
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlock copolymer hierarchical structures from the interplay of multiple assembly pathways†
Alessandro
Ianiro
 *ab,
Meng
Chi
a,
Marco M. R. M.
Hendrix
ab,
Ali Vala
Koç
a,
E. Deniz
Eren
a,
Michael
Sztucki
c,
Andrei V.
Petukhov
da,
Gijsbertus
de With
a,
A. Catarina C.
Esteves
a and
Remco
Tuinier
*abd
*ab,
Meng
Chi
a,
Marco M. R. M.
Hendrix
ab,
Ali Vala
Koç
a,
E. Deniz
Eren
a,
Michael
Sztucki
c,
Andrei V.
Petukhov
da,
Gijsbertus
de With
a,
A. Catarina C.
Esteves
a and
Remco
Tuinier
*abd
aLaboratory of Physical Chemistry, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: alessandro.ianiro@unifr.ch; r.tuinier@tue.nl
bInstitute for Complex Molecular Systems (ICMS), Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cESRF, The European Synchrotron, 71 Avenue des Martyrs, 38000, Grenoble, France
dVan ‘t Hoff Laboratory for Physical and Colloid Chemistry, Department of Chemistry and Debye Institute, Utrecht University, Padualaan 8, 3584 CH, Utrecht, The Netherlands
First published on 3rd March 2020
Abstract
Structurally complex hierarchical block copolymer assemblies can be formed in solution via the interplay of multiple assembly pathways. Crystallizable block copolymers can undergo self-assembly, crystallization or phase separation in solution. By selecting appropriate solvency conditions (solvent and temperature) it is possible to induce an interplay between these processes resulting in a complex association behavior. This leads to the formation of assemblies with up to four levels of hierarchical organization. Furthermore, varying the copolymer composition enables to tune the formation mechanism and the morphology of the aggregates.
Living organisms often synthesize hierarchical materials with attractive features.1,2 Nacre3 and bone4 are explicative examples of how hierarchy can be exploited to introduce functional properties, making the whole greater than the sum of the parts. In natural materials, controlled synthesis of building blocks and level-by-level assembly of the structure take place simultaneously and are directed by the biological matrix.2 Conversely, artificial hierarchical materials are often prepared using procedures in which synthesis and assembly are separated, as the sophisticated strategy adopted by nature is still challenging to replicate.
Block copolymers (BCPs) are versatile synthetic self-assembling building blocks for the preparation of different kinds of colloidal and bulk materials.5–7 Bulk and solution self-assembly of BCPs result in the formation of structures with typically up to two hierarchical levels, as shown by several theoretical and experimental studies.8–11 Therefore, new strategies are desired to control block copolymer self-assembly across multiple hierarchical levels in order to make materials with novel properties.
Despite the fact that BCP self-assembly in solution has been extensively studied, a large part of these studies does not take into account that different association pathways are possible. In weakly selective solvents, where there is a small solubility difference between the blocks, BCP dispersions might undergo phase separation. Such a process is intrinsically different from self-assembly, as it involves a disordered aggregation of copolymer molecules. It has recently been shown12 that if the blocks are only weakly incompatible, phase separation is preferred over self-assembly in these solvents. This has important consequences on the formation mechanisms of self-assembled structures in solution,13 where gradual changes in the solvent selectivity are often used to promote self-assembly. After phase separation, internal long-range periodicity might evolve within the copolymer aggregates due to block segregation, similarly to what happens in the bulk.8
Block copolymer phase separation has another interesting feature: it can often be (reversibly) controlled with temperature.14,15 This is possible because weak amphiphilicity implies that the copolymer is overall poorly soluble but not far from the solution temperature, and thus the phase boundaries between soluble and insoluble states can be crossed with relatively narrow temperature variations.
