structure (Fig. 1B, left panel). The collapse of the nanocapsules occurs due to the drying process in the sample preparation and the vacuum used during the measurement.| NC-sample | Average diameter in toluenea | Average diameter in watera | PDI in toluene (90°) | ξ-potential/mV |
|---|---|---|---|---|
| a Determined by multiangle DLS (at scattering angles of 30 to 150°). | ||||
| TAD-TAD | 84 ± 8 nm | 155 ± 16 nm | 0.220 | −37 ± 5 |
| Trp-TAD | 119 ± 12 nm | 163 ± 16 nm | 0.100 | −41 ± 5 |
| BSA-TAD | 238 ± 24 nm | 350 ± 40 nm | 0.171 | — |
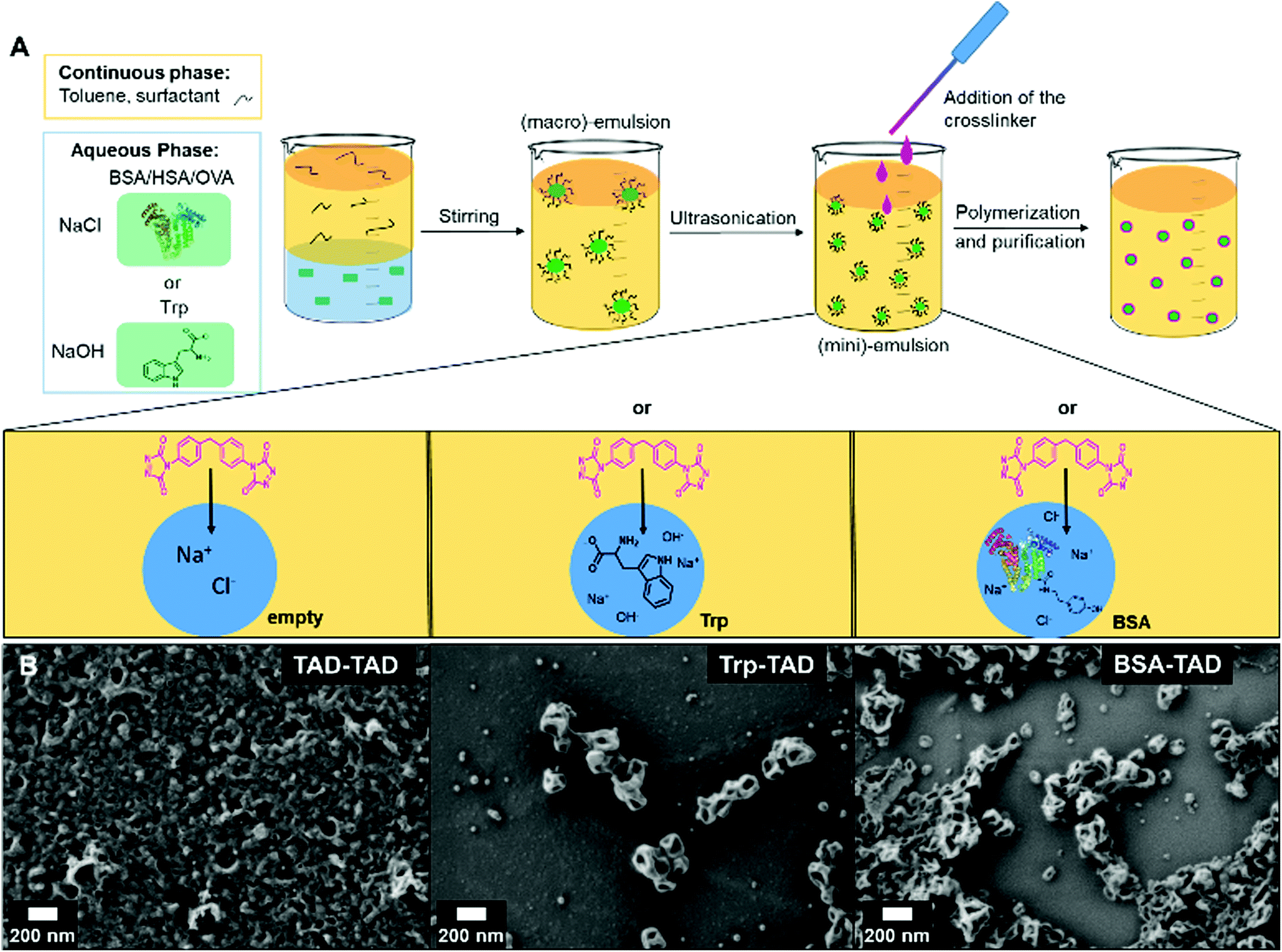 | ||
| Fig. 1 (A) Scheme of the protein nanocapsule synthesis by an inverse miniemulsion procedure and MDI-TAD as crosslinker. Trp-TAD (tryptophan) and BSA-TAD nanocapsules, as control pure TAD nanocapsules were used; (B) scanning electron microscopy (SEM) images of the TAD nanocapsules. | ||
Consistent with Fig. 1, pure TAD nanocapsules appear smaller in size (84 nm) but reveal a significantly higher polydispersity index (PDI of 0.220, see Table 1). After redispersion in water, the sizes increased. This change occurs by the formation of a hydration shell or by swelling of the polymer in water. The measured ξ-potentials were negative in all cases.
