
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of miscible supramolecular network blends using DDA·AAD hydrogen-bonding interactions of pendent side-chains†
Heather M.
Coubrough
a,
Matthew
Reynolds
b,
James A.
Goodchild
 b,
Simon D. A.
Connell
b,
Johan
Mattsson
b and
Andrew J.
Wilson
*ac
b,
Simon D. A.
Connell
b,
Johan
Mattsson
b and
Andrew J.
Wilson
*ac
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK. E-mail: a.j.wilson@leeds.ac.uk
bSchool of Physics & Astronomy, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
First published on 12th May 2020
Abstract
The formation of polymer blends can result in materials with superior properties through combination of homo- or co-polymers with divergent functionalities. However, the contrasting physical properties of different polymers often result in phase separation. Herein we induce miscible blend formation of immiscible poly(methyl methacrylate) and polystyrene polymers through triple donor–donor–acceptor and acceptor–acceptor–donor (DDA·AAD) hydrogen bonding between complementary heterodimers on pendent side-chains. RAFT polymerization is used to synthesize a series of poly (methylmethacrylate) and polystyrene co-polymers bearing complementary side-chain hydrogen bonding motifs. Mixing of these polymers promoted miscible blend formation as demonstrated by atomic force microscopy and differential scanning calorimetry. The effectiveness of blend formation was shown to depend upon the extent of incorporation of hydrogen-bonding motif bearing co-monomer; lower degrees of incorporation lead to ineffective blending, whereas higher degree of incorporation, suppress phase separation and promote miscibility.
Introduction
Polymer blends are physical mixtures of two or more polymers that can be either homopolymers or copolymers.1–4 A major incentive to develop blends is to tune the properties of the individual polymer components, leading to, for instance, enhanced: impact strength, mechanical properties, processability and resistance or stability towards a range of conditions e.g. solvent moisture or temperature.1–5 Modulating properties in this manner has industrial utility e.g. in consumer plastics or resins, and in automotive and aircraft industries,1,3,4,6 where for instance, friction and wear applications require better performance. Polymer blends also offer economic and environmental advantages in terms of the recycling of polymer waste and in developing biodegradable polymers e.g. to counter the low viscosity, poor toughness and challenges in recycling poly(lactic acid).7–10 For blending to be viable, the polymers must be compatible, and form stable mixtures. If the polymers are not compatible, phase-separation occurs and where this is significant or changes with time, the system can exhibit undesirable or ill-defined mechanical properties.11 In general, the behaviour of polymer blends depends on the degree of mixing of the components and their mutual interaction, as well as the individual properties of the components. Blends can be classed as miscible, immiscible or partly miscible. A difference in properties renders many pairs of polymers immiscible on a molecular level (e.g. (poly)methyl methacrylate (PMMA) and (poly)styrene (PS)). Indeed, in the majority of cases, the mixing of two polymers results in phase separation.5,12 Although, the entropy of mixing is always positive for polymer blends leading to a negative contribution to the mixing free energy, the contribution is negligible for long-chain polymers and hence favourable enthalpic interactions are required to promote miscibility.13 Most combinations of polymers lead to enthalpic contributions to the mixing free energy which are positive and dominate the negative entropic contributions, resulting in phase separation. This is expected given that the properties of individual polymers are influenced by the non-covalent chemistry between polymer chains and differ from backbone to backbone, rendering the design of miscible blends challenging. A generic solution to this challenge would comprise a diverse tool-kit of complementary recognition motifs which can be introduced into immiscible polymer backbones to facilitate tuneable mixing. In this work, we exemplify this alternative and powerful route to achieve blending between otherwise immiscible polymer components. We achieve this by introduction of complementary triple hydrogen bonding motifs into the two immiscible polymers.Supramolecular polymers have had a profound influence on materials research,14–16 including the development of self-healing materials,17 supramolecular adhesives18 and biomaterials.19,20 Hydrogen bonding is particularly appealing for supramolecular polymer assembly.15,21,22 and depends upon the availability of suitable recognition modules i.e. hydrogen-bonding motifs (HBMs) motifs.23–29 To obtain linear supramolecular polymers high affinity HBMs are required,30 whereas for side-chain-functionalized31 or cross-linked polymers32,33 the use of multiple lower affinity HBMs can be used for instance, nucleobases34,35 and the diamidopyridine/diaminotriazine–thymine dyad (DAP/DAT·T).36–42 In the context of supramolecular blends, the use of this latter approach exploiting backbones bearing side-chains functionalized with HBMs is desirable. Previously Park et al. reported the development of supramolecular polymers using quadruple hydrogen bonding between complementary functionalized macromolecules.33,43 Supramolecular blends comprising poly(methyl methacrylate) (PMMA) and poly(styrene) (PS) functionalized with acceptor–donor–acceptor (ADA) and donor–acceptor–donor (DAD) HBMs respectively (Fig. 1) were described previously.44 14 mol% phenylureidopyrimidine (PUPY; ADA and 6 mol% diamidopyridine (DAP; DAD) was sufficient to promote formation of a miscible polymer blend. However, the ratio of HBM to un-functionalized monomer was high as a consequence of the relatively weak PUPY·DAP affinity (Ka ∼ 56 M−1 in CHCl3).45 In this work we used a higher affinity (Ka ∼ 3 × 104 M−1)46 pair of HBMs (Fig. 1): ureidoimidazole (UIM) and amidoisocytosine (AIC)47,48 with donor–donor–acceptor (DDA) and acceptor–donor–donor (ADD), which, have been used to prepare supramolecular polyurethanes.49 The use of the UIM·AIC pair allows generation of miscible blends with lower molar ratios of HBM functionalized co-monomers: 2% UIM in PMMA to 5.0% AIC in PS. Such a step allows the use of functionalized co-polymers that are chemically closer to their respective un-functionalized polymers and hence possess more analogous material characterization properties. Moreover the use of orthogonal50 recognition motifs opens the door to the preparation of self-sorting51,52 materials53–55 comprising a greater number of components and opportunities to prepare miscible blends to order i.e. where the desired affinity of the HBMs can be selected so as to maximise miscibility for a given pair of polymers.
