
An anti-pressure, fatigue-resistant and rapid self-healing hydrogel based on a nano-micelle assembly†
Zhiang
Shao,  a
a
a
Abstract
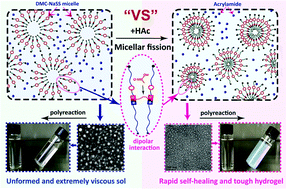
Herein, we found that a trace amount of acetic acid can strengthen DMC-NaSS micelles, resulting in a novel self-healing hydrogel with multiple non-covalent crosslinks. The hydrogel has both excellent self-healing capabilities and mechanical properties.
 Please wait while we load your content...
Please wait while we load your content...