As analyzed by nuclear magnetic resonance (NMR) spectroscopy, tryptophan and TAD successfully reacted at the interface of the miniemulsion (Fig. 2), as indicated by occurrence of the TAD indole reaction. The marked signals a, a′, b and b′ (green box Fig. 2) can be attributed to the indole-ring of the added tryptophan as TAD is able to perform an electrophilic aromatic substitution at the indole ring, leading to bio-orthogonally formed Trp-TAD adducts.15,20 In comparison to this specific reaction, the product formed in TAD-TAD emulsion (red spectrum) is less defined as demonstrated by a variety of signals at 8 and 10 ppm visible, indicating hydrolysis products occurring due to the handling of TAD in an aqueous environment. In turn, in the Trp-TAD emulsion these signals were missing. The signal at a chemical shift of 10.5 ppm in the red spectrum (marked with c) is specific for the proton of the NH-group of the urazole pre-species, which forms by means of decomposition of TAD molecules as depicted in the red box (Fig. 2). In a TAD-TAD emulsion, the TAD crosslinker is able to hydrolyze in aqueous environment to an amine species, which is then able to react with remaining TAD molecules.32 These observations point to the fact that Trp-TAD emulsions undergo a more specific reaction than TAD-TAD emulsions and therefore react with the offered amino acid in a bio-orthogonal way.
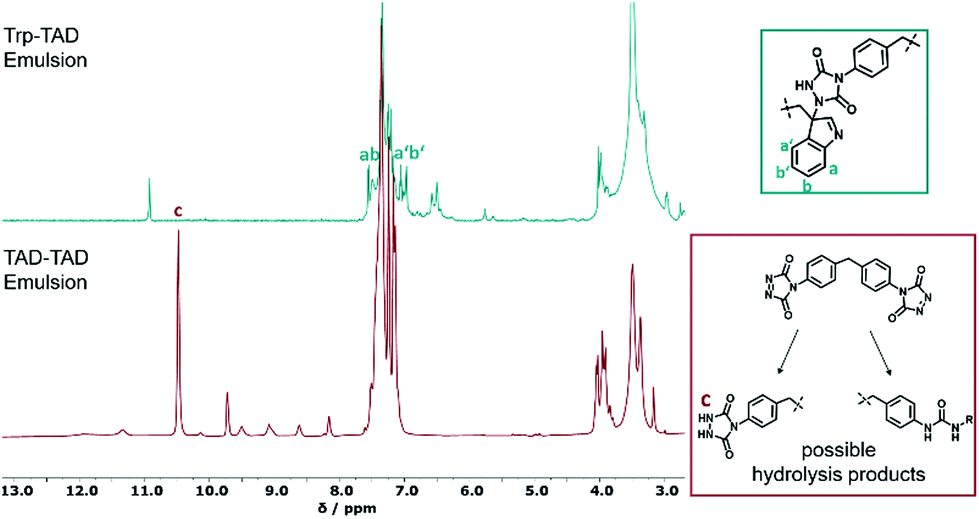 | ||
| Fig. 2 1H-NMR spectra of a Trp-TAD emulsion (green) in comparison to a TAD-TAD emulsion (red) in d6-DMSO after reaction. Next to the spectra, possible products formed during the reaction are shown (for full spectra see Fig. S2†). | ||
Encapsulation of magnetite (Fe3O4) nanoparticles into TAD-crosslinked BSA and Trp nanocapsules
For imaging purposes, magnetite (Fe3O4) nanoparticles were encapsulated into the BSA- and Trp-TAD nanocapsules (Fig. 3). These capsules were then again characterized by multiangle dynamic light scattering as well as transmission electron microscopy (TEM) to verify the presence of magnetite.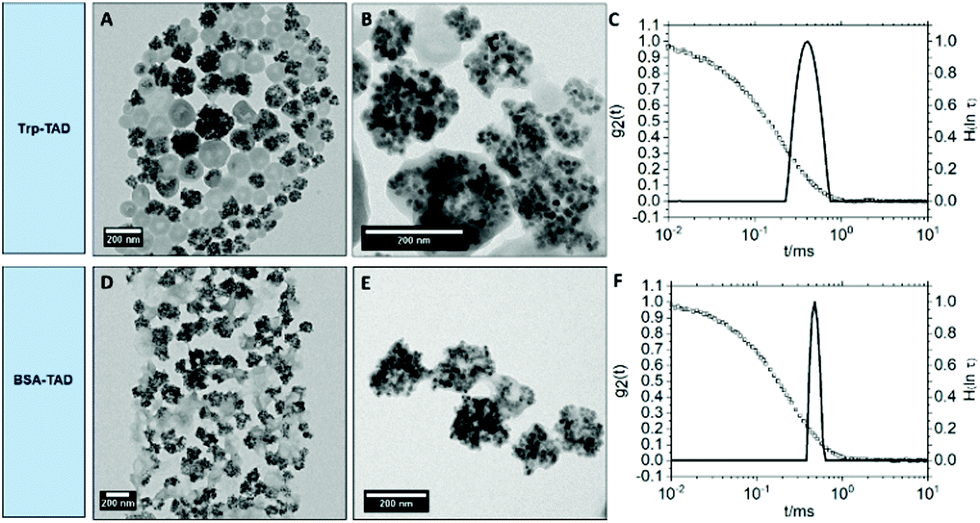 | ||
