
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer-functionalized polymer nanoparticles and their behaviour in suspensions†
Waraporn
Wichaita
 ab,
Young-Gon
Kim
a,
Pramuan
Tangboriboonrat
b and
Héloïse
Thérien-Aubin
*a
ab,
Young-Gon
Kim
a,
Pramuan
Tangboriboonrat
b and
Héloïse
Thérien-Aubin
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany. E-mail: therien@mpip-mainz.mpg.de
bDepartment of Chemistry, Faculty of Science, Mahidol University, Rama 6 Road, Phyathai, Bangkok 10400, Thailand
First published on 13th February 2020
Abstract
Soft polymer nanoparticles can be functionalized with end-tethered polymer chains to control their solvent compatibility and stability. Controlling and understanding the behaviour of such functionalized latex suspensions are critical for their comprehensive applications. To investigate the effect of the nanoparticle architecture on their rheological behaviour, a library of polystyrene nanoparticles functionalized with a canopy of end-tethered poly(methyl acrylate) chains with different degrees of polymerization and grafting densities was prepared. When the end-tethered polymer chains were long enough, the suspensions of polymer-functionalized nanoparticles underwent a liquid to gel transition when the concentration of the nanoparticles was increased. The architecture of the polymer canopy was the determining factor for the mechanical properties of the resulting gels; nanoparticles with moderate grafting density where the polymer chains adopt a relaxed polymer brush conformation led to the formation of the strongest and most robust gels. In comparison with suspensions prepared with polymer functionalized nanoparticles, particles with a soft and swollen core formed gels with higher yield stress at a lower solid content.
Introduction
Nanoparticles (NPs) with a grafted layer of polymers have become ubiquitous building blocks for applications from the biomedical field to the production of sustainable energy.1–4 In these systems, end-tethered polymer chains are often used to modify the properties of NPs such as stability, solvent compatibility, dispersibility, and assembly. Polymer chains tethered to latex nanoparticles represent a particular challenge since both the canopy of end-tethered chains and the core of the nanoparticles are soft and deformable. Thus, designing an ideal polymer layer to optimize the behaviour of the NPs for a specific application is non-trivial and new design principles are needed to rationally engineer such polymer canopies. Polymer NPs functionalized with a canopy made of end-tethered polymer chains combine the complex rheological behaviour of polymer chains and polymer colloids. Furthermore, to facilitate the processing of such materials, it is essential to understand how the architecture of such particles influences their behaviour in flow.Polymer solutions can be complex non-Newtonian viscoelastic fluids, especially in the semi-dilute and concentrated regime, when the concentration of the polymer in solution is above the critical overlap concentration. The behaviour of the polymer chains in solution depends on not only the concentration but also the architecture, topology, and flexibility of the chains.5,6 Branched polymers, such as star-, H-, or comb-shaped macromolecules, exhibit larger shear viscosities than their linear analogs.7,8 The viscosity of solutions of branched polymers increases as the number of branches increases, while the effect of the branch length becomes more marginal as the number of arms increases.9 One of the key features of branched systems is the restricted arm interpenetration and arm entanglement as the branching degree increases.10 In comparison with solutions of linear polymer chains, the overlap concentration of solutions of dendrimers or of solutions of polymers having a branched architecture occurs at higher concentrations because of the limited effective chain entanglement.11
In comparison with polymer solutions, the rheological behaviour of colloidal dispersions is controlled by even more factors; the viscoelasticity not only depends on the volume fraction occupied by the colloids and the colloid–colloid interaction,12 but is also affected by the reorganization and reorientation of the particles in suspension and their effects on the local flow patterns,13,14 and the formation (or destruction) of colloidal mesostructures.15,16 Furthermore, when dealing with soft and deformable colloids in suspension, both the shape and the volume occupied by the particles can change under the application of shear, leading to strong flow-dependent behaviour.17–19 The softness of the nanoparticles affects the mechanical properties of the resulting gels and glasses and the concentration needed to observe a liquid to gel transition.20,21 Soft and deformable particles are expected to form stronger gels than hard spheres.22
The behaviour of nanoparticles in suspension could be tuned by the functionalization of the surface of the nanoparticles with a canopy of end-tethered polymer chains.3,23,24 The behaviour of such nanoparticles dispersed in a polymer matrix has been shown to depend on the composition of the canopy as determined by the degree of polymerization (N) and grafting density (σ) of the chains since these parameters influence both the particle/particle interaction and the particle/environment interaction.25,26 The influence of N and σ on the stability of polymer functionalized nanoparticles dispersed in a solvent or in a solid matrix has been widely studied for nanoparticles with a rigid core. In such systems, the dynamics of the nanoparticle suspensions and the particle/particle interactions in suspension were mostly governed by the architecture of the polymer canopy27via the excluded volume interactions and chain configuration entropy,28,29 and fluctuations of the polymer canopy influenced the fragility of the particle assemblies.30 Furthermore, for similar polymer canopies, the degree of swelling of the polymer canopy changes the chain conformation in the canopy and the resulting behaviour of the suspensions.31
However, when the core particle is soft and deformable the influence of the architecture of the canopy (N and σ) has not been addressed, and significant differences could be expected. On the one hand, gel nanoparticles display a more polymer-like behaviour when they are highly swollen because of their compressibility and improved interpenetration. On the other hand, the swelling of the polymer canopy could promote the canopy interpenetration or the solvation shell surrounding the NPs can create new slipping planes and act as a lubricating agent for the nanoparticle suspensions.
Here, to understand the relationship between the rheological and mechanical properties of the suspension and the structure of soft-core/soft-corona polymer nanoparticles dispersed in a good solvent, we designed a library of soft and swollen crosslinked polystyrene (PS) nanoparticles functionalized with a canopy of end-tethered poly(methyl acrylate) (PMA) chains. Using surface-initiated atom transfer radical polymerization (SI-ATRP) the length of the polymer chains was precisely controlled, and the grafting density was tuned during the synthesis of the PS core. The rheological properties of suspensions of these PS@PMA NPs in different solvents were investigated to gain insights into how the architecture of the canopy of end-tethered polymer chains and how the combination of a swollen gel core with the swollen polymer canopy affect the NP suspension.
Experimental
Synthesis of a polystyrene nanoparticle core (PS-SS-Br NPs)
First, 6.5 mL of styrene (St, 1 eq.), 82 μL of divinylbenzene (DVB, 0.01 eq.) and 0.65 mL of hexadecane (HD, 0.04 eq.) were mixed with 0.0552 g of a 2,2′-azobis(2-methylbutyronitrile) initiator (V-59, 0.005 eq.). Then, 48 mL of sodium dodecyl sulfate (SDS) solution (10 mM) was added and the mixture was pre-emulsified for 15 min by magnetic stirring at 600 rpm. The solution was sonicated for 2 min (20 kHz, 70% A, 10 s on/2 s off) to obtain a miniemulsion before being transferred to a round bottom flask and heated to 80 °C. After 2.5 h of polymerization, 4 mL of a 250 mM solution of SDS was added and the system was purged by bubbling Ar for 10 min. Then, to create a thin layer of an inimer (MA-SS-Br, Fig. S1, ESI†) containing polymer network at the surface of the NP, a mixture of 89–98 mol% of St (1.33 mL), 1 mol% DVB (16.6 μL), 1–10 mol% of MA-SS-Br (0.0326–0.326 mL) and V-59 (0.0115 g) was subsequently added dropwise with a syringe pump at a rate of 1 mL h−1. The reaction mixture was left stirring overnight before being filtered. The PS NPs in the aqueous suspension were then precipitated in 200 mL of MeOH and air-dried. To completely eliminate HD and SDS from the PS NPs, the NPs were dispersed in THF and precipitated in MeOH three times. Finally, the NPs were air-dried.To determine the number of initiating sites at the surface of the NP, the sulfur content in the NPs was analyzed. The NPs were dispersed in water and stabilized with cetyltrimethyl ammonium chloride (CTAC). A PS-SS-Br NP suspension in DCM was added dropwise to 10 mL of an aqueous solution containing 5.0 mg of CTAC and sonicated. Then, DCM was evaporated under mild vacuum (200 mbar, 40 °C). The sulfur content in the resulting aqueous suspension of PS NPs was measured by inductively coupled plasma atomic emission spectrometry (ICP-AES) with an ACTIVA M spectrometer (Horiba Jobin Yvon) equipped with a Meinhardt-type nebulizer and a cyclone chamber, and processed with ACTIVAnalyst 5.4.