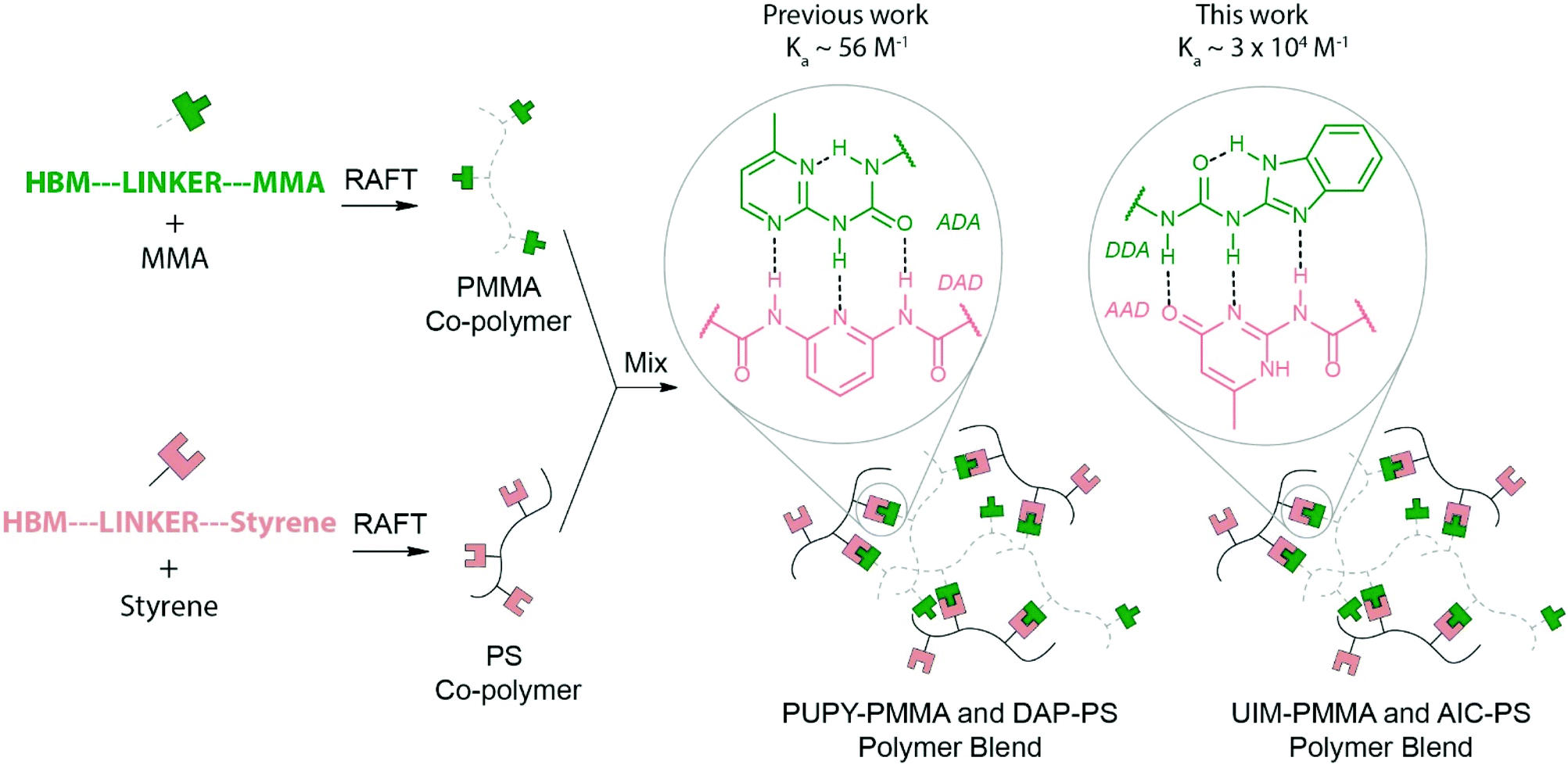 | ||
| Fig. 1 Schematic outlining the strategy for the formation of a supramolecular blend between PUPY-PMMA (ADA, green) and DAP-PS (DAD, pink) in previous work or UIM-PMMA (ADD, green) and AIC-PS (DAA, pink) co-polymers in this work. | ||
Results and discussion
The strategy to prepare self-assembled polymer blends with DDA·AAD complementary hydrogen bonding partners is outlined in Fig. 1. UIM-MMA (green) and AIC-styrene (pink) co-monomers 1 and 2 were synthesized (Scheme 1), then co-polymerized using reversible addition fragmentation chain-transfer (RAFT) to form functionalized co-polymers. These co-polymers were subsequently mixed to create a supramolecular polymer blend by evaporation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of the polymers in dichloromethane and then characterized by differential scanning calorimetry (DSC) and atomic force microscopy (AFM).
:1 mixtures of the polymers in dichloromethane and then characterized by differential scanning calorimetry (DSC) and atomic force microscopy (AFM).
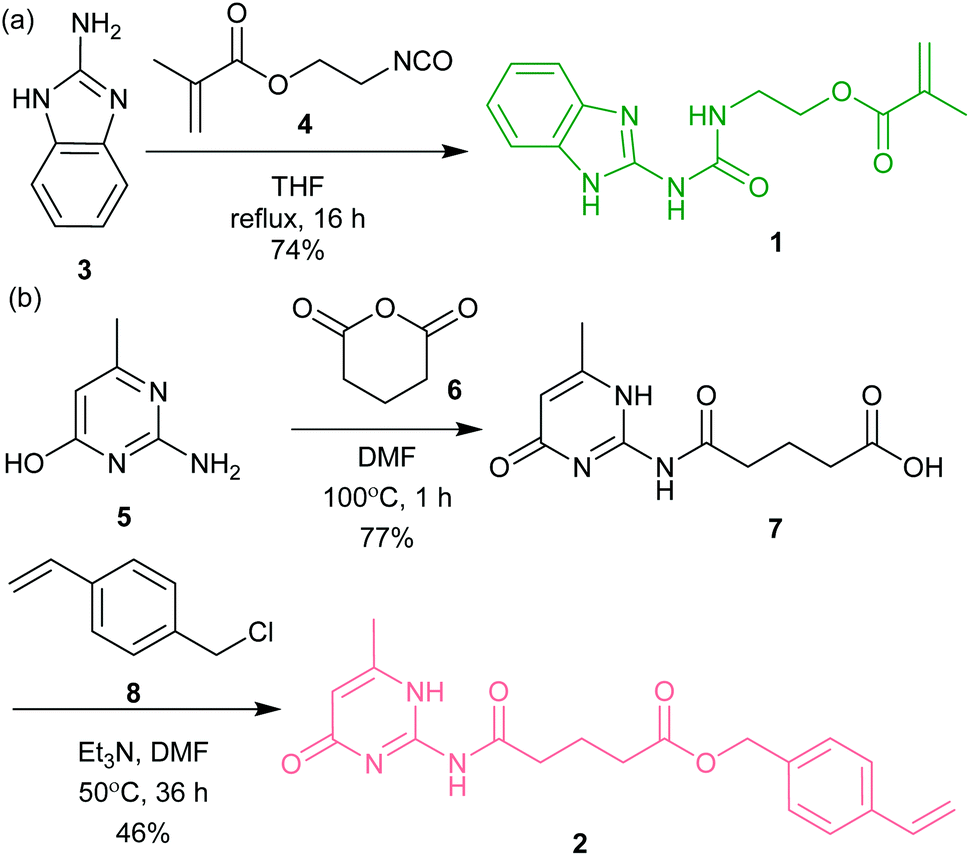 | ||
| Scheme 1 The synthetic route for preparation of (a) ureidoimidazole methacrylate monomer 1 (UIM-MMA) and (b) amidoisocytosine styrene monomer 2 (AIC-styrene). | ||
Co-monomer synthesis
The polymerizable hydrogen-bonding monomers are comprised of three parts: a hydrogen-bonding moiety and a polymerizable moiety, separated by a linker (Scheme 1). The linker was necessary to limit any electronic influence of proximal functionality on the reactivity of the alkene during RAFT polymerization. This was motivated by previous studies in which direct attachment of the hydrogen-bonding motif to styrene resulted in preferential polymerization over un-functionalized styrene, presumably due stabilization of the reactive radical by the electron-withdrawing hydrogen-bonding motif.19 Methacrylate functionalized ureidoimidazole monomer (UIM-MMA) 1 was obtained in good yield in one step from 2-aminobenzimidazole 3 and 2-isocyanatoethyl methacrylate 4 (Scheme 1a). The styrene-functionalized amidoisocytosine monomer (AIC-S) 2 was obtained in two synthetic steps (Scheme 1b). The first step was the reaction of 2-amino-4-hydroxy-6-methylpyrimidine 5 with glutaric anhydride 6 to form amide 7 with the hydrogen bonding moiety and linker. The intermediate was then reacted with 4-vinyl-chloride 8 to produce AIC-S 2. Prior to copolymerization of the functionalized monomers 1 and 2, with methylmethacrylate or styrene as appropriate, we assessed the impact on the addition of the polymerizable groups on the ability to participate in the expected UIM·AIC triple hydrogen bonding interaction relative to the previously described UIM·AIC complex.46 Due to low solubility of the UIM-MMA 1 in aprotic solvents, such as CDCl3, 1H NMR analysis was not possible. Consequently, we relied on a molecular modelling study of monomer 1 with 2 carried out using a hybrid Monte Carlo Molecular Mechanics (MCMM) conformational search (see ESI and Fig. ESI 1†). All the conformers retained, with a relative energy within 10 kJ mol−1 of the lowest energy conformer (399 in total), displayed the desired DDA·AAD intermolecular hydrogen bonding interaction, as well as the expected intramolecular hydrogen bonds. These analyses indicate that the polymerizable functional groups in UIM-MMA 1 and AIC-S 2 are unlikely to significantly influence dimerization of the UIM·AIC motifs through triple hydrogen bonding.Polymer synthesis