The presence of crystalline blocks might also give rise to crystallization phenomena.16 Only recently, scientists started to pay attention to the effect of crystalline blocks on self-assembly, both in bulk17,18 and in solution.16,19,20
Thus, dispersed crystalline block copolymers can potentially follow different organization pathways such as self-assembly, crystallization from solution, phase-separation followed by internal block segregation, and phase separation followed by crystallization (of one or more blocks). In this work, we show that by controlling the solvency conditions (solvent selectivity and temperature) it is possible to induce and stir an interplay between these multiple association pathways. Particularly interesting is tuning the solvent selectivity such that liquid–liquid phase separation can be induced over a narrow temperature window, in proximity of the crystallization temperature. This additional association pathway results in the emergence of a complex associative behaviour, that leads to the formation of exotic BCP architectures with up to four levels of hierarchical organization.
To demonstrate this proof of principle we used crystalline-crystalline block copolymers made of poly(ethylene oxide) (PEO) and poly-ε-caprolactone (PCL). Both PEO and PCL homopolymers are crystallizable. The crystallization of PEO–PCL block copolymers is mostly regulated by the relative block chain length:20 in symmetric PEO–PCL the blocks crystallize simultaneously, while for asymmetric compositions the longer block crystallizes first.20
In certain solvents, such as hexanol,19 amyl–acetate19 and water–acetone mixtures,21 PEO–PCL block copolymers form lamellar crystals upon crystallization of either PEO or PCL, depending on the solvent.
In this work ethanol is selected as a solvent for its weak selectivity for the PEO blocks. In ethanol PEO–PCL block copolymers show a thermo-responsive phase behavior, with a demixing temperature that depends on the copolymer composition.22 The behavior of PEO–PCL in ethanol is studied using Small- and Wide-Angle Synchrotron X-ray Scattering (SAXS/WAXS), Differential Scanning Calorimetry (DSC), and Scanning Electron Microscopy (SEM). Synchrotron X-ray scattering measurements are especially suited for this study because they provide in situ, time-dependent and temperature-dependent structural information over a broad range of length scales. Furthermore, they provide an ensemble-averaged information. The DSC analysis provides energetic information and is useful to study crystallization phenomena, while SEM imaging enables direct visualization of the copolymer structures at the length scale of few tens micrometers, not easily accessible with SAXS/WAXS.
All characterizations were performed on 50 mg ml−1 dispersions upon cooling from T = 70 °C to T = −20 °C. Unless specified otherwise, data were obtained with a cooling rate vc = 10 °C min−1. Details of the sample preparation, characterization and data analysis procedures are described in the ESI.† It is noted that each SAXS/WAXS experiment reported in this work is composed of many scattering curves. For clarity, these are combined into temperature-dependent 3D plots and contour maps. In the contour maps, the scattered intensities are represented using variations of a color scale. Such a representation enables an accessible visual comparison between data from different experiments.
Ethanol dispersions of three different PEO–PCL block copolymers, having the same PEO block but containing different PCL mass ratios (fPCL = MPCLn/MPEOn) have been studied. The copolymers were labeled BCP1 (Me-EO45-CL30, fPCL = 1.71), BCP2 (Me-EO45-CL13, fPCL = 0.74) and BCP3 (Me-EO45-CL6, fPCL = 0.34). The SAXS/WAXS measurements (Fig. 1A, C and E) reveal that above the phase separation temperatures (Tp ≈ 34, 6.6 and −2.3 °C for BCP1, BCP2, and BCP3, respectively), the copolymers are molecularly dissolved. The radius of gyration Rg (Fig. 2A) of the dissolved copolymers, estimated from fitting the scattering data at various temperatures with a theoretical model (form factor) for polymer coils with excluded volume23 (not shown), slightly increase with decreasing temperature until phase separation takes place. Such a behavior is interpreted as an increase in the segregation state of the two blocks mediated by temperature, rather than a solvent-induced expansion of the copolymer chains.