Synthesis of the end-tethered canopy of poly(methyl acrylate) on the surface of the PS core (PS@PMA)
In a typical reaction, 0.4 mL of methyl acrylate, 0.1 mL of a solution containing Cu(II)Br2 (2000 ppm) and PMDETA (Cu(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand = 1:10 molar ratio) in DMF were added into a vial containing a suspension of 50 mg PS-SS-Br NPs dispersed in 4 mL of anisole. PDMS (0.1 mL) was added to the suspension. The mixture was stirred and purged with argon for 30 min. Then, a 0.5 mL solution of ascorbic acid (1600 ppm) in DMF was added. This resulting suspension was degassed with argon for another 10 min, and the vial was then placed into an oil bath at 60 °C and allowed to react. Once the appropriate monomer conversion was reached, the reaction mixture was diluted with THF, precipitated in MeOH and dried overnight under vacuum. The reaction was repeated with various amounts of the monomer, Cu(II)/ligand and bromoisobutyrate grafted moieties.
:ligand = 1:10 molar ratio) in DMF were added into a vial containing a suspension of 50 mg PS-SS-Br NPs dispersed in 4 mL of anisole. PDMS (0.1 mL) was added to the suspension. The mixture was stirred and purged with argon for 30 min. Then, a 0.5 mL solution of ascorbic acid (1600 ppm) in DMF was added. This resulting suspension was degassed with argon for another 10 min, and the vial was then placed into an oil bath at 60 °C and allowed to react. Once the appropriate monomer conversion was reached, the reaction mixture was diluted with THF, precipitated in MeOH and dried overnight under vacuum. The reaction was repeated with various amounts of the monomer, Cu(II)/ligand and bromoisobutyrate grafted moieties.
To characterize the end-tethered PMA (Fig. 1), the chains were cleaved from the PS@PMA NPs. The disulfide bonds between the PS core and the PMA chains were cleaved by reduction with dithiothreitol. A suspension of PS@PMA NPs was prepared by mixing 0.1 g of PS@PMA NPs with 10 mL of DCM for 24 h. Then, DL-dithiothreitol (DTT, 10 mg) and two drops of 1,8-diazabicyclo[5.4.0]undec-7-ene (ca. 20 mg) were added to the NP suspension and stirred for 24 h. After evaporation of the solvent, the dried and cleaved polymer/particle mixture was mixed in 10 mL of THF with an additional 5 mg of DTT and stirred overnight. The suspension was then filtered through a syringe filter (PTFE, ∅ = 0.2 μm). The solutions were centrifuged at 29068g for 20 min to eliminate the residual PS core and the molecular weight of PMA in solution was measured by SEC in THF.
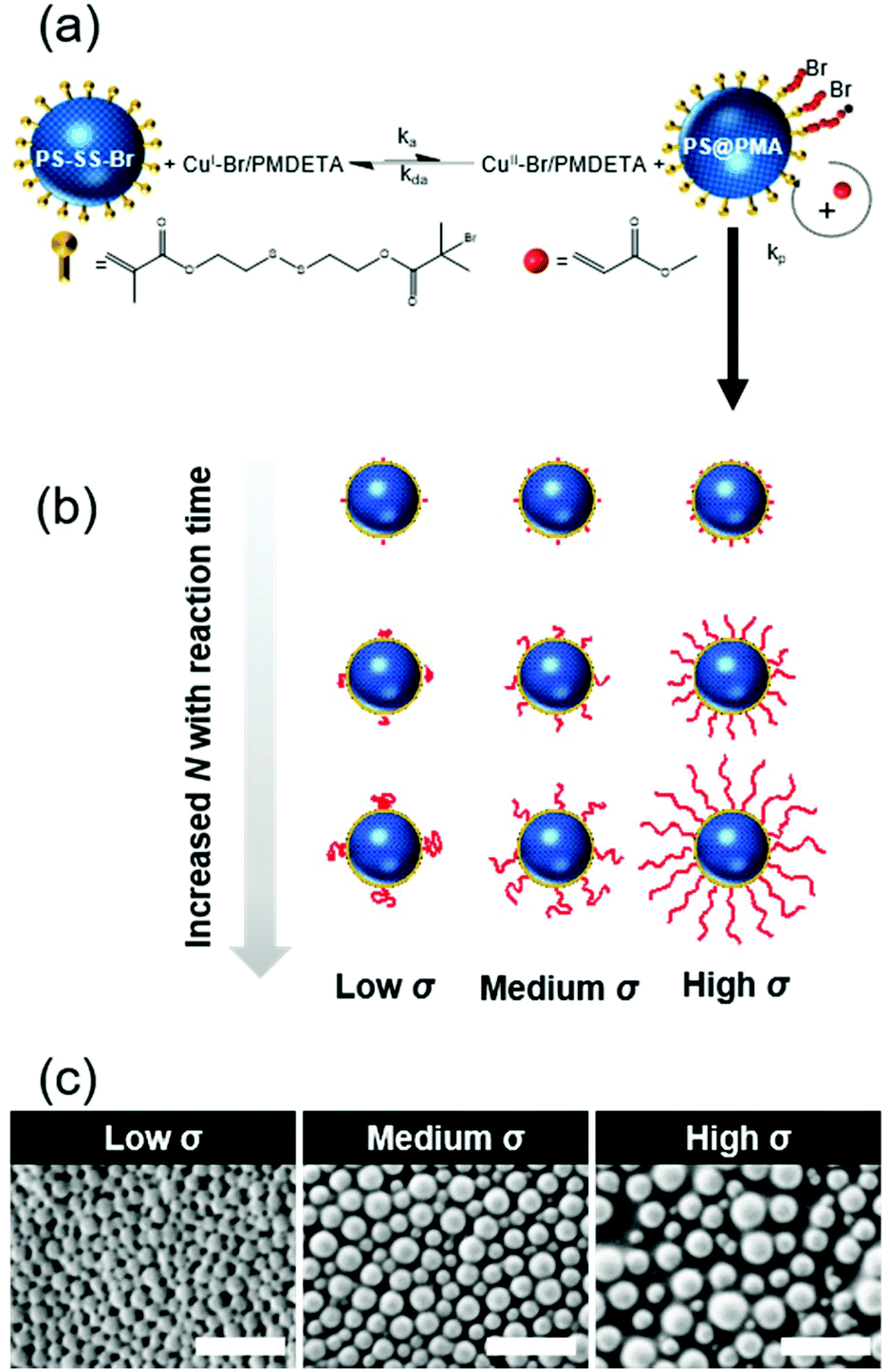 | ||
| Fig. 1 Preparation of a library of PS@PMA nanoparticles. (a) Synthetic scheme; (b) NPs with different grafting densities (σ) and chain lengths (N); and (c) SEM of the resulting PSx@PMA20k NPs. Scale bars are 500 nm. | ||
Rheological behaviour of PS@PMA suspensions
Suspensions of PS and PS@PMA NPs and a solution of free PMA (Mn of 40 K) were prepared in anisole at a concentration ranging from 0.1 to 15 wt%. The rheological properties of the suspensions were measured with a Bohlin Gemini 200 rotational rheometer at 25 °C equipped with a cone and plate geometry of 40 mm diameter and 4° cone angle. The gap was set at 150 μm. Continuous shear experiments were performed by varying the shear rate between 0.1 and 1000 s−1. The dynamic behaviour of the suspensions was studied using oscillatory shear experiments; for frequency-sweep experiments, the strain applied to the system was fixed to 10% and the frequency varied from 0.1 to 100 rad s−1 and in strain-sweep experiments, the frequency was fixed to 10 rad s−1 and the strain varied from 0.1 to 1000%.Results and discussion