| Fig. 3 TEM micrographs of Trp-TAD nanocapsules (A and B) and BSA-TAD nanocapsules (D and E) with encapsulated magnetite nanoparticles in toluene and size distribution of the different nanocapsules at an exemplary scattering angle of 90° in toluene represented by the distribution of relaxation times H(lnτ) (solid lines) together with the respective autocorrelation functions g2(t) (filled squares) (C and F). | ||
The TEM micrographs in Fig. 3 show that the Fe3O4 nanoparticles were encapsulated into Trp-TAD nanocapsules (A and B) as well as into BSA-TAD nanocapsules (D and E) with a high number of particles per capsule. They can be separated from empty nanocapsules by using a neodym magnet. The encapsulation proved a high loading capacity of the TAD crosslinked nanocapsules. Both nanocapsule types slightly changed their shape due to encapsulation of magnetite nanoparticles. Light scattering measurements revealed narrow size distributions also for nanocapsules with cargo (Fig. 3 and Table 2).
BSA-, HSA-, OVA-TAD nanocapsules
Proteins such as HSA and OVA have been extensively exploited for the use in different nanocarrier preparations and are widely applied in therapeutic materials.33We therefore synthesized TAD crosslinked nanocapsules with these proteins and compared them to TDI crosslinked protein nanocapsules. DLS and ξ-potential analysis revealed good consistency in the characteristics of the different nanocapsules (Table 3).
| NC-sample | Average diameter in toluenea | Average diameter in watera | PDI in toluene (90°) | ξ–potential/mV |
|---|---|---|---|---|
| a Determined by DLS at 90°. | ||||
| BSA-TAD | 115 ± 55 nm | 193 ± 150 nm | 0.182 | −34 ± 5 |
| HSA-TAD | 145 ± 69 nm | 192 ± 91 nm | 0.144 | −25 ± 5 |
| OVA-TAD | 189 ± 82 nm | 214 ± 135 nm | 0.334 | −38 ± 5 |
| BSA-TDI | 122 ± 53 nm | 199 ± 75 nm | 0.144 | −27 ± 5 |
| HSA-TDI | 152 ± 51 nm | 178 ± 46 nm | 0.141 | −16 ± 5 |
| OVA-TDI | 108 ± 45 nm | 200 ± 54 nm | 0.161 | −30 ± 5 |
However, OVA-TAD nanocapsules had the largest nanocapsule diameters in toluene as well as in water and showed a broader size distribution in comparison to nanocapsules with other proteins. The ξ-potential of all nanocapsules was negative.