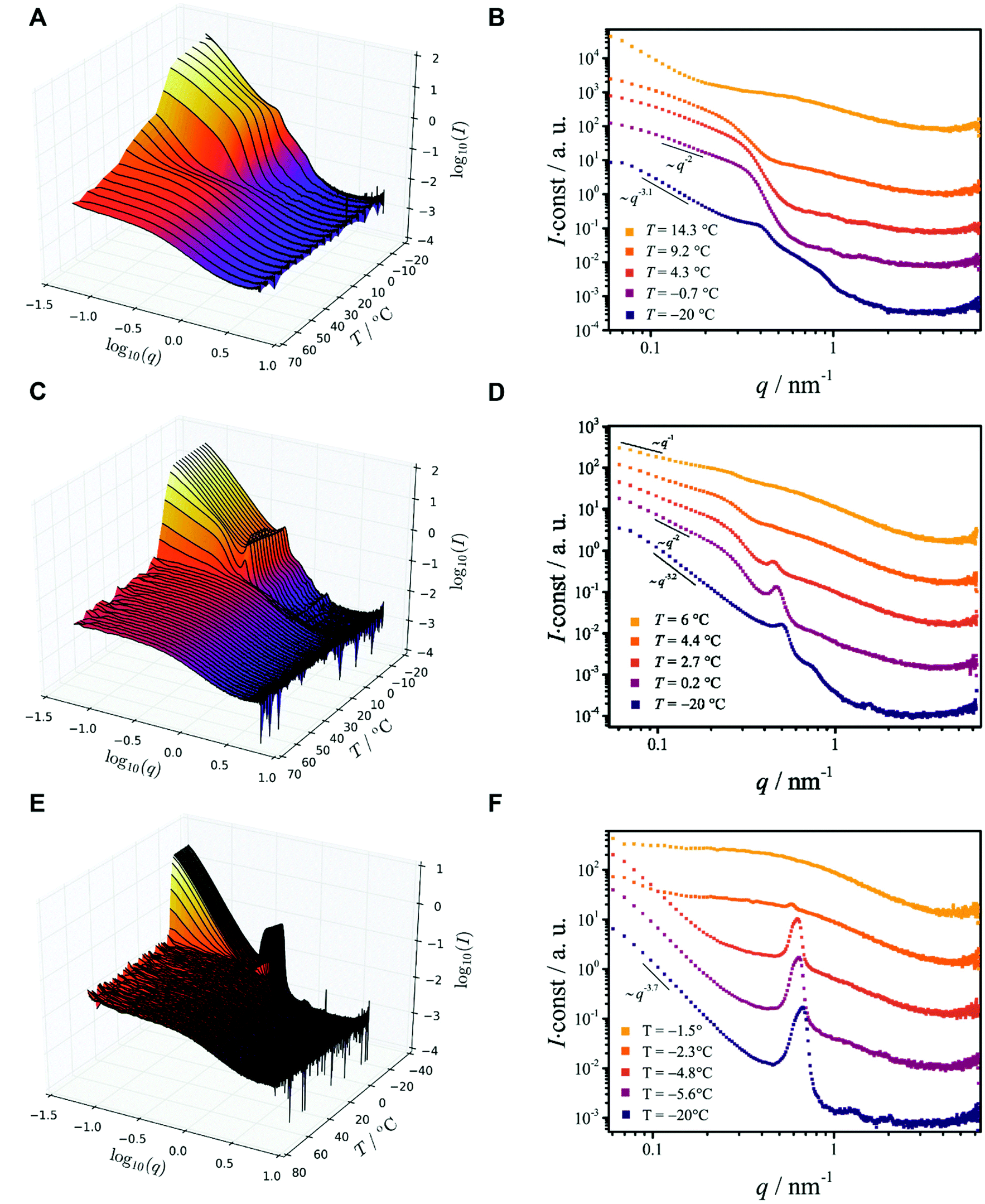 | ||
| Fig. 1 SAXS data from the block copolymers in ethanol. (A, C and E) 3-D representation of the SAXS data and (B, D and F) 2D representations of selected scattering curves. (A and B) are the scattering data from BCP1; (C and D) are the scattering data from BCP2; (E and F) are the scattering data from BCP3. | ||
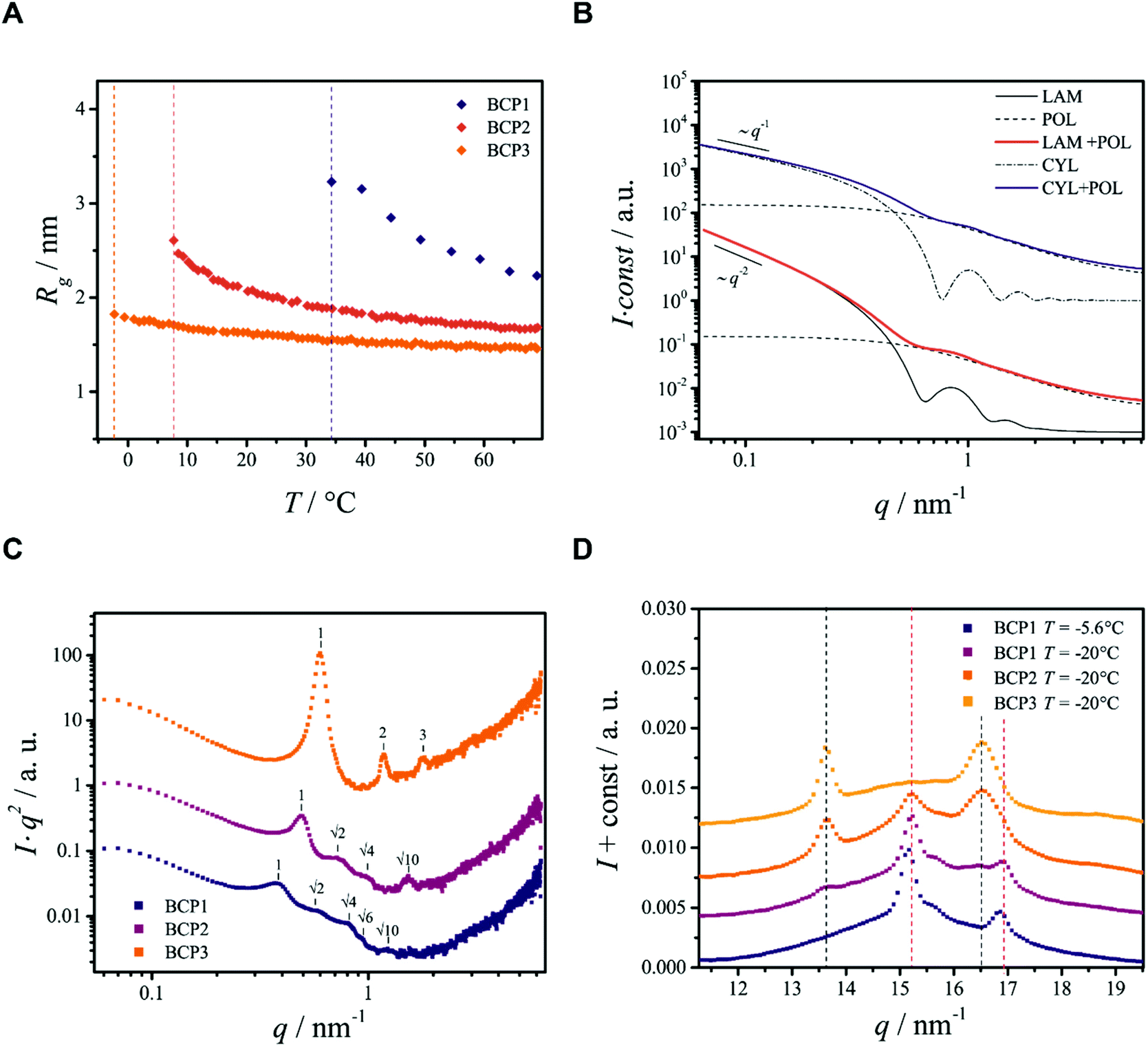 | ||
| Fig. 2 Analysis of selected SAXS data from the block copolymers in ethanol. (A) Radius of gyration Rg as a function of temperature T for the three BCPs studied. Vertical dashed-lines indicate the phase separation temperature Tp. (B) Calculated theoretical scattering curves used for the interpretation of the scattering data from BCP1 and BCP2. The models describe the scattered intensities of mixtures of molecularly dissolved BCPs23 (POL) with lamellae25 (LAM) or cylinders26 (CYL). At small q the typical scaling of the scattered intensity can be recognized. (C) Kratky plots obtained from SAXS data at T = −20 °C, used to enhance the visibility of the diffraction peaks. (D) Selected WAXS data. Vertical black and red dashed lines indicate the position of PEO and PCL reflections respectively, determined in Fig. S4B.† | ||
Phase separation of BCP1, the block copolymer with the longest PCL blocks (fPCL = 1.71), is a two step-process similar to that of pure PCL (see ESI†). The onset of phase separation (Tp ≈ 34 °C) is associated with a collapse of the copolymer chains (Fig. 1A) and with an increase of the scattered intensity at small values of the scattering vector q. Crystallization takes place at