A library of core–corona nanoparticles with a core of polystyrene grafted with a canopy of poly(methyl acrylate) chains (PSσ@PMAN) with different chain lengths (N) and grafting densities (σ) was prepared (Table 1). The synthesis of the particles proceeded via a three-step process. First, miniemulsion polymerization32 was used to prepare PS nuclei used to form core–shell NPs through a starve-fed emulsion polymerization process33 to create a thin layer of a copolymer of styrene and the inimer at the surface of the PS NP. The grafting density was tuned by controlling the concentration of the inimer in the outer layer of the PS core. Finally, each inimer was used to initiate the polymerization of methyl acrylate by surface-initiated atom transfer radical polymerization (SI-ATRP).34 In the dry state, at room temperature, the resulting NPs were composed of a rigid glassy core surrounded by a soft rubbery corona (Fig. 1c). PS and PMA were selected because they are strongly immiscible (χPS/PMA = 0.03·N = 169·χPS/PMMA)35,36 as observed experimentally with binary mixtures of PS and PMA and PS/PMA block copolymers readily phase segregating.35,37 Thus, the choice of PS and PMA prevented any specific interaction between the PS core and the PMA canopy potentially influencing the behaviour of the resulting NPs suspensions.| Sample | Reaction time (h) | M n PMA (103 g mol−1) | N | Effective grafting density σ (nm−2) | ||
|---|---|---|---|---|---|---|
| H2O | DMSO | Anisole | ||||
| PSh@PMA3k | 0.5 | 3.1 | 36 | 2.5 | 2.5 | 0.82 |
| PSh@PMA6k | 1 | 6.0 | 70 | |||
| PSh@PMA10k | 2 | 10.0 | 128 | |||
| PSh@PMA15k | 2.5 | 15.0 | 171 | |||
| PSh@PMA20k | 3 | 19.0 | 220 | |||
| PSh@PMA30k | 1 | 35.0 | 406 | |||
| PSh@PMA40k | 3 | 42.0 | 493 | |||
| PSh@PMA50k | 6 | 54.0 | 623 | |||
| PSm@PMA3k | 0.5 | 2.6 | 31 | 0.80 | 0.80 | 0.22 |
| PSm@PMA6k | 0.5 | 6.9 | 79 | |||
| PSm@PMA20k | 3 | 20.0 | 228 | |||
| PSm@PMA30k | 2 | 30.0 | 349 | |||
| PSm@PMA40k | 3 | 42.0 | 493 | |||
| PSm@PMA50k | 4.5 | 51.0 | 593 | |||
| PSl@PMA3k | 0.5 | 3.8 | 44 | 0.17 | 0.13 | 0.04 |
| PSl@PMA6k | 1 | 8.5 | 99 | |||
| PSl@PMA20k | 2 | 23.0 | 267 | |||
| PSl@PMA30k | 1.5 | 32.0 | 377 | |||
| PSl@PMA40k | 2 | 41.0 | 474 | |||
| PSl@PMA50k | 2.5 | 52.0 | 610 | |||
Furthermore, using SI-ATRP to grow PMA from the PS surface allowed to precisely tune the length of the grafted chains, since the polymerization of the methyl acrylate was controlled as demonstrated by the linear increase of monomer conversions with the polymerization time (Fig. S2, ESI†). The experimental molecular weights of the PMA brushes determined by SEC (Mn,SEC) were in good agreement with the expected values based on the monomer conversion measured by NMR spectroscopy and the number of initiating sites measured by ICP (Table S2, ESI†). The chemical composition in PS and PMA within the NPs could be controlled by the architecture of the PMA canopy. The chemical composition of the NPs was quantified by FTIR spectroscopy using a series of binary mixture of pure PS NPs and linear PMA in known weight ratios as a calibration (Fig. S3, ESI†). The contents of PMA in each sample scaled as expected with the different σ and N (Fig. 2a). The combined analysis of the NPs by ICP, GPC, NMR and FTIR spectroscopy showed that the length and grafting density of the PMA chains in the PS@PMA NPs were precisely controlled. This allowed to tune the architecture of the PMA canopy.
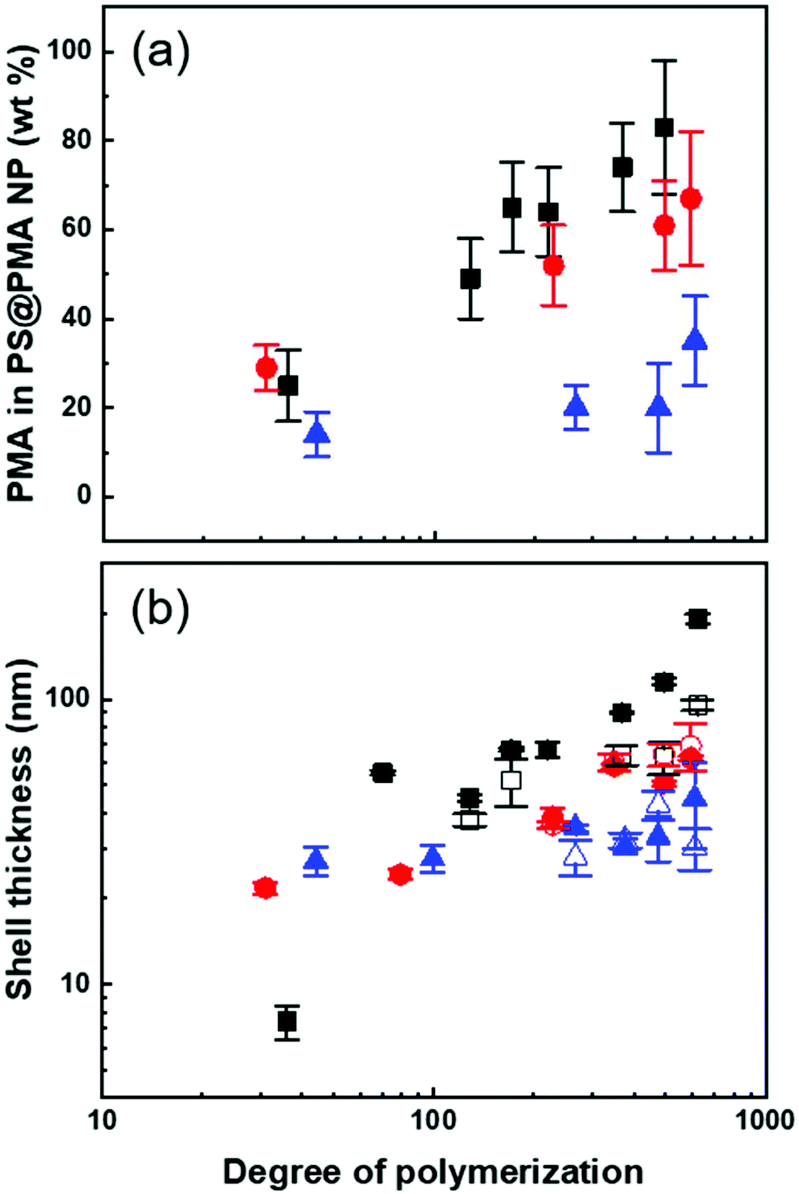 | ||
| Fig. 2 Effect of the architecture of the PMA canopy on (a) the mass fraction of PMA in the NPs measured by FTIR, and (b) the shell thickness of the PMA layer for NPs in dilute suspensions in anisole (solid symbol) and in DMSO (open symbol) measured by DLS for PS@PMA NPs with increasing grafting density (▲) PSl@PMA (0.17 chains per nm2), (●) PSm@PMA (0.80 chains per nm2) and (■) PSh@PMA (2.5 chains per nm2). | ||
The PS@PMA NPs with their well-defined composition were used to investigate the effect of the NP architecture on the behaviour of their resulting suspensions. The thickness of the canopy of PMA was dependent on the solvent quality and decreased when the solvent quality became poorer.38 In water, poor solvent for both PS and PMA (Table S1, ESI†), both PS and PMA were shrunken, and the variation in the diameter with the increase in the N of PMA was limited (Table S2, ESI†). The difference in the swelling of the PS core in the various solvents used resulted in a variation of the effective grafting density observed for a given NPs (Table 1). In anisole, good solvent for both PS and PMA, and in DMSO, poor solvent for PS but good solvent for PMA (Table S1, ESI†), the PS@PMA NPs swelled