For creation of polymer blends it was important that incorporation of the hydrogen bonding motifs should not occur in preference to the un-functionalized monomer during polymerization. Whilst a post-polymerization strategy might have proven effective, a “controlled” copolymerization was selected to limit inconsistencies in macromonomer chain lengths and minimize differences in the number and distribution of functionalized monomers within a chain. RAFT polymerization is known as a robust controlled technique which can accommodate a variety of functionalities.56 Consequently, RAFT was employed for the copolymerization of the functionalized monomers with the corresponding un-functionalized monomers to create a series of PMMA and PS copolymers.Using a range of UIM-MMA 1: MMA 9 feed ratios (Table 1), a series of PMMA-based (co)polymers 10–13 were produced following the conditions outlined in Scheme 2a. The commercially available chain transfer agent (CTA) 14 (4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid) was chosen for the synthesis of low molar mass dispersity PMMA-based (co)polymers, together with azobutyronitrile (AIBN) 15 as a radical initiator. A selection of polymer lengths were obtained and in general a small broadening of polydispersity Đ were observed for the copolymers. Polymer 12 was longer, consistent with a longer reaction time. Using 1H NMR analysis, we confirmed that molar incorporation of monomer 1 into the copolymers broadly followed the molar feed ratio, based on the ratio of UIM and MMA integrals. From this, we infer that the hydrogen-bonding motif is randomly distributed within the copolymer backbone rather than clustered in blocks. The synthesized polymers and the corresponding characterization data are collated in Table 1.
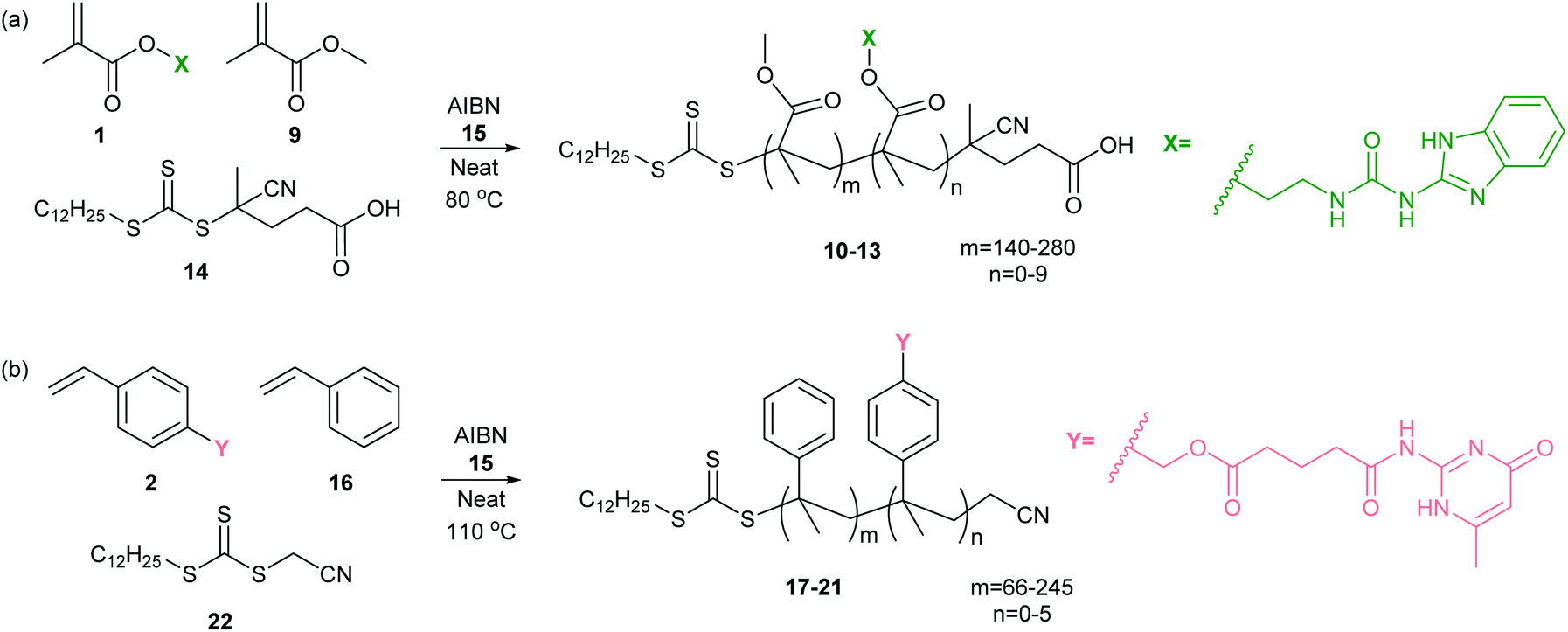 | ||
| Scheme 2 Synthesis of co-polymers 10–13 and 17–21 using RAFT (a) MMA 9 and MMA-UIM co-monomer 1 and (b) styrene 16 and PS-AIC co-monomer 2. | ||
| (Co)polymer | Monomer | HBM | HBM molar feed (%) | mol% HBM incorporation | wt% HBM incorporation | M n (103 g mol−1) | M w (103 g mol−1) | Đ | HB unit per chain | T g (°C, DSC) |
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 9 | N/A | 0 | 0 | 0 | 28.0 | 32.9 | 1.17 | 0 | 114 |
| 11 | 9 | 1 | 3 | 2.5 | 3.0 | 15.1 | 20.3 | 1.34 | 3.5 | 111 |
| 12 | 9 | 1 | 3 | 2.0 | 1.0 | 39.0 | 50.2 | 1.29 | 7.5 | 123 |
| 13 | 9 | 1 | 6 | 5.5 | 4.9 | 19.1 | 25.2 | 1.31 | 9.0 | 113 |
| 17 | 16 | N/A | 0 | 0 | 0 | 25.6 | 34.0 | 1.33 | 0 | 98 |
| 18 | 16 | 2 | 1 | 1.5 | 1.2 | 16.0 | 28.3 | 1.77 | 2.0 | 96 |
| 19 | 16 | 2 | 3 | 2.0 | 2.8 | 8.1 | 16.2 | 2.01 | 1.5 | 98 |
| 20 | 16 | 2 | 6 | 5.0 | 6.8 | 8.1 | 16.6 | 2.04 | 3.5 | 98 |
| 21 | 16 | 2 | 6 | 5.5 | 6.0 | 11.4 | 20.8 | 1.82 | 5.5 | 100 |
Using a range of AIC-S 2: styrene 16 feed ratios we also prepared a series of PS copolymers 17–21 (Table 1). Cyanomethyl dodecyl trithiocarbonate 22 was the chosen CTA for polystyrene (PS)-based copolymer preparation.57 Using heat to directly initiate styrene 16 polymerization, we found the conversion was low relative to the PMMA-based copolymers, requiring longer reaction times and resulting in higher Đ (data not shown). We therefore used AIBN 15 as a radical initiator with the aim of increasing the rate of initiation and decreasing the polydispersity of the PS-based copolymers. A pronounced decrease in degree of polymerization and increase in Đ was observed indicating the AIC hydrogen bonding motif monomer 2 to be capable of facilitating termination. Using 1H NMR analysis, we confirmed that molar incorporation of monomer 2 into the copolymers broadly followed the molar feed ratio, based on the ratio of AIC and PS integrals. The synthesized polymers and characterization data are collated in Table 1. As for the MMA copolymers we infer a random distribution of hydrogen bonding monomers within the polymer chain. For clarity: in the following sections, the polymers are named according to the mol% incorporation of HBM.