Cellular interaction of protein nanocapsules with cancer cells
For cellular experiments, nanocapsules consisting of a shell of BSA, OVA or HSA were crosslinked with TDI or TAD and the fluorescent dye SR101 (sulforhodamine 101) were prepared. HeLa cells were exposed to protein nanocapsules (75 μg mL−1) for 2 h or 24 h. The amount of fluorescent positive cells was determined via flow cytometry and visualized by cLSM (Fig. S7†). The cell viability was analyzed by CellTiter-Glo® Luminescent Cell Viability Assay and was found to be nearly 100% for every sample and both incubation times (Fig. S8†). In addition, it is known that the behavior of a nanocarrier in a physiological environment (e.g. blood) is strongly dominated by the surface interaction with blood proteins forming a protein corona, which further affects the biodistribution, toxicity and cellular uptake of the nanocarrier.34–36 To explore the effect of protein corona formation for the here presented protein nanocapsules, cellular uptake experiments were either performed under serum free conditions (−HS) or in cell culture medium supplemented with human serum (+HS). The results are summarized in Fig. 4. | ||
| Fig. 4 Cellular uptake of protein nanocapsules crosslinked with TDI or TAD. HeLa cells were incubated with BSA, OVA and HSA nanocapsules at 75 μg mL−1 for 2 h (a) or 24 h (b) under serum free conditions (−HS) or in cell culture medium with 10% human serum (+HS). | ||
We found that the cellular interaction of all protein nanocapsules with HeLa cells was strongly influenced by the crosslinker type (TDI vs. TAD). Under serum free conditions, independently of the protein type (BSA vs. OVA vs. HSA), nanocapsules crosslinked with TAD interacted stronger with HeLa cells compared to nanocapsules crosslinked with TDI in a time dependent manner (Fig. 4). After 2 h of incubation, up to only 10% HeLa cells showed significant uptake of TDI-crosslinked protein nanocapsules, while for TAD-cross-linked nanocapsules it was more than twice as much (20–30%). This difference in uptake was again observed when the cells were exposed to the nanocapsules for 24 h. In strong contrast to that, the cellular interaction was strongly reduced in cell culture medium with human serum compared to serum free condition. Here, we detected minor differences between the TDI and TAD crosslinked nanocapsules.
Impact of protein structure on cellular uptake of TAD and TDI crosslinked nanocapsules
One explanation for the difference in the cellular uptake behaviour under serum free conditions could be related to a difference in the protein structure for the nanocapsules crosslinked with TDI vs. TAD. As an example, Fleischer et al.37 studied the cellular interaction of BSA coated polystyrene nanoparticles and showed that the structure of BSA was preserved once it adsorbed onto negatively charged nanoparticles. In contrast, upon adsorption of BSA on positively charged nanoparticles, BSA was denatured.Changes in the secondary structure of BSA eventually had a major impact on cellular binding. Negatively charged nanoparticles coated with native BSA especially bound to albumin receptors present on the cell surface of monkey kidney epithelial cells (BS-C-1), whereas positively charged nanoparticles coated with denatured BSA interacted with the scavenger receptors. This highlights the distinct role of the protein structure and cell surface receptor interaction. Thus, the protein structure of the here presented protein nanocapsules could be different depending on the chosen crosslinker as also different amino acid side chains are involved in the crosslinking process.