and a variation of the thickness of the canopy with its architecture was observed. When pure PS NPs were dispersed in anisole, they experienced a swelling of ca. 100%, whereas when the same NPs were dispersed in DMSO they only experienced a swelling of less than 15% (Table S2, ESI†). PS@PMA NPs were dispersed in the same solvents and the difference in the size of the swollen PS@PMA NPs were compared to the size of the pure PS NPs in the same solvent to calculate the thickness of the PMA canopy. At low grafting density, only a marginal increase in diameter was observed because the canopy of PMA was collapsed on the surface and occupied a limited volume (Fig. 2 and Table S2, ESI†). At higher σ, an increase in the diameter of the NP was observed in water due to the crowding of neighboring polymer chains forcing PMA to adopt a stretched conformation.
The thickness of the layer of tethered polymer chains on a substrate varies with N, the repulsion between the monomers, the solvent type, σ and blob size.39,40 When the substrate is the curved surface of a NP the behaviour of the end-tethered chains is more complex than that on a flat substrate due to the curvature dependence of the conformation of the polymer chain since the local polymer concentration decreases with an increase in the distance from the surface of the NPs.38,41 Here, the solvated radius of the NPs was used to calculate the thickness of the PMA corona (Fig. 2b). The correlation between the thickness of the brush layer (T) and N in various solvents scales with:
| T ∼ k(N)α | (1) |
The results of the fit of eq. 1 to the thickness of the PMA canopy (Table S3, ESI†) show that in anisole and in DMSO, α increased as σ increased due to the steric constraints of tethered polymer chains. At a high grafting density, the variation in the thickness of the PMA canopy suggested that due to a high local polymer concentration and high solvent quality, the brushes adopted a stretched conformation. For NPs with moderate grafting density, the brushes were extended, but not fully stretched, indicative of the semi-dilute brush regime.44 For samples with low σ, the polymer brushes adopted a more collapsed conformation. When the NPs were dispersed in water, no clear trend in the variation of the thickness of the PMA canopy with N and σ could be observed. The effects of N and σ on the thickness of the PMA canopy suggest that, in anisole, the tethered chains were forced to adopt a stretched conformation on the surface of NPs with high and medium σ due to the steric hindrance, while at lower σ the tethered chain adopted a more relaxed conformation.
The behaviour of the suspension of PS@PMA NPs, naked PS NPs and a binary mixture of PS NPs and free PMA40k in anisole was investigated under a continuous shear. The solution of free PMA40k chains and the solution containing a binary mixture of PS NPs and PMA40k chains both displayed mostly Newtonian behaviours (Fig. 3a) because of the limited interaction and entanglements in these systems. In contrast, suspensions of PS NPs and PS@PMA NPs exhibited a shear-thinning behaviour attributed to the presence of highly swollen PS or PS@PMA in anisole leading to the formation of either jammed suspensions or interdigitated networks.18,45
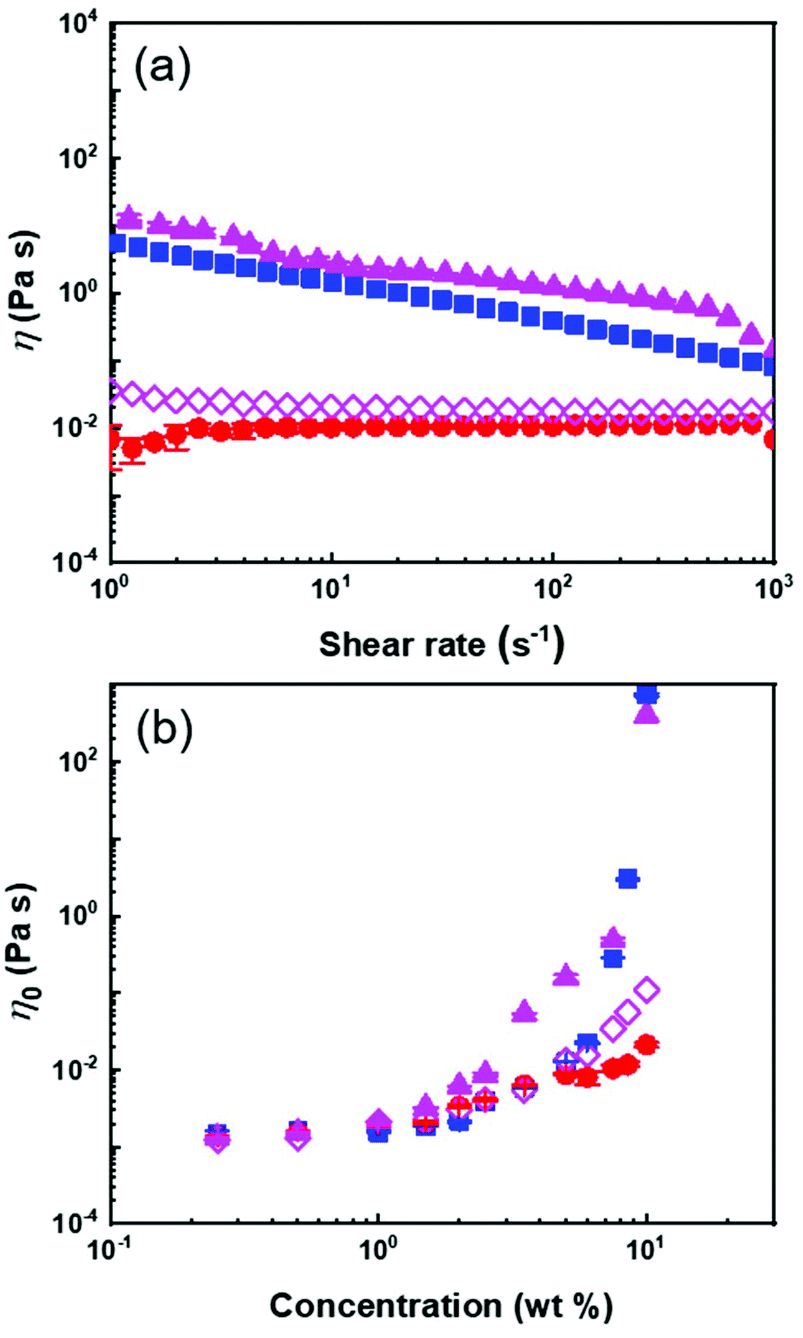 | ||
| Fig. 3 (a) Dynamic viscosity of 10 wt% suspensions and (b) the effect of the NP concentration of the zero-shear viscosity of suspensions of (■) PS NPs, (▲)PSh@PMA40k NPs, (●) free PMA40k and (◇) a binary mixture of PS NPs and free PMA40k in anisole. | ||
The influence of the concentration on the viscosity of the NP suspensions was measured between 0 and 10 wt% in anisole. In continuous shear experiments, the viscosity of the PS@PMA suspension was affected by the number of NPs in the suspension and the architecture of the PMA canopy. The suspensions of PS@PMA displayed two regimes of distinct shear-dependent behaviour (Fig. S5, ESI†); at a low concentration, the suspension behaved as a simple Newtonian liquid over the entire range of the shear rate studied, while at a higher concentration (ca. 5 wt% for PSh@PMA40k) the suspensions behaved like shear-thinning fluids.