Blend characterization
The heat flow measured on cooling for the two control polymers 10 (PMMA) and 17 (PS) together with a 1:1 blend of the two polymers is shown in Fig. 2a (see Fig. ESI 2† for all individual polymers). For both PMMA and PS, the glass transitions are clearly observed as exothermic steps upon cooling and the Tg values determined as the inflection points thus characterize the transition midpoints. We obtain a Tg value of 98 °C for PS and 114 °C for PMMA, respectively, which are consistent with previously reported literature values;58 the Tg-values are marked with vertical dashed lines. The 1:1 blend of the two polymers (10 and 17) shows two distinct Tg's characterised by steps at 114 and 98 °C, respectively.
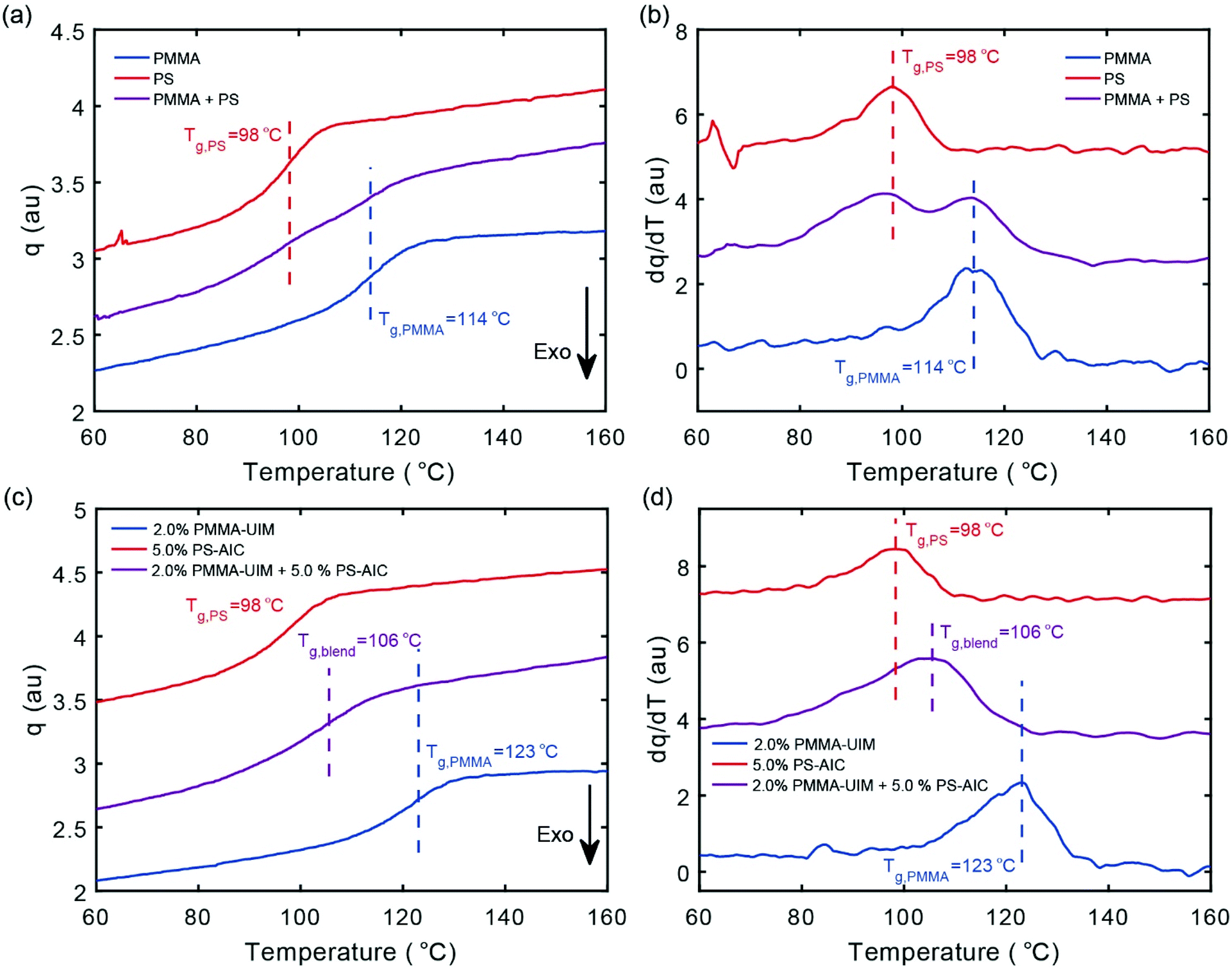 | ||
| Fig. 2 DSC thermogram of temperature against (a, c) heat flow and (b, d) first derivative of heat flow on cooling of (a, b) control sample: polymer 10 (PMMA) with polymer 17 (PS) and (c, d) blended sample: PMMA co-polymer 12 (2% PMMA-UIM). PS co-polymer (5.0% PS-AIC) 20 in red, PMMA in blue and blend in purple. | ||
To further clarify the transition behaviour, the temperature derivative of the data in Fig. 2a are shown in Fig. 2b, where it is clear that the blend data correspond to two separate glass-transitions. Moreover, the transitions of the pure polymer components are unchanged by the formation of the blend (Fig. 2a and b), supporting the immiscibility of PMMA and PS, which is well known59 (data for the corresponding heating runs are shown in the Fig. ESI 3†).
The temperature dependent heat flow of the co-polymer 12 (2% PMMA-UIM) and the functionalized co-polymer 20 (5.0% PS-AIC) together with a 1:1 blend of the two components are shown in Fig. 2c; the corresponding temperature derivatives are shown in Fig. 2d. The Tg-value of co-polymer 20 (5.0% PS-AIC) is 98 °C and thus very similar to that of the un-functionalized PS 17, demonstrating that the modification of PS does not significantly change its Tg-value. In contrast, the Tg-value for co-polymer 12 (2.0% PMMA-UIM) is 123 °C, compared with the 114 °C of the un-functionalized PMMA 10; thus the functionalization increases the Tg-value by 8 °C. We have shown using DSC (Fig. ESI 2†) that the degree of copolymerization with functionalized groups does not greatly alter the calorimetric response either for PMMA or PS; however, polymer 12 (2% PMMA-UIM) does show a Tg increase of 8 °C compared to the other co-polymers. It is not fully clear why this is, but the observation might be reconciled by that the fact that the co-polymer 12 (2% PMMA-UIM) has a higher degree of polymerization in comparison to un-functionalized PMMA 10 (Table 1).