HSA and BSA are both carrier proteins and have a structural homology of 76%. OVA belongs to the superfamily of the serpins, and has therefore a different structure, size and also a different function.38,39 The isocyanate containing crosslinker TDI favorably reacts with mostly nucleophilic groups, like amino groups, which are well accessible through the lysine residues of the proteins. TAD on the other hand reacts with aromatic amino acids like tyrosine and tryptophan residues, which are located more in the interior of the protein.
In Fig. 5, the crystal structures of BSA, HSA and OVA are compared to each other and the corresponding amino acids, which react with the different crosslinkers are highlighted.
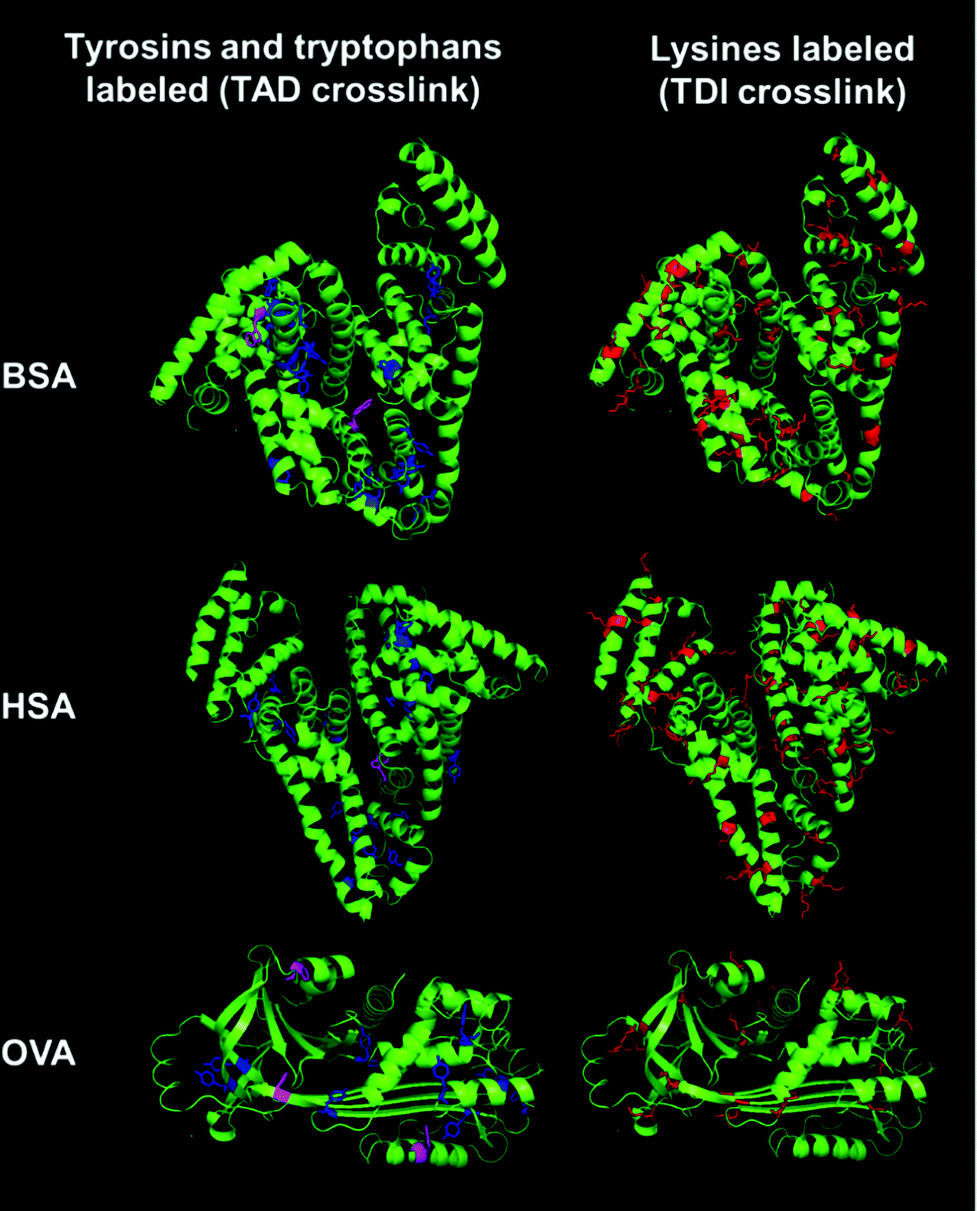 | ||
| Fig. 5 Crystal structures of BSA (PDB ID: 3v03), HSA (PDB ID: 1BM0) and OVA (PDB ID: 1OVA). To visualize the localization of the different amino acids the crystal structures of BSA, HSA and OVA are shown. Lysine residues are labeled in red in the protein structures. Tyrosine residues are labeled in blue and tryptophan residues are labeled in magenta.40–42 | ||
In OVA as well as in BSA and HSA many lysine residues are present (Fig. 5 labeled in red). These lysine residues are usually located at the exterior of the protein, enabling hydrogen bonding and ionic interactions. The amount of lysines in BSA and HSA is around 10% (59 and 57 lysines respectively), while OVA has about 5% of lysines (20 lysines). The groups are well accessible and, therefore, many reactions are possible. Tyrosine (in blue) and tryptophan residues (in magenta) are usually found buried inside the protein due to their hydrophobicity. BSA, HSA and OVA all have approximately the same amount of tyrosine residues (around 3% – 20 tyrosines in BSA, 18 in HSA and 10 tyrosines in OVA). BSA has two tryptophan residues per unit, while HSA just has one. In contrast to that, in the protein structure of OVA, three tryptophan residues can be found, while OVA has just two-thirds of the size of BSA and HSA. Comparing the locations of the different amino acids in the proteins, TDI and TAD might lead to variable degrees of denaturation of the proteins, exhibiting different structures to the outer surface of the generated nanocapsules, which finally leads to a different cell uptake behavior, depending on the crosslinking type. Changes in the protein structure can usually be analyzed using e.g. CD spectroscopy. However, it is difficult to use this method for dispersions, because the absorption measured can be affected by the scattering of the nanocapsules as well as of other components, like e.g. dyes, salts or surfactants present in the dispersion.