The zero-shear viscosity (η0) obtained for the suspensions prepared with the different NPs systematically increased with an increase in the concentration of NPs in the suspension (Fig. 3b). The viscosity of the suspensions at a given concentration increases with an increase in N. However, the effect of σ was as straightforward (Fig. S6†); for 10 wt% suspensions of NPs with relatively long brushes (N ca. 500), the viscosities of high and medium grafting density samples were similar but larger than those of samples with the lowest grafting density. The influence of the architecture of the PMA canopy was especially evident when comparing suspensions containing the same number concentration of NPs (or the same number concentration of PS core) (Fig. S7b†); for the same number of NPs in the suspensions, η0 increased with σ, while the influence of the architecture was less critical when comparing suspensions containing the same amounts of methyl acrylate (Fig. S7c†).
The transition between the dilute and semi-diluted regimes (C′) was defined as the inflection point in the variation of η0 with NP concentration (Fig. 3b).29 The C′ of the PS suspension, caused by space jamming, was ca. 6 wt% and the C′ of PS@PMA NPs decreased with the increase in the N and σ of the PMA chains (Fig. 4). In comparison with PS NPs, PS@PMA NPs could, in addition to space jamming, also undergo interpenetration of the PMA canopy and entanglement of the PMA chains can occur for the chains with larger N, leading to an increase in viscosity. However, the addition of free PMA chains decreased the viscosity of the suspension (Fig. 3).
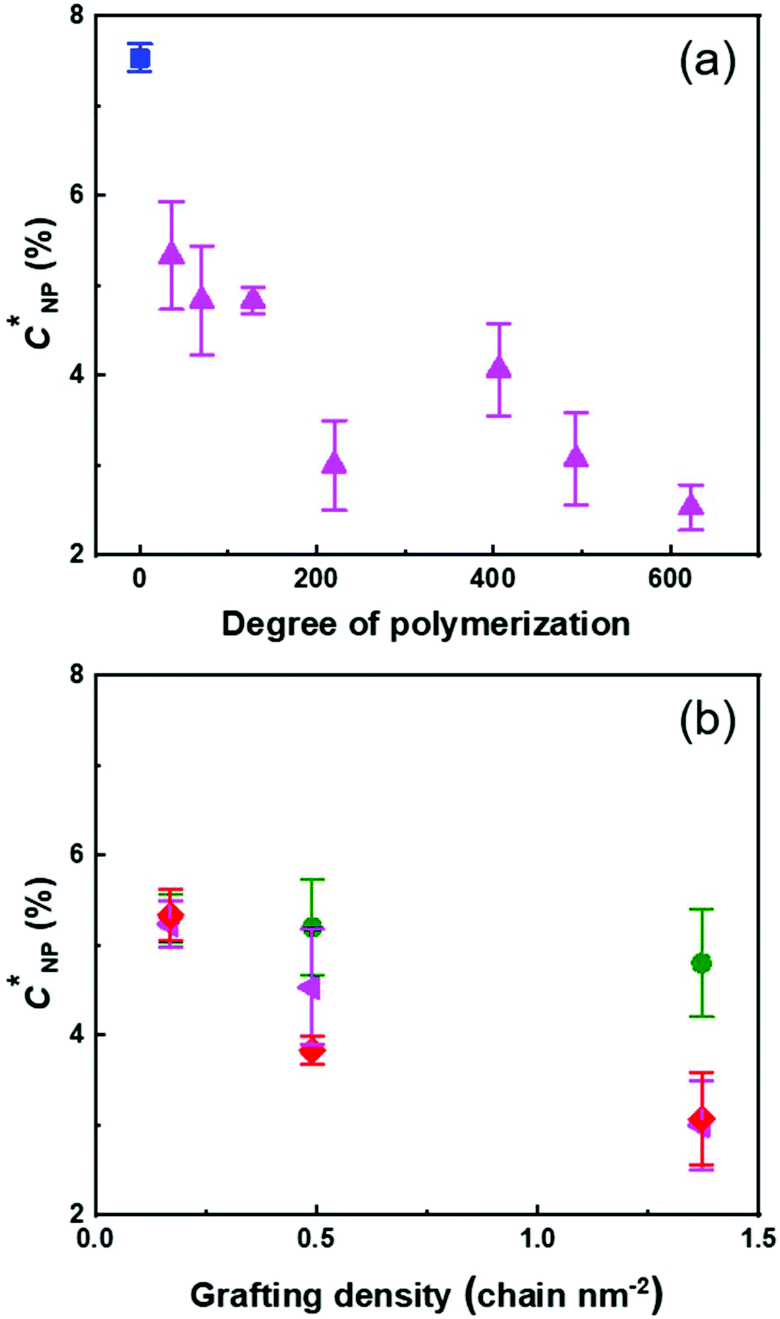 | ||
| Fig. 4 Effect of the architecture of the PMA canopy on the NP concentration at the transition between the dilute and semi-dilute regimes for a suspension of PS@PMA in anisole. (a) Effect of the PMA chain length for (▲) PSh@PMAx and (■) PS NPs, and (b) the effect of grafting density for (●) PSx@PMA6k, (◀) PSx@PMA20k and (◆) PSx@PMA40k. | ||
Fig. 4 shows that the concentration of NPs in the suspension required to observe the transition between the dilute and semi-dilute regimes decreased with increasing N and increasing σ. The interpenetration of the PMA canopy led to an earlier onset of the transition between the viscoelastic regimes. The rheological behaviour of the suspensions, at any given N and σ, was mainly influenced by the methyl acrylate content in the suspension (Fig. S8, ESI†), and the observed C′ decreased linearly with the number of methyl acrylate units decorating the PS core. The relation between C′ and the NP architecture was further evidenced when the concentration of C′ was calculated in terms of the number of NPs in the suspension ([NP]′ in Fig. S8c, ESI†). In this case, as both N and σ increased, a decrease in [NP]′ was observed. However, the total amount of the polymer on the NP was the key factor influencing [NP]′, and similar effects on [NP]′ were observed for NPs with a few long PMA chains or multiple short PMA chains, an increase in either N or σ promoting the NP–NP interactions and reducing the number of NPs needed to achieve the same effect of the flow behaviour of the suspensions.