Significantly, the 1:1 blend of the two polymer components shows a broad glass-transition signature with a peak temperature of Tg = 106 °C, situated between the Tg's of the two blend components, a result which is consistent with a significant increase in compatibility and thus blend miscibility. In a previous study on blends exploiting a ADA·DAD hydrogen-bonding interaction,44 14 mol% (16 wt%) of the ADA and 6 mol% (5 wt%) of the DAD hydrogen bonding motifs were required for blending of the respective PMMA and PS co-polymers. Here, a similar effect was achieved using a AAD·DDA interaction with only 2.0 mol% (1 wt%) and 5 mol% (7 mol wt%) of the UIM and AIC motifs respectively. Hence the stronger dimerization affinity translates to supramolecular polymer blends and consequently could result in the creation of supramolecular polymers with lower percentages of hydrogen bonding motifs with material properties close to those of un-functionalized polymers. However, the fact that the amount of HMB required in the polystyrene compound is comparable between the two studies emphasises that (inter-polymer) hydrogen-bond affinity is not the sole determinant for effective miscibility (see further discussion below). We also note, that the low-T flank of the broad transition coincides quite well with the transition of the pure 5.0% PS-AIC component, as shown most clearly in the derivative data representation of Fig. 2b on cooling (see Fig. ESI 3† for heating). We note that two separate glass transitions can sometimes be observed even for polymer blends with a good compatibility.60 This could be explained by the fact that a polymer chain will over some length-scale be primarily surrounded by itself, leading to an effective “self-concentration”. For some polymer combinations this can thus lead to a blend with two separate glass transitions and two corresponding Tg values even for a compatible polymer combination.60 These effects have been observed e.g. for blends of polyethylene oxide and PMMA,61 and for polyisoprene and poly(4-tert-butylstyrene).62 Thus, whilst functionalization of PMMA and PS with HBMs results in a significant increase in compatibility as demonstrated by calorimetry, quantification of the effectiveness of this approach requires complementary characterisation which we achieve using atomic force microscopy (AFM), as discussed below.
To further explore the limitations on the formation of compatible blends using the AIC·UIM motif we carried out further DSC experiments on blends made from polymers with different HBM incorporation (see ESI† for details). We observed that at lower levels of HBM incorporation, a significant increase of the blend compatibility did not occur. For both co-polymer 11 (1.5% PMMA-UIM) mixed with functionalized co-polymer 18 (1.5% PS-AIC) and co-polymer 11 (1.5% PMMA-UIM) mixed with functionalized co-polymer 19 (2.0% PS-AIC), two distinct transitions were observed in the DSC results on the blended sample (Fig. ESI 4 and 5† respectively). In both cases, the high-Tg component putatively associated with the PMMA-based polymer component increases (by ∼10 °C), whereas the low Tg component stays roughly unchanged. More surprisingly, indications of miscible blend formation with higher levels of HBM incorporation were less pronounced using DSC; for co-polymer 13 (5.5% PMMA-UIM) mixed with functionalized co-polymer 21 (5.5% PS-AIC), two Tg's were observed, with the second again showing an increase relative to co-polymer 13 (5.5% PMMA-UIM) on its own (Fig. ESI 6†).
The ability to blend at the molecular level through hydrogen bonding is influenced by a number of factors: (i) the strength of hydrogen bonds; (ii) the number of HBMs on each chain; (iii) the ability of the HBMs to (self)-stack (and or their crystallinity). The observations collectively can be explained as follows: as expected at lower levels of HBM incorporation there are insufficient numbers of HBMs on each polymer to promote good mixing, whilst at higher levels of HBM incorporation there maybe too many and these could self-stack through π–π interactions or self-associate through intra-polymer hydrogen-bonding, to compete with inter-polymer hydrogen-bonding and heterodimerization. 1H NMR analyses on blended samples indicate a change in hydrogen-bonding on mixing, however infrared analyses were unable to provide further experimental insight and a more detailed study will be pursued in due course (see ESI Fig. ESI 7 and 8†). The apparent increase in Tg in the PMMA-related transitions may arise as a consequence of forming a partially miscible blend whereby there is some hydrogen-bonding between PS and PMMA domains, but also stacking and aggregation. In addition, as noted above, two separate glass transitions can be observed also in some miscible blends. Hence we explored this further using AFM analyses on mixtures of polymers, as discussed in the next section. Finally, the increased degree of polymerization of the co-polymer 12 (2% PMMA-UIM) points to a role for degree of polymerization in controlling blending at higher molecular weights where the number of non-covalent “contact points” between chains could be greater.
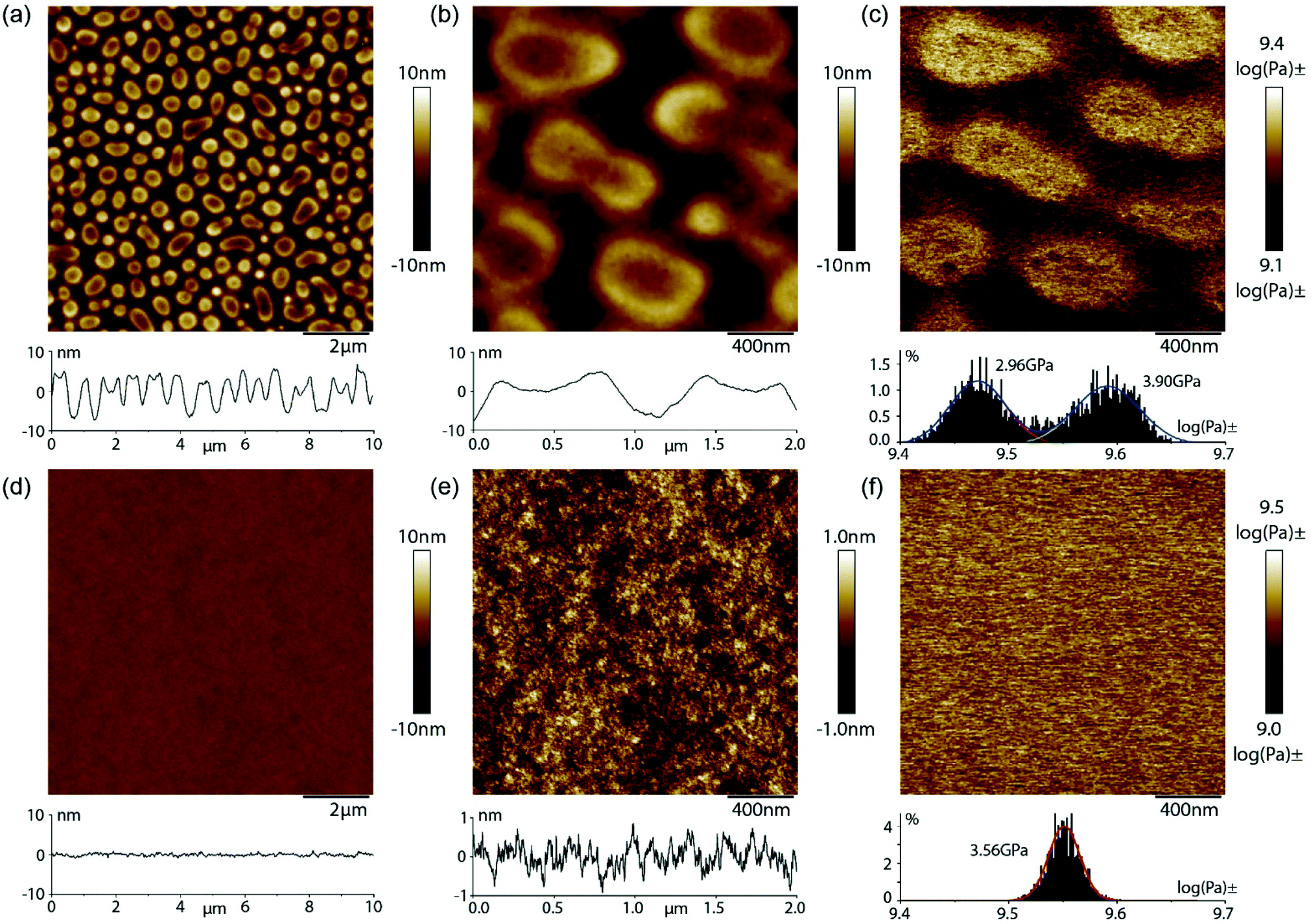 | ||
| Fig. 3 AFM analyses of blended comprising 1:1 mixture of un-functionalized PMMA 10 and PS 17 (a–c) or AIC-PMMA 12 and AIC-PS 20 (d–f) showing height images at 2 mM an (a, d) and 400 nM (b and e) together with rheological analysis (c and f). | ||