lower temperature (Txtl ≈ 18.6 °C) and leads to the formation of two-dimensional structures, which can be recognized from the I ∼ q−2 scaling of the small-q scattered intensity (Fig. 1B) and from comparison with a theoretical model (Fig. 2B). When the temperature reaches T = −20 °C, the 2-D structures form big aggregates with an irregular shape and rough surface, as testified by a scaling of the scattered intensity I ∼ q−3.1 (Fig. 1B). This is confirmed by the SEM image in Fig. 3A, showing a freeze-dried BCP1 sample previously equilibrated at T = −20 °C in ethanol. The SEM image (Fig. 3A) shows the formation of flower-like aggregates in coexistence with sponge-like particles.
 | ||
| Fig. 3 SEM micrographs of freeze-dried (A) BCP1, (B and C) BCP2 and (D) BCP3 samples, previously equilibrated at T = −20 °C in ethanol. | ||
The appearance of shallow diffraction peaks in the small-angle scattering at T = −20 °C (Fig. 1B) suggests that the copolymer chains rearrange forming poorly ordered periodic structures. The position of the small-angle diffraction peaks (Fig. 2C) with respect to the position of the most intense peak q* is related to the symmetry of the formed phase. In the case of BCP1, the relative peak positions (approximately related as 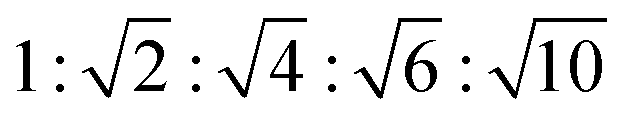 ) suggest a cubic bicontinous arrangement24 (Fig. 2C). The rearrangement observed at low T is induced by a secondary crystallization event, with onset at T ≈ −8 °C, which is visible in the DSC curves (Fig. S5C in ESI†).