When the concentration of PS@PMA increased above C′, a transition between a liquid-like state and a gel-like state was observed for some NP architectures (Fig. 5). Concentrated suspensions of PS NPs formed gels at a concentration greater than ca. 8 wt%, while suspensions of PS@PMA NPs with long chains (N > 200 units) formed gels at concentrations ranging from 2 to 15 wt%. However, suspensions of PS@PMA NPs with short chains (N < 175 units) did not form gels even at concentrations as high as 20 wt%. Similarly, binary mixtures of PS NPs and free PMA chains did not form gel-like suspensions even at high concentrations.
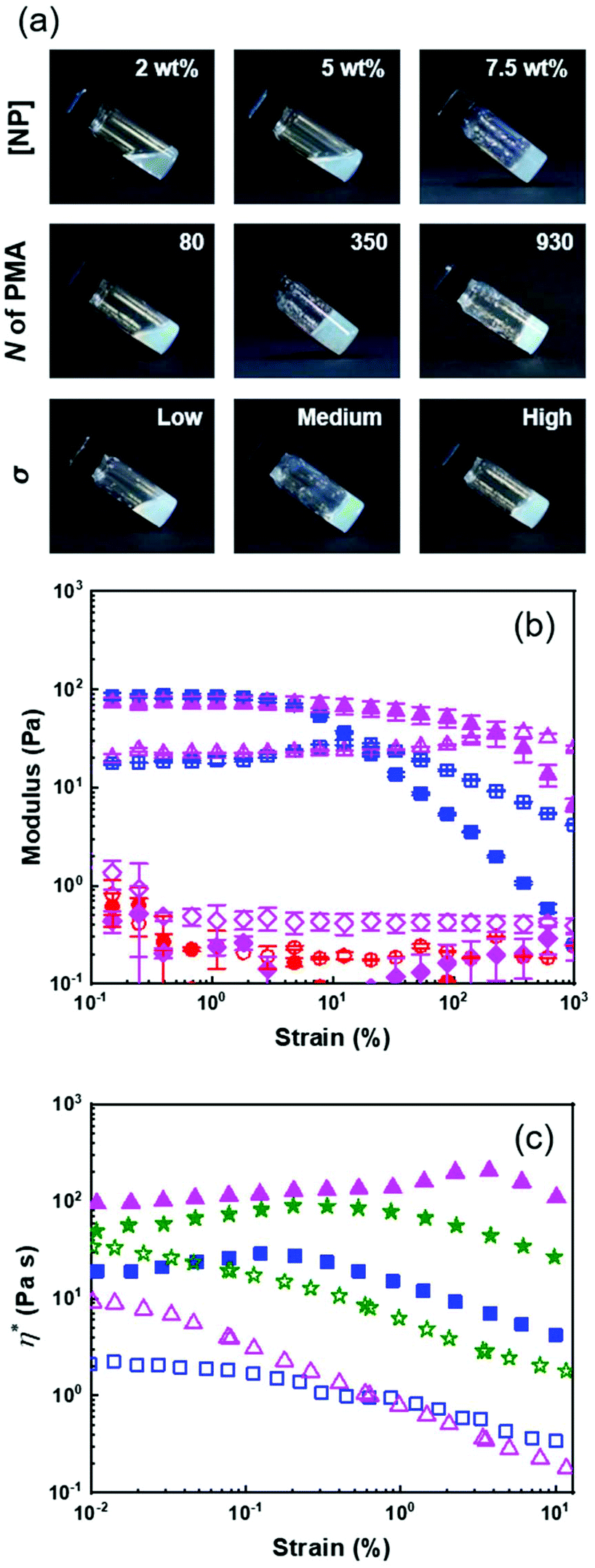 | ||
| Fig. 5 Formation of colloidal gels in concentrated suspensions of PS@PMA NPs. (a) influence of the NP concentration (PSm@PMA20k) and architecture of the PMA canopy (PSm@PMAx and PSx@PMA20k). (b) Elastic (solid symbol) and viscous (open symbol) moduli of 10 wt% suspensions of (■) PS NPs, (▲)PSh@PMA40k NPs, (●) free PMA40k and (◆) a binary mixture of PS NPs and free PMA40k. (c) Complex viscosity of 10 wt% suspensions of (■) PS NPs, (▲)PSh@PMA40k NPs, and (★)PSm@PMA40k NPs in anisole (filled symbol) and DMSO (open symbol). | ||
Fig. 5b shows that at 10 wt% in anisole, solutions of free PMA and binary mixtures of PS NPs and free PMA displayed viscous behaviour while concentrated suspensions of pure PS NPs and of grafted PS@PMA behaved as viscoelastic solids (G′ > G′′). Furthermore, gels formed with pure PS NPs displayed a lower yield strain than the gels made of PS@PMA NPs. While PS@PMA NPs formed viscoelastic solids, the binary mixture of PS NPs and free PMA, with the same chemical composition, did not. The formation of colloidal gels by the PS NP suspensions could only be ascribed to core–core interactions and colloidal jamming. The addition of free PMA chains to the PS NPs suspension prevented the formation of such networks leading to a transition between a viscoelastic solid and a viscous liquid. Similarly, the grafting of short PMA chains also prevented efficient PS–PS interactions required to form a colloidal gel. However, when the PMA chains grafted to the PS core were much longer than the critical entanglement length (Ne,PMA = 125)46 of PMA, the interpenetration, and potential entanglement, of the PMA canopies led to the formation of a network of PS@PMA NPs, where the PS core in the PS@PMA NPs acted as junction points. In addition to anisole, which is a good solvent for both the PS core and the PMA canopy, suspensions of PS@PMA were also prepared in DMSO, a selective solvent for PMA. When the same NPs were dispersed in DMSO and in anisole at the same concentration (Fig. 5c), the suspensions in anisole systematically displayed more solid-like behaviour and a larger complex viscosity than the suspensions in DMSO. Even when taking into account the volumes occupied by the NPs in different solvents (Fig. S10, ESI†), only weak gels were formed in DMSO although the polymer canopies were similarly swollen in both solvents (Fig. 2 and Table S3†). The swelling of the core in anisole in comparison with that in DMSO was the only variable parameter and the easier gelation in anisole must be ascribed to the interplay between the PS core and the PMA canopy.