In contrast, the AFM images of the blend comprising the co-polymer 12 (2% PMMA-UIM) and co-polymer 20 (5.0% PS-AIC) demonstrates a very high degree of miscibility on the nanoscale. The surface of the 103 nm thick film is flat and featureless (Fig. 3d) and magnification of the height scale reveals a fine textured surface with height variations around 1 nm (Fig. 3e). The highly compatible nature of the blend is further illustrated by the modulus measurement which demonstrates a single phase and a mean modulus of 3.6 GPa, which is situated between the moduli of pure PMMA and PS, as observed in Fig. 3c. High magnification views of the blend sample are shown in Fig. ESI 10a† along with simultaneously acquired nanomechanical channels of elastic DMT modulus (Fig. ESI 10b†), surface deformation (Fig. ESI 10c†), and adhesion (Fig. ESI 10d†). The contrast in the modulus image is very weak, but shows regions of contrast variation on a scale <50 nm. These structural fluctuations are more clearly observed in the surface deformation and adhesion channels. Thus, very weak and small length-scale fluctuations are observed on small scales, but no indication of any significant phase segregation is observed in support of the DSC results demonstrating a significantly increased blend compatibility. We also obtained AFM data on the blend comprising the co-polymer 13 (5.5% PMMA-UIM) mixed with functionalized co-polymer 21 (5.5% PS-AIC), (see Fig. ESI 11†); these also indicate good miscibility on the nanoscale (see ESI†). Interestingly, however, the mean modulus of 4.9 GPa is higher than either pure PMMA or PS and point to more complex behaviour as is suggested by the DSC data.
Conclusions
We have designed analogues of styrene and methyl methacrylate polymerizable monomers functionalized with the HBMs ureidoimidazole (UIM) and amidoisocytosine (AIC) bearing donor–donor–acceptor (DDA) and acceptor–acceptor–donor (ADD) arrays respectively. RAFT polymerization was used to prepare a series of PMMA and PS co-polymers with varying degrees of complementary functionalized HBMs. AFM and DSC analyses of the resultant supramolecular assemblies demonstrate the power of the approach for making miscible blends. Miscible polymer blends were obtained for mixtures of functionalized co-polymers of PMMA and PS, while PMMA and PS in the absence of hydrogen bonding arrays were shown to be immiscible, thus emphasizing the requirement of hydrogen bonding motifs to induce blend formation. This work, taken together with previously reported HBM functionalized side-chain supramolecular polymers,44 highlights the ability of hydrogen bonding to overcome the inherent phase separation behaviour of immiscible polymers. More broadly these preliminary studies exemplify the power of an expanded supramolecular toolkit with which miscible blends can be prepared to order, with HBMs selected based on the strength of dimerization and knowledge of the immiscibility of the component polymers, to ensure maximum similarity and therefore properties to the respective un-functionalized polymers. Such studies together with the development of multi-component blends that exploit self-sorting51,52 will represent the focus of our future studies.Experimental
General considerations for synthesis
Solvents and reagents were purchased from Sigma Aldrich, Fisher Scientific or fluorochem and used without further purification unless otherwise stated. Where anhydrous solvents were required, dichloromethane, chloroform, tetrahydrofuran and acetonitrile were obtained from the in-house solvent purification system Innovative Inc. PureSolv®. Anhydrous dimethyl formamide and N,N-diisopropylethylamine were obtained from Sigma Aldrich equipped with Sure/Seal™. All non-aqueous reactions were carried out under a nitrogen atmosphere. For reactions under non-anhydrous conditions, the solvents used were HPLC quality and provided by Fisher or WVR. Water in aqueous solutions and used for quenching was deionised. Mixtures of solvents are quoted as ratios and correspond to a volume: volume ratio. Analytical thin layer chromatography was performed on Merck Kieselgel 60 F254 0.25 mm pre-coated aluminium plates. Product spots were visualised under UV light (λmax = 254 nm) or using a suitable stain. Flash chromatography was carried out using Merck Kieselgel 60 silica gel using pressure by means of head bellows or using disposable RediSepRf silica flash columns on an automated Biotage Isolera One system. Nuclear magnetic resonance spectra were obtained at 298 K (unless stated) using a Bruker AV500 spectrometer operating at 11.4 T (500 MHz for 1H and 125 MHz 13C) as stated. Infra-red spectra were obtained using a Bruker Alpha Platinum ATR where absorption maxima (νmax) are quoted in wavenumbers (cm−1) and only structurally relevant absorptions have been included. High Resolution Mass Spectra (HRMS) were recorded on a BrukerDaltonicsmicroTOF using electrospray ionisation (ESI). Liquid Chromatography and Mass Spectrometry (LC-MS) was performed using an Agilent Technologies 1200 series LC and a Bruker HCT ultra ion-trap MS. Gel permeation chromatography (GPC) measurements were conducted using Waters Acquity APC system fitted with three 2.4 μm columns plus a guard column and a refractive index detector. DMF and THF eluent was used at a flow rate of 0.5 mL min−1 and temperature of 40 °C. A series of 6 near-monodisperse poly(methylmethyacrylate) standards in DMF and near-monodisperse polystyrene standards in THF (Mw ranging from 2000 to 100000 g mol−1) were employed as calibration standards in conjunction with RI detector for determining molecular weights.
Monomer synthesis
:95 methanol:dichloromethane) to yield (4-ethenylphenyl)methyl 4-[(6-methyl-4-oxo-1,4-dihydropyrimidin-2-yl) carbamoyl]butanoate as an off-white solid (1.14 g, 3.20 mmol, 46%); 1H NMR (500 MHz, CDCl3): δ 7.04 (d, J 8.2, 2H, Ar–H), 7.31 (d, J 8.2, 2H, Ar–H), 6.71 (dd, J1 17.3, J2 11.2, 1H, CHCHH′), 5.99 (s, 1H, Py–CH), 5.75 (d, J 17.3, 1H, CHCHH′), 5.27 (d, J 11.2, 1H, CHCHH′), 5.11 (s, 2H, Bn–CH2), 2.60–2.51 (m, 2H, OCOCH2), 2.49 (t, J 7.2, 2H, NHCOCH2), 2.12–1.99 (m, J 7.2, 2H, CH2CH2CH2) ppm; 13C NMR (125 MHz, CDCl3): δ 172.7, 166.1, 162.7, 149.3, 137.6, 135.3, 128.4, 126.4, 114.4, 108.0, 66.2, 35.9, 33.1, 23.9, 19.8 ppm; νmax (solid state) = 3118, 3061, 2945, 2888, 1731, 1664 cm−1; ESI-HRMS m/z found 356.1621 [M + H]+ C19H21N3O4 requires 356.1566.
006, Mw (g mol−1) 32854 (GPC); Đ: 1.17 (GPC); 1H NMR (500 MHz, CDCl3) δ 3.58 (polymer CH3O), 2.00–1.66 (polymer CH2), 1.06–0.70 (polymer CH3) ppm.