) suggest a cubic bicontinous arrangement24 (Fig. 2C). The rearrangement observed at low T is induced by a secondary crystallization event, with onset at T ≈ −8 °C, which is visible in the DSC curves (Fig. S5C in ESI†).
The WAXS patterns (Fig. 2D and Fig. S6C in ESI†) reveals that the second thermal transition originates from the (partial) crystallization of the PEO blocks. In fact, at T > −8 °C only the diffraction signal from crystalline PCL domains is detected in the SAXS data, while at T < −8 °C reflections from PEO also become visible (Fig. 2D). The phase separation temperature of BCP2, determined by SAXS (Tp ≈ 7 °C), is lower than the crystallization temperature determined by DSC (Txtl ≈ 9 °C). This is an apparent effect because crystallization is per se a phase separation process. Likely, this is caused by kinetic effects due to differences in the way heat is transferred to the sample in the two measurements setup, and to the stochastic nature of the process. These data suggest that BCP2 in ethanol undergoes a crystallization-induced phase separation, like pure PEO (see ESI†).
The evolution of the demixing process (Fig. 1C and D) resembles that of BCP1 (Fig. 1A and B), but the formation of cylindrical or ribbon-like structures at T ≈ 6 °C precedes the formation of two-dimensional structures, which aggregate at lower temperature forming irregular clusters with a rough surface (Fig. 1D). This is corroborated by the change of the scaling exponent of the small-q scattered intensities from I ∼ q−1 (T ≈ 6 °C) to I ∼ q−2 (T ≈ 0 °C) and finally to I ∼ q−3.2 (T = −20 °C), shown in Fig. 1D, and compared with theoretical models for the form factor of different morphologies in Fig. 2B.