Both the modulus and the yield strain of the PS@PMA NP suspensions were affected by the architecture of the PMA canopy. Fig. 6a shows that, at a fixed σ, as N of the canopy increased, the modulus of a 10 wt% suspension increased and the behaviour of the suspension transitioned from liquid-like to gel-like. This transition was observed at a concentration much lower than what was observed with hard NPs functionalized with polymer chains. This was even more directly observed by comparing the mechanical properties of PS@PMA NPs in suspensions in anisole producing suspensions of the soft core surrounded by a soft canopy and suspensions in DMSO where a hard core is functionalized with the same polymer canopy (Fig. S10†). The suspensions in anisole of PS@PMA NPs functionalized with short PMA chains (N < 150 units) did not form gels in the concentration range studied. The stiffness of the suspension, defined as G* (G′ + iG′′) in the low strain regime, increased as a function of N (Fig. 7 and Fig. S9, ESI†). However, when the chain length was kept constant and σ increased (Fig. 6b) the properties of the colloidal gels prepared with the NPs varied in a more complex manner. For suspensions of PSx@PMA20k NPs (short brushes of N ca. 225 units) at 10 wt% in anisole only PSm@PMA20k and PSl@PMA20k displayed a gel-like behaviour, the suspension of NPs with the highest grafting density, PSh@PMA20k, remained a viscous liquid. The colloidal gel of PSm@PMA20k NPs was the stiffest (higher G*) and strongest (higher yield strain). However, for PSx@PMA40k NPs having longer PMA chains (N ca. 500 units) the formation of colloidal gels was observed for the three different σ (Fig. S10, ESI†) and the gels prepared with the PSl@PMA40k NPs were both the weakest and the most fragile, those prepared with PSh@PMA40k NPs showed the highest yield strain, and those prepared with PSm@PMA40k NPs were the strongest. These results show that high σ can prevent the efficient interdigitation of the PMA canopy and the resulting effective NP–NP interactions due to the large local PMA concentration in the canopies with high σ. Increasing the length of the polymer brush alleviated this effect likely because the effective local concentration of the polymer decreased as the distance between the surface of the PS core and the surface of the PMA canopy increased.
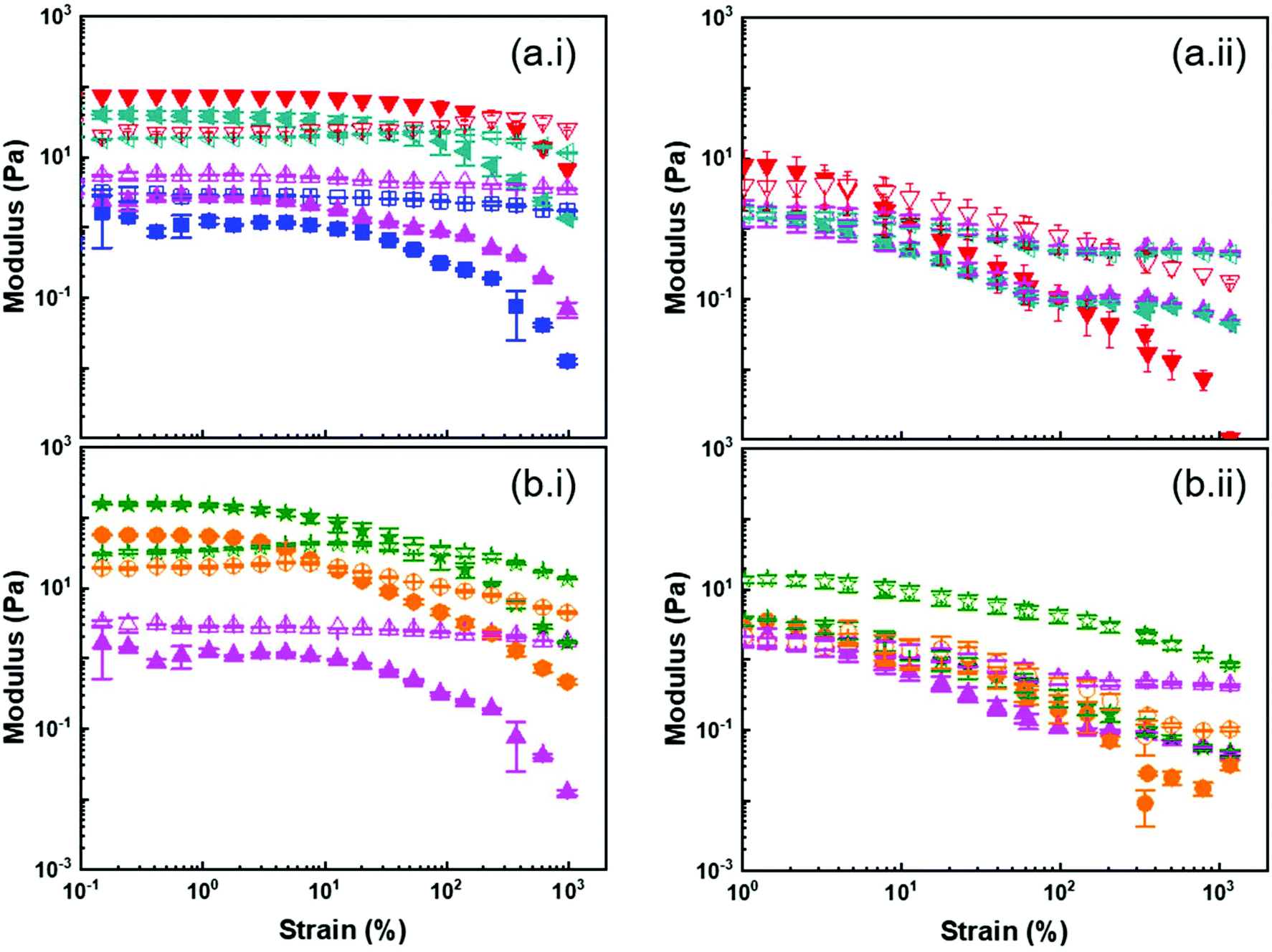 | ||
| Fig. 6 Visco-elastic behaviour (G′ (solid symbol) and G′′ (open symbol)) of 10 wt% suspensions of PSx@PMAN in anisole (i) and DMSO (ii). (a) Effect of the PMA brush length for (■)PSh@PMA10k NPs, (▲) PSh@PMA20k, (◀) PSh@PMA30k, and (▼) PSh@PMA40k. (b) Effect of grafting density for (●) PSh@PMA20k, (★) PSm@PMA20k and (▲) PSl@PMA20k. | ||
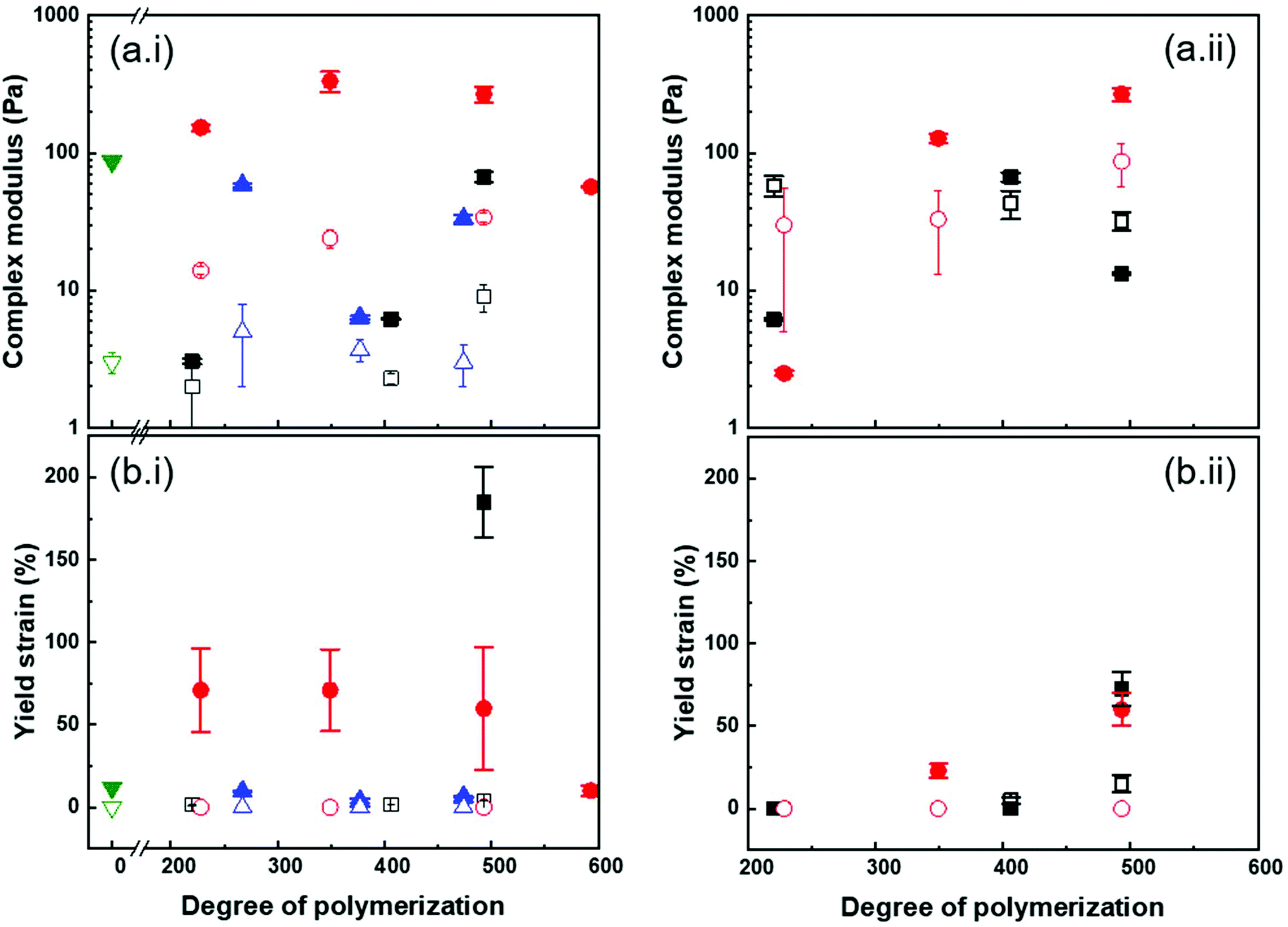 | ||
| Fig. 7 Effect of the architecture of the PMA canopy on the properties of the colloidal gels formed with (i) C = 10 wt% or (ii) ϕcalca. 0.5 of (■)PSh@PMA, (●) PSm@PMA, (▲) PSl@PMA and (▼) PSh@PMA40k in suspensions in anisole (closed symbol) or DMSO (open symbol). | ||
Fig. 7 shows that the gelation of PS@PMA NPs with longer PMA chains, for which interdigitation of the PMA canopy was favoured, led to an increase in the cohesion of the network formed. Yet, increasing the grafting density over a certain point decreased the efficiency of the canopy interdigitation. However, when the gels were prepared with a constant number of NPs in concentration (Fig. S11†), an increase in both N and σ led to an increase in the modulus and yield strain of the colloidal gels. The mechanical properties of the colloidal gels suggested that both the number of PS cores acting as either a crosslinking point or a filler in the polymer network and the fraction of PMA in the samples and thus the canopy interdigitation were critical in determining the final behaviour of the suspension.
At a constant mass fraction of NPs, suspensions of PS@PMA NPs in anisole were systemically stiffer and tougher than the suspensions of the same NPs in DMSO. While the canopies of the PS@PMA NPs were undergoing similar swelling in both solvents, it was not the case for the PS core. Taking into account the volume occupied by each NP in the different solvents, the concentration of NPs in suspension in DMSO needed to be higher than in anisole to reach the same volume fraction. Even when comparing suspensions with similar volume fractions of NPs, the suspensions in anisole behaved in a more solid-like manner and these suspensions were tougher, with significantly larger yield strain than the suspensions prepared in DMSO (Fig. 7). The difference of yield strain cannot be