Adsorption of blood proteins to protein nanocapsules
As shown in Fig. 4 protein corona formation had a major impact on the cellular uptake towards HeLa cells. Therefore, we aimed to deeply investigate the interaction of the protein nanocapsules with blood proteins. All nanocapsules were incubated with human serum for 1 h at 37 °C. Protein corona coated nanocapsules were isolated via centrifugation and further analyzed by SDS-PAGE (Fig. S4–6†), Pierce Assay (Fig. S3†) and mass spectrometry (Fig. 6). A full list of all identified proteins is provided in a separate supporting Excel Sheet. | ||
| Fig. 6 Protein corona profile of TDI (a) and TAD (b) nanocapsules. Protein nanocapsules were incubated with human serum for 1 h at 37 °C and the protein corona was analyzed by LC-MS and classified into eight different groups according to function. The amount of each protein was calculated in % based on the total amount of all identified proteins. The two different crosslinkers TDI (a) or TAD (b) were used for the nanocapsule preparation. | ||
All corona proteins identified by mass spectrometry were classified into eight different protein groups depending on their biological function and are summarized in Fig. 6. We found that regardless of the protein type and crosslinker, there was no specific enrichment of one or two major protein classes in the protein corona of all nanocapsules (see also protein corona profiles Fig. S4–S6†). This is in strong contrast to other nanocarrier systems such as nanocapsules prepared from hydroxyethyl starch (HES)43,44 or polystyrene nanoparticles.30,45 Generally, for those systems, one or two protein groups (e.g. lipoproteins or serum albumin) are the dominant class proteins with an overall amount between 50–70%. Next to this, the absolute amount of corona proteins (Fig. S3†), which bound to all protein nanocapsules was comparable (<500 μg m−2 surface area) and significantly lower compared to polystyrene nanoparticles (∼2–3 mg m−2) or HES nanocapsules (∼1 mg m−2). This could possibly be a result of different hydrophilicities of the materials and the fact that proteins are natural molecules also occurring in the human body. This was already found for other nanocarriers made from “body-similar” components.30,46,47 However, having a closer look at the individual protein classes, we found that the crosslinker type (TAD vs. TDI) had an influence on the relative amount of specific protein classes. As an example, the amount of immunoglobulins was higher for TDI (∼20%) compared to TAD nanocapsules (5–9%). In contrast to that, proteins involved in the coagulation cascade especially interacted with TAD nanocapsules (13–26%) compared to TDI nanocapsules (6–11%). This effect was observed for all protein nanocapsules leading to the conclusion that the crosslinker type and not the base material (BSA vs. OVA vs. HSA) shaped the protein corona profile.
In general, the surface hydrophilicity has a strong influence on protein adsorption. As TDI is more hydrophobic than TAD, this could have an impact on the surface properties the protein nanocapsules and eventually mediate a specific protein interaction. In literature, it was shown that with increasing surface hydrophobicity the amount of immunoglobulins in the protein corona increased.45 This goes along with the here presented data as the amount of immunoglobulins was higher for TDI nanocapsules compared to TAD nanocapsules.
The influence of the crosslinker type on the protein interaction effect became very prominent for certain individual proteins, which is shown in Fig. 7. For all protein nanocapsules crosslinked with TDI, Ig kappa and Ig gamma-1 (two examples of proteins belonging to the group of immunoglobulins) were enriched on the surface in comparison to TAD cross-linked nanocapsules (Fig. 7a and b). Next to this, for all TAD nanocapsules, the amount of kininogen-1 and platelet factor 4 (two examples of proteins belonging to the group of coagulation) was higher in comparison to all TDI nanocapsules (Fig. 7c and d). Immunoglobulins belong to the protein class of opsonins. These proteins are known to mediate interactions with phagocytic cells.48,49 Therefore, adsorption of immunoglobulins on the surface of nanocarriers can contribute to a decreased blood circulation time. In the case of TAD nanocapsules, lower amounts of immunoglobulins were detected in the protein corona, which could contribute to a prolonged blood circulation time. However, to answer this question, further in vivo biodistribution studies are needed.