The SEM analysis again confirms such observations, showing that BCP2 forms palmate particles made of ribbon- or leaf-shaped building blocks. Such particles coexist with much bigger planar structures, in agreement with the SAXS/WAXS data. A clear diffraction peak is observed for T ≲ 4 °C in the SAXS. This peak suggests that the BCP2 ribbon or leaf-shaped aggregates are composed of a stack of multiple lamellar layers (Fig. 1D). The position of this diffraction peak shifts from q ≈ 0.45 nm−1 to q ≈ 0.5 nm−1 with decreasing temperature (Fig. 1D), indicating that the size of the repeating copolymer domains decreases from ≈14 to ≈12.5 nm. Such a decrease is consistent with a crystallization-induced ordering of the copolymer chains. At T = −20 °C, the appearance of other shallow diffraction peaks (Fig. 1D and 2C) at q*, 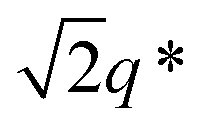 ,
, 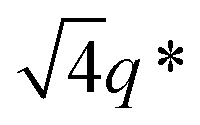 and
and 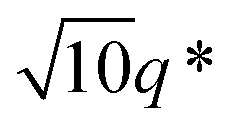 suggests that the copolymers arrange into a cubic bicontinous phase. The DSC data show that BCP2 crystallizes in a single event (Fig. S5D in ESI†). The wide-angle scattering analysis (Fig. 2D and Fig. S6D†) at T = −20 °C reveals that both blocks crystallize.
suggests that the copolymers arrange into a cubic bicontinous phase. The DSC data show that BCP2 crystallizes in a single event (Fig. S5D in ESI†). The wide-angle scattering analysis (Fig. 2D and Fig. S6D†) at T = −20 °C reveals that both blocks crystallize.
Also BCP3, with the shorter PCL block (fPCL = 0.34), undergoes crystallization-induced phase separation, with a demixing temperature Tp ≈ −20 °C again apparently lower than the crystallization temperature (Txtl ≈ 3 °C). The phase separation of BCP3 is sharp and produces lamellar crystals (Fig. 1F). The repeating spacing of such crystals decreases from ≈11 nm (T = −4.8 °C) to ≈9 nm (T = −20 °C), similarly to BCP2 (Fig. 1F). At T = −20 °C, the appearance of other diffraction peaks at q*, 2q*, 3q* (Fig. 1F and 2C) confirm the lamellar arrangement of the copolymer molecules. This is further confirmed by the SEM analysis (Fig. 3D). The crystallization of BCP3 also takes place in a single event (Fig. S5E†); only the PEO blocks crystallize according to the WAXS analysis (Fig. 2D and Fig. S6E†).
Due to the different phase behaviors of the individual blocks (see ESI†) and to the different PCL block lengths, the three copolymers studied exhibit fundamentally different structure formation mechanisms in ethanol (Fig. 4). It can be concluded that the copolymer phase behavior is directed by the longer block. A longer PCL block gives rise to a two-step process with liquid–liquid phase separation preceding a crystallization dominated structural rearrangement. The process leads to the formation of flower-like particles. A longer PEO block results in crystallization-induced phase separation which leads to the formation of lamellar structures. A quasi-symmetric composition seems to infer intermediate characteristics to the demixing process. Therefore, the relative block length governs the interplay between block segregation and crystallization during macroscopic phase separation. Varying this parameter enables to tune the formation mechanism and the morphology of the assembled structures.