135, Mw (g mol−1) 20306 (GPC); Đ: 1.34 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.39 (br. s, 2H, co-monomer imidazole–H), 7.10 (br. s, 2H, co-monomer imidazole–H), 4.22–3.94 (m, 4H, co-monomer CH2CH2O), 3.86–3.31 (polymer CH3O), 2.10–1.60 (polymer CH2), 1.10–0.58 (polymer CH3) ppm.
577 (GPC); Đ: 1.71 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.40 (2H, br s, co-monomer imidazole–H), 7.16 (2H, br s, co-monomer imidazole–H), 4.24–4.00 (4H, m, co-monomer CH2CH2O), 3.59 (polymer CH3O), 2.20–1.55 (polymer CH2), 1.10–0.50 (polymer CH3) ppm.
114, Mw (g mol−1) 25166 (GPC); Đ: 1.32 (GPC); 1H NMR (500 MHz, DMSO-d6) δ 7.36 (br s, 2H, co-monomer imidazole–H), 7.05 (br s, 2H, co-monomer imidazole–H), 4.11–3.89 (m, 4H, co-monomer CH2CH2O), 3.77–3.36 (polymer CH3O), 2.05–1.60 (polymer CH2), 1.10–0.58 (polymer CH3) ppm.
562, Mw (g mol−1) 33969 (GPC); Đ: 1.33 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.32–6.84 (polymer Ar–H), 6.84–6.24 (polymer Ar–H), 2.00–1.65 (polymer CH–Ar), 1.65–1.18 (polymer CH2CH–Ar) ppm.
958, Mw (g mol−1) 28297 (GPC); Đ: 1.77 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.22–6.86 (polymer Ar–H), 6.71–6.26 (polymer Ar–H), 6.02–5.96 (br s, 1H, co-monomer pyridyl–H), 5.10–4.95 (br s, 2H, co-monomer Bn–CH2), 2.71–2.55 (m, 2H, co-monomer OC(O)CH2), 2.55–2.34 (m, 2H, co-monomer NHC(O)CH2), 2.34–2.14 (m, 3H, co-monomer pyridyl–CH3), 2.14–2.00 (m, 2H, co-monomer CH2CH2CH2), 2.00–1.70 (polymer CH), 1.68–1.16 (polymer CH2) ppm.
173 (GPC); Đ: 2.01 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.22–6.86 (polymer Ar–H), 6.71–6.26 (polymer Ar–H), 6.03–5.94 (br s, 1H, co-monomer pyridyl–H), 5.10–4.93 (br s, 2H, co-monomer Bn–CH2), 2.66–2.53 (m, 2H, co-monomer OC(O)CH2), 2.51–2.34 (m, 2H, co-monomer NHC(O)CH2), 2.25–2.12 (m, 3H, co-monomer pyridyl–CH3), 2.12–1.99 (m, 2H, co-monomer CH2CH2CH2), 1.99–1.68 (polymer CH), 1.68–1.16 (polymer CH2) ppm.
551 (GPC); Đ: 2.04 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.22–6.86 (polymer Ar–H), 6.71–6.26 (polymer Ar–H), 6.03–5.96 (br s, 1H, co-monomer pyridyl–H), 5.10–4.93 (br s, 2H, co-monomer Bn–CH2), 2.67–2.53 (m, 2H, co-monomer OC(O)CH2), 2.53–2.39 (m, 2H, co-monomer NHC(O)CH2), 2.20–2.11 (m, 3H, co-monomer pyridyl–CH3), 2.11–1.97 (m, 2H, co-monomer CH2CH2CH2), 1.97–1.68 (polymer CH), 1.68–1.16 (polymer CH2) ppm.
400, Mw (g mol−1) 20843 (GPC); Đ: 1.82 (GPC); 1H NMR (500 MHz, CDCl3) δ 7.22–6.86 (polymer Ar–H), 6.80–6.25 (polymer Ar–H), 6.05–5.90 (br s, 1H, co-monomer pyridyl–H), 5.13–4.91 (br s, 2H, co-monomer Bn–CH2), 2.73–2.53 (m, 2H, co-monomer OC(O)CH2), 2.52–2.32 (m, 2H, co-monomer NHC(O)CH2), 2.26–2.11 (m, 3H, co-monomer pyridyl–CH3), 2.11–2.00 (m, 2H, CH2CH2CH2), 2.00–1.68 (polymer CH), 1.68–1.16 (polymer CH2) ppm.
:1 mixtures of co-polymers in dichloromethane solution (20 mg mL−1) of each component by mass. The samples were dried at room temperature for 24 hours, and subsequently annealed in a vacuum oven at 120 °C for 24 hours in order to completely remove any dichloromethane prior to measurements. After drying, the samples with typical weight of ∼5–15 mg were placed in aluminium pans (hermetic Tzero pans from TA instruments) that were hermetically sealed. Experiments were performed between 20 °C and 180 °C under a helium atmosphere, using heating/cooling scanning rates of 10 °C min−1.
:1 mixture (20 mg of each component in 2 mL of dichloromethane) onto a silicon wafer. Topographical and nanomechanical surface imaging of the polymer blends was carried out under ambient laboratory conditions using Peak Force Quantitative Nanomechanical Mapping (PF-QNM) mode on a Bruker Icon AFM running Nanoscope v9.4 control software. The TESPA-V2 probes (Bruker), with a nominal spring constant of 37 N m−1 were selected to give a strong mechanical signal in samples with modulus around 1 GPa. At the start of each experiment the cantilevers were first calibrated using a sapphire surface to obtain the deflection sensitivity, high speed QNM sensitivities at each tapping frequency, and the sync distance delay times. The spring constant was then calibrated using both the thermal noise method and Sader method, the latter required as a check due to the small amplitude of the thermal noise peak with this stiff lever, thus greater error. Spring constants were measured in the range 40–42 N m−1. The relative calibration method was used to achieve quantitative nanomechanics by calibrating against a known standard, a phase separated blend of PS/LDPE. The tip radius parameter was adjusted to achieve a PS modulus of 3.0 GPa, giving a modulus for the LDPE phase as 60–70 MPa. Images were acquired at a PF tapping frequency of 1 kHz, at 640 or 768 pixel resolution. Peak force was adjusted to achieve a sample deformation of 2 nm. Over time the calibration tends to drift due to changes to the probe hence the contact radius, so the calibration standard was frequently re-imaged, which was used to both recalibrate for subsequent imaging, and also to correct the measurements just prior to re-calibration. Film depths were measured by gently scoring the surface with a new razor blade, producing a clean square bottomed scratch about 10 μm in width, which was then imaged using AFM. AFM images were processed and analysed using Nanoscope Analysis v1.9 (Bruker).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC (EP/KO39292/1 and EP/M009521/1). H. C. thanks the University of Leeds for a Leeds Anniversary Research Scholarship. The authors thank Dr Martin Walko for assistance in acquiring NMR and IR spectra.Notes and references
- D. R. Paul and J. W. Barlow, J. Macromol. Sci., C, Polym. Rev., 1980, 18, 109–168 CrossRef.
- J. Runt and J. Huang, in Handbook of Thermal Analysis and Calorimetry, ed. S. Cheng, Elsevier Science B.V., 2002, vol. 3, pp. 273–294 Search PubMed.
- M. T. Demeuse, in High Temperature Polymer Blends, ed. M. T. DeMeuse, Woodhead Publishing, 2014, pp. 1–13, DOI:10.1533/9780857099013.1.
- L. A. Goettler and J. J. Scobbo, in Polymer Blends Handbook, ed. L. A. Utracki and C. A. Wilkie, Springer Netherlands, Dordrecht, 2014, pp. 1433–1458, DOI:10.1007/978-94-007-6064-6_17.