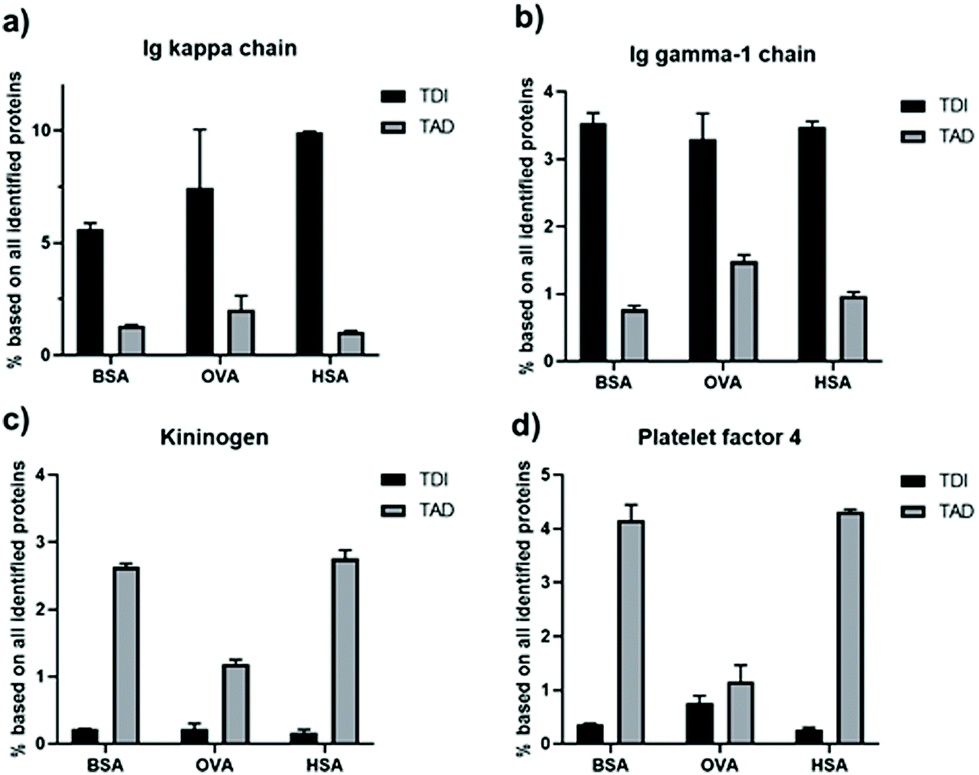 | ||
| Fig. 7 Relative abundance of single proteins in the protein corona of TDI and TAD protein nanocapsules determined by LC-MS given in %. The amount of Ig kappa chain (a), Ig gamma-1 chain (b), kininogen-1 (c) and platelet factor 4 (d) in the protein corona of the different nanocapsules was compared. | ||
Conclusion
We report the synthesis of nanocapsules crosslinked by 4,4′-(4,4′-diphenylmethylene)-bis-(1,2,4-triazoline-3,5-dione) (MDI-TAD) in an inverse miniemulsion process. MDI-TAD reacts with aromatic amino acids and also with full proteins in a bio-orthogonal way leading to nanocapsules with narrow size distributions and defined morphologies. Magnetite nanoparticles could be encapsulated into Trp-TAD and BSA-TAD nanocapsules with a high loading capacity. The average diameter and ξ-potential of TAD-crosslinked nanocapsules and TDI-crosslinked nanocapsules consisting of different albumins, were similar. However, in our study we showed that the crosslinker type used for the preparation of protein nanocapsules is a key factor that mediates cell and protein interaction. TAD crosslinked nanocapsules interacted stronger with HeLa cells than TDI nanocapsules under serum free conditions. This could be caused by a change in surface hydrophilicity of the differently crosslinked nanocapsules or due to significant differences in the degree of denaturation of the crosslinked proteins depending on the chosen crosslinker type. Moreover, the protein corona profile was influenced by the crosslinker type. The protein corona of TDI crosslinked nanocapsules was enriched with immunoglobulins, whereas the corona of TAD nanocapsules mainly consisted of proteins involved in the coagulation cascade. Therefore, our study sheds light onto the biological properties of protein based nanocarriers and this knowledge is needed for the successful application of nanocarriers as drug delivery vehicle in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Deutsche Forschungsgemeinschaft (SFB 1066). Furthermore, the authors thank C. Rosenauer, G. Glaßer and C. Sieber for technical support. Open Access funding provided by the Max Planck Society.References
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20 CrossRef CAS PubMed.
- J. Reinholz, K. Landfester and V. Mailänder, Drug Delivery, 2018, 25, 1694–1705 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed.
- K. Piradashvili, M. Fichter, K. Mohr, S. Gehring, F. R. Wurm and K. Landfester, Biomacromolecules, 2015, 16, 815–821 CrossRef CAS PubMed.
- D. Paßlick, K. Piradashvili, D. Bamberger, M. Li, S. Jiang, D. Strand, P. R. Wich, K. Landfester, M. Bros and S. Grabbe, J. Controlled Release, 2018, 289, 23–34 CrossRef PubMed.
- J. Kreuter, J. Controlled Release, 1991, 16, 169–176 CrossRef CAS.
- K. Landfester and V. Mailänder, Expert Opin. Drug Delivery, 2013, 10, 593–609 CrossRef CAS PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- K. Piradashvili, J. Simon, D. Paßlick, J. R. Höhner, V. Mailänder, F. R. Wurm and K. Landfester, Nanoscale Horiz., 2017, 2, 297–302 RSC.
- M. Neves-Petersen, S. Petersen and G. P. Gajula, in Molecular Photochemistry - Various Aspects, ed. S. Saha, 2012, ch. 7, pp. 125–133 Search PubMed.
- C. M. Madl and S. C. Heilshorn, Adv. Funct. Mater., 2018, 28, 1706046 CrossRef PubMed.
- D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef PubMed.
- S. Vandewalle, R. De Coen, B. G. De Geest and F. E. Du Prez, ACS Macro Lett., 2017, 6, 1368–1372 CrossRef CAS.
- S. B. Hanay, D. F. Brougham, A. A. Dias and A. Heise, Polym. Chem., 2017, 8, 6594–6597 RSC.
- S. B. Hanay, B. Ritzen, D. Brougham, A. A. Dias and A. Heise, Macromol. Biosci., 2017, 17 Search PubMed.
- D. Kaiser, J. M. Winne, M. E. Ortiz-Soto, J. r. Seibel, T. A. Le and B. J. Engels, J. Org. Chem., 2018, 83, 10248–10260 CrossRef CAS PubMed.