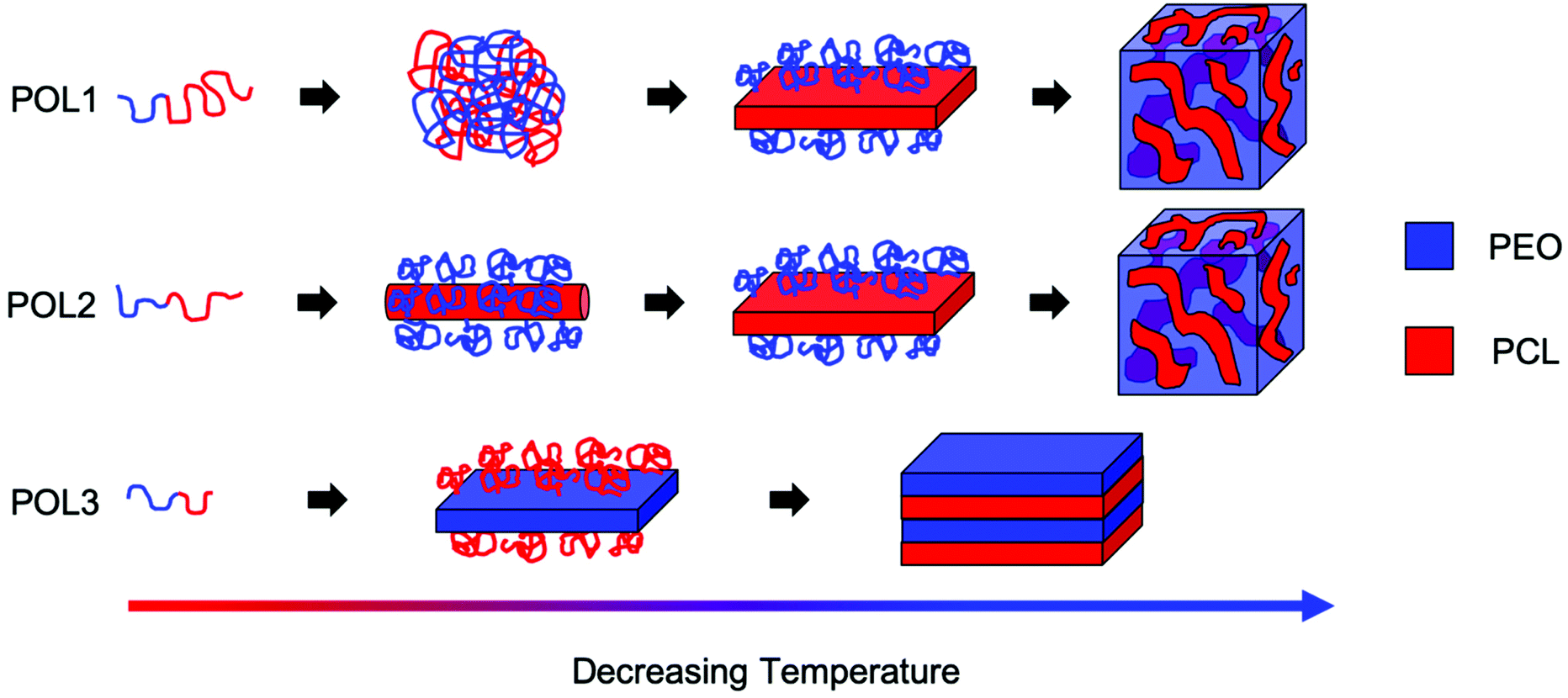 | ||
| Fig. 4 Schematic representation of the arrangement of the PEO (blue) and PCL (red) chains at the different steps of the structure formation process for the different copolymers studied. | ||
The structures of BCP1 and BCP2 aggregates are hierarchical and consist of four levels of organization. Microscale aggregates with complex shapes are composed of smaller micrometer-sized two-dimensional sub-units. These sub-units are composed of nanophase separated domains originating by the tendency of the blocks to segregate. In turn, these domains are composed of sub-domains with different chain packings (amorphous or crystalline). Finally, the sub-domains with different chain packings are composed of the individual block copolymer molecules. The formation of these hierarchical structures is achieved through the interplay of macroscopic phase separation, block segregation and crystallization of both blocks (see ESI† for further discussion). To the best of our knowledge, this work represents the first case where the interplay between block confinement and crystallization is studied during liquid–liquid phase separation.