- L. A. Utracki and M. M. Dumoulin, in Polypropylene Structure, blends and composites: Volume 2 Copolymers and Blends, ed. J. Karger-Kocsis, Springer Netherlands, Dordrecht, 1995, pp. 50–94, DOI:10.1007/978-94-011-0521-7_3.
- L. M. Robeson, Polym. Eng. Sci., 1984, 24, 587–597 CrossRef CAS.
- I. M. Arcana, B. Bundjali, I. Yudistira, B. Jariah and L. Sukria, Polym. J., 2007, 39, 1337–1344 CrossRef CAS.
- A. Soroudi and I. Jakubowicz, Eur. Polym. J., 2013, 49, 2839–2858 CrossRef CAS.
- K. Hamad, M. Kaseem, Y. G. Ko and F. Deri, Polym. Sci., Ser. A, 2014, 56, 812–829 CrossRef CAS.
- F. Wu, M. Misra and A. K. Mohanty, RSC Adv., 2019, 9, 2836–2847 RSC.
- J. L. Koenig, in Spectroscopy of Polymers, ed. J. L. Koenig, Elsevier Science, New York, 2nd edn, 1999, pp. 77–145, DOI:10.1016/B978-044410031-3/50003-7.
- W. J. MacKnight and F. E. Karasz, in Comprehensive Polymer Science and Supplements, ed. G. Allen and J. C. Bevington, Pergamon, Amsterdam, 1989, pp. 111–130, DOI:10.1016/B978-0-08-096701-1.00207-X.
- S.-W. Kuo, J. Polym. Res., 2008, 15, 459–486 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823–6835 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860–4860 RSC.
- C. A. Anderson, A. R. Jones, E. M. Briggs, E. J. Novitsky, D. W. Kuykendall, N. R. Sottos and S. C. Zimmerman, J. Am. Chem. Soc., 2013, 135, 7288–7295 CrossRef CAS PubMed.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- W. H. Binder and R. Zirbs, Adv. Polym. Sci., 2007, 207, 1 CrossRef CAS.
- S. K. Yang and S. C. Zimmerman, Isr. J. Chem., 2013, 53, 511–520 CrossRef CAS.
- S. C. Zimmerman and P. S. Corbin, Struct. Bonding, 2000, 96, 63–94 CrossRef CAS.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC.
- A. J. Wilson, Soft Matter, 2007, 3, 409–425 RSC.
- P. K. Baruah and S. Khan, RSC Adv., 2013, 3, 21202–21217 RSC.
- D. A. Leigh, C. C. Robertson, A. M. Z. Slawin and P. I. T. Thomson, J. Am. Chem. Soc., 2013, 135, 9939–9943 CrossRef CAS PubMed.
- W. P. J. Appel, M. M. L. Nieuwenhuizen, M. Lutz, B. F. M. de Waal, A. R. A. Palmans and E. W. Meijer, Chem. Sci., 2014, 5, 3735–3745 RSC.
- S. Rayavarapu, S. Kheria, D. R. Shinde, R. G. Gonnade and G. J. Sanjayan, Org. Biomol. Chem., 2017, 15, 10087–10094 RSC.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193–207 RSC.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS PubMed.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 11582–11590 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- M. J. Kunz, G. Hayn, R. Saf and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 661–674 CrossRef CAS.
- H. Xu, T. B. Norsten, O. Uzun, E. Jeoung and V. M. Rotello, Chem. Commun., 2005, 5157–5159 RSC.
- M. Weck, Polym. Int., 2007, 56, 453–456 CrossRef CAS.
- P. Arumugam, H. Xu, S. Srivastava and V. M. Rotello, Polym. Int., 2007, 56, 461–466 CrossRef CAS.
- Y. Ishihara, H. Bazzi, V. Toader, F. Godin and H. Sleiman, Chem. – Eur. J., 2007, 13, 4560–4570 CrossRef CAS PubMed.
- F. Herbst and W. H. Binder, Polym. Chem., 2013, 4, 3602–3609 RSC.
- J.-H. Wang, O. Altukhov, C.-C. Cheng, F.-C. Chang and S.-W. Kuo, Soft Matter, 2013, 9, 5196–5206 RSC.
- T. Park, S. C. Zimmerman and S. Nakashima, J. Am. Chem. Soc., 2005, 127, 6520–6521 CrossRef CAS PubMed.
- A. Gooch, N. S. Murphy, N. H. Thomson and A. J. Wilson, Macromolecules, 2013, 46, 9634–9641 CrossRef CAS PubMed.
- A. Gooch, A. M. McGhee, L. C. Renton, J. P. Plante, C. I. Lindsay and A. J. Wilson, Supramol. Chem., 2009, 21, 12–17 CrossRef CAS.
- M. L. Pellizzaro, A. M. McGhee, L. C. Renton, M. G. Nix, J. Fisher, W. B. Turnbull and A. J. Wilson, Chem. – Eur. J., 2011, 17, 14508–14517 CrossRef CAS PubMed.
- A. Gooch, A. M. McGhee, M. L. Pellizzaro, C. I. Lindsay and A. J. Wilson, Org. Lett., 2011, 13, 240–243 CrossRef CAS PubMed.
- A. M. McGhee, C. Kilner and A. J. Wilson, Chem. Commun., 2008, 344–346 RSC.
- A. Gooch, C. Nedolisa, K. A. Houton, C. I. Lindsay, A. Saiani and A. J. Wilson, Macromolecules, 2012, 45, 4723–4729 CrossRef CAS.
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC.
- H. M. Coubrough, S. C. C. van der Lubbe, K. Hetherington, A. Minard, C. Pask, M. J. Howard, C. Fonseca Guerra and A. J. Wilson, Chem. – Eur. J., 2019, 25, 785–795 CrossRef CAS PubMed.
- M. L. Pellizzaro, K. A. Houton and A. J. Wilson, Chem. Sci., 2013, 4, 1825–1829 RSC.
- H. Shigemitsu, T. Fujisaku, W. Tanaka, R. Kubota, S. Minami, K. Urayama and I. Hamachi, Nat. Nanotechnol., 2018, 13, 165–172 CrossRef CAS PubMed.
- J. Ruiz-Olles and D. K. Smith, Chem. Sci., 2018, 9, 5541–5550 RSC.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- J. M. G. Cowie and V. Arrighi, Polymers, Chemistry and Physics of Modern Materials, CRC Press, Boca Raton, 2007 Search PubMed.
- K. Tanaka, A. Takahara and T. Kajiyama, Macromolecules, 1996, 29, 3232–3239 CrossRef CAS.
- T. P. Lodge and T. C. B. McLeish, Macromolecules, 2000, 33, 5278–5284 CrossRef CAS.
- T. P. Lodge, E. R. Wood and J. C. Haley, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 756–763 CrossRef CAS.
- J. Zhao, M. D. Ediger, Y. Sun and L. Yu, Macromolecules, 2009, 42, 6777–6783 CrossRef CAS.
- D. Hoffman, Master's, Michigan Technological University, 2010 Search PubMed.
- B. J. Briscoe, L. Fiori and E. Pelillo, J. Phys. D: Appl. Phys., 1998, 31, 2395–2405 CrossRef CAS.
- M. Lorenzoni, L. Evangelio, C. Nicolet, C. Navarro, A. S. Paulo, G. Rius and F. Pérez-Murano, J. Micro/Nanolithogr., MEMS, MOEMS, 2015, 14(1–6), 6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01913h |
| This journal is © The Royal Society of Chemistry 2020 |