- R. Cookson, S. Gilani and I. Stevens, J. Chem. Soc. C, 1967, 1905–1909 RSC.
- H. A. Houck, K. De Bruycker, C. Barner-Kowollik, J. M. Winne and F. E. Du Prez, Macromolecules, 2018, 51, 3156–3164 CrossRef CAS.
- N. Van Herck and F. E. Du Prez, Macromolecules, 2018, 51, 3405–3414 CrossRef CAS.
- H. A. Houck, K. De Bruycker, S. Billiet, B. Dhanis, H. Goossens, S. Catak, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Chem. Sci., 2017, 8, 3098–3108 RSC.
- K. De Bruycker, S. Billiet, H. A. Houck, S. Chattopadhyay, J. M. Winne and F. E. Du Prez, Chem. Rev., 2016, 116, 3919–3974 CrossRef CAS PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- M. Bausch and B. David, J. Org. Chem., 1992, 57, 1118–1124 CrossRef CAS.
- R. Cookson, I. Stevens and C. Watts, Chem. Commun., 1966, 744a–744a RSC.
- H. Schlaad, H. Kukula, J. Rudloff and I. Below, Macromolecules, 2001, 34, 4302–4304 CrossRef CAS.
- M. B. Bannwarth, S. Ebert, M. Lauck, U. Ziener, S. Tomcin, G. Jakob, K. Munnemann, V. Mailander, A. Musyanovych and K. Landfester, Macromol. Biosci., 2014, 14, 1205–1214 CrossRef CAS PubMed.
- F. Rusconi, É. Valton, R. Nguyen and E. Dufourc, Anal. Biochem., 2001, 295, 31–37 CrossRef CAS PubMed.
- M. Tonigold, J. Simon, D. Estupiñán, M. Kokkinopoulou, J. Reinholz, U. Kintzel, A. Kaltbeitzel, P. Renz, M. P. Domogalla, K. Steinbrink, I. Lieberwirth, D. Crespy, K. Landfester and V. Mailänder, Nat. Nanotechnol., 2018, 13, 862–869 CrossRef CAS PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372 CrossRef PubMed.
- J. C. Silva, M. V. Gorenstein, G. Z. Li, J. P. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 CrossRef CAS PubMed.
- L. Vlaminck, K. De Bruycker, O. Türünç and F. Du Prez, Polym. Chem., 2016, 7, 5655–5663 RSC.
- A. O. Elzoghby, W. M. Samy and N. A. Elgindy, J. Controlled Release, 2012, 157, 168–182 CrossRef CAS PubMed.
- J. Simon, T. Wolf, K. Klein, K. Landfester, F. R. Wurm and V. Mailänder, Angew. Chem., Int. Ed., 2018, 57, 5548–5553 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546 CrossRef CAS PubMed.
- M. Mahmoudi, F. Quinlan-Pluck, M. P. Monopoli, S. Sheibani, H. Vali, K. A. Dawson and I. Lynch, ACS Chem. Neurosci., 2013, 4, 475–485 CrossRef CAS PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017–14026 CrossRef CAS PubMed.
- P. G. Gettins, Chem. Rev., 2002, 102, 4751–4804 CrossRef CAS PubMed.
- E. L. Gelamo and M. Tabak, Spectrochim. Acta, Part A, 2000, 56, 2255–2271 CrossRef.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439–446 CrossRef CAS PubMed.
- P. E. Stein, A. G. Leslie, J. T. Finch and R. W. Carrell, J. Mol. Biol., 1991, 221, 941–959 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS.
- J. Simon, S. Christmann, V. Mailänder, F. R. Wurm and K. Landfester, Isr. J. Chem., 2018, 58, 1363–1372 CrossRef CAS.
- B. Kang, P. Okwieka, S. Schöttler, S. Winzen, J. Langhanki, K. Mohr, T. Opatz, V. Mailänder, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 7436–7440 CrossRef CAS.
- J. Simon, T. Wolf, K. Klein, K. Landfester, F. R. Wurm and V. Mailänder, Angew. Chem., Int. Ed., 2018, 57, 5548–5553 CrossRef CAS PubMed.
- B. Kang, P. Okwieka, S. Schöttler, O. Seifert, R. E. Kontermann, K. Pfizenmaier, A. Musyanovych, R. Meyer, M. Diken and U. Sahin, Biomaterials, 2015, 49, 125–134 CrossRef CAS PubMed.
- C. Weber, M. Voigt, J. Simon, A.-K. Danner, H. Frey, V. Mailänder, M. Helm, S. Morsbach and K. Landfester, Biomacromolecules, 2019, 20, 2989–2999 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py00087f |
| This journal is © The Royal Society of Chemistry 2020 |