Conclusions
This study shows that tuning the solvency conditions in order to favor the interplay between block copolymer phase separation, crystallization and segregation results in the emergence of a complex (out-of-equilibrium) associative behavior. This provides a way to control the association of block copolymers across multiple length-scales in solution by simply varying temperature. By means of a model experimental system consisting of PEO–PCL block copolymers in ethanol, we show that varying the block copolymer composition allows to change the specific characteristics of the assembly pathway, resulting in the formation of different kinds of materials with up to four levels of hierarchical organization. Although the physical properties of these hierarchical materials are yet to be determined, we expect our approach to be useful for the synthesis of novel functional materials, including photonic crystals, high performance structural materials as well as to prepare templates for inorganic nanostructures.This strategy can be potentially applied to any block copolymer with crystallizable and weakly incompatible blocks dispersed into a weakly selective solvent.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Bhushan, Philos. Trans. R. Soc., A, 2009, 367, 1445–1486 CrossRef CAS PubMed.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS.
- J. Sun and B. Bhushan, RSC Adv., 2012, 2, 7617–7632 RSC.
- S. Weiner and W. Traub, FASEB J., 1992, 6, 879–885 CrossRef CAS PubMed.
- P. Alexandridis, Curr. Opin. Colloid Interface Sci., 1996, 1, 490–501 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. H. Gröschel and A. H. E. Müller, Nanoscale, 2015, 7, 11841–11876 RSC.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- E. B. Zhulina and O. V. Borisov, Macromolecules, 2012, 45, 4429–4440 CrossRef CAS.
- E. B. Zhulina, M. Adam, I. Larue, S. S. Sheiko and M. Rubinstein, Macromolecules, 2005, 38, 5330–5351 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- W. T. Chuang, U. S. Jeng, H. S. Sheu and P. Da Hong, Macromol. Res., 2006, 14, 45–51 CrossRef CAS.
- A. Ianiro, H. Wu, M. M. J. van Rijt, M. P. Vena, A. D. A. Keizer, A. C. C. Esteves, R. Tuinier, H. Friedrich, N. A. J. M. Sommerdijk and J. P. Patterson, Nat. Chem., 2019, 11, 320–328 CrossRef CAS PubMed.
- T. Ougizawa and T. Inoue, Polym. J., 1986, 18, 521–527 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- R. M. Van Horn, M. R. Steffen and D. O'Connor, Polym. Cryst., 2018, 1, e10039 Search PubMed.
- D. J. Quiram, R. A. Register and G. R. Marchand, Macromolecules, 1997, 30, 4551–4558 CrossRef CAS.
- Y.-L. Loo and R. A. Register, Dev. Block Copolym. Sci. Technol., 2004, 213–243 CAS.
- R. M. Van Horn, J. X. Zheng, H. Sun, M. Hsiao, W. Zhang, X. Dong, J. Xu, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2010, 43, 6113–6119 CrossRef CAS.
- N. Brigham, C. Nardi, A. Carandang, K. Allen and R. M. Van Horn, Macromolecules, 2017, 50, 8996–9007 CrossRef CAS.
- A. Ianiro, J. Patterson, Á. González García, M. M. J. van Rijt, M. M. R. M. Hendrix, N. A. J. M. Sommerdijk, I. K. Voets, A. C. C. Esteves and R. Tuinier, J. Polym. Sci., Part B: Polym. Phys., 2018, 330–339 CrossRef CAS.
- A. Ianiro, I. Jimenez-Pardo, A. C. C. Esteves and R. Tuinier, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2992–2999 CrossRef CAS.
- B. Hammouda, in Polymer Characteristics, Springer Berlin Heidelberg, Berlin, Heidelberg, 1993, pp. 87–133 Search PubMed.
- K. Holmberg, Handbook of Applied Susrface and Colloid Chemistry, John Wiley & Sons, West Sussex, 2002, vol. 2 Search PubMed.
- F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487–502 CrossRef CAS.
- A. Fournet and G. Guinier, Small-Angle Scattering of X-Rays, John Wiley & Sons, Inc., New York, 1955 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary characterizations and discussions, supplementary figures. See DOI: 10.1039/d0py00081g |
| This journal is © The Royal Society of Chemistry 2020 |