ascribed to the difference in the canopy architecture. Even if the variation of the grafting density during the swelling of the PS core was taken into account the samples prepared PSh@PMA dispersed in anisole should be directly comparable to the suspension of PSm@PMA in DMSO. The respective effective grafting density was 0.80 chains per nm2 for PSm@PMA in DMSO and 0.82 chains per nm2 for PSh@PMA in anisole. Since the PMA canopies had similar structures in anisole and in DMSO, the conformation of the chain in the canopy was similar in both solvents; thus the ease of interpenetration between adjacent NPs should be similar in both solvent, since the main difference between the two systems was only the swelling of the PS core. The results suggest that having NPs with a soft core functionalized with a polymer canopy favours the formation of tough and strong gels even at low concentrations.
Conclusions
In summary, a library of PS@PMA core-canopy particles with different canopy architectures, i.e., chain length (N) and grafting density (σ), was prepared and their behaviour in suspensions was analyzed using rheology. The conformation of the end-tethered polymer chains transitioned from collapsed chains, to brushes, to stretched brushes and was influenced by the degree of polymerization, grafting density and solvent quality. Using selective solvents uniquely for the canopy or a common solvent for the core and the canopy, suspensions with different behaviours were obtained, those with a “hard core” and those with a “soft core”.Polymer-functionalized soft core NPs displayed similar trends as expected for more traditional hard core NPs. As expected, suspensions of soft PS NPs and soft PS@PMA NPs displayed a non-Newtonian behaviour. The soft PS NPs formed jammed suspensions while for suspensions of soft PS@PMA NPs the interdigitation of the PMA canopy could be the main contributor to the shear thinning behaviour. The viscosity of the suspension was governed by both the concentration of the NPs in the suspension and the architecture of the PMA canopy. The concentration at which the transition between the dilute and semi-dilute regimes occurred shifted to a lower concentration with increasing N and σ.
As the concentration increased, the PS@PMA suspensions transitioned from a viscous liquid to a gel in which the PS core was a junction point for the entangled grafted PMA chains. This transition was observed at lower concentrations for NPs dispersed in a solvent where both the core and the canopy were swollen. The key factor affecting the sol–gel transition of the PMA functionalized PS NPs was the number of MA units decorating the PS core, but the properties of the resulting gels were largely influenced by the architecture of the PMA canopy and the swelling of the core. More effective interdigitation between the NPs occurred at high N and moderate σ due to the increased opportunity for canopy interpenetration. Gels were formed with either naked PS NPs or PS@PMA NPs with long PMA chains, where the interdigitation of the PMA canopy could be accompanied by chain entanglements, but not with PSx@PMAN NPs with short polymer brushes (N < 200 methyl acrylate units) or binary mixtures of PS NPs and free PMA chains. It was found that the mechanical properties of the suspension, both moduli and yield strain, were affected by both the corona architecture and concentration of NPs in the suspension. The direct comparison of soft-PS@soft-PMA NP and hard-PS@soft-PMA NP suspensions revealed that the soft NPs formed gels at lower concentrations and that the gels at similar NP contents were stronger and tougher when the core of PS@PMA was softer in comparison with suspensions prepared with a hard core. These results provide information to tailor the behaviour of the polymer latex by controlling the architecture of a layer of the grafted polymer to tune the rheological and mechanical properties of the latex suspensions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Dr Katharina Landfester for her continuous support. The authors acknowledge the financial support of the Max Planck Society and the support of the Max Planck Center for Complex Fluid Dynamics. The authors also thank Andreas Hanewald for technical assistance with the rheological measurements. W. W. is grateful for the scholarship from the Science Achievement Scholarship of Thailand. Open Access funding was provided by the Max Planck Society.Notes and references
- S. Srivastava, J. L. Schaefer, Z. Yang, Z. Tu and L. A. Archer, Adv. Mater., 2014, 26, 201–234 CrossRef CAS PubMed.
- S. Choudhury, R. Mangal, A. Agrawal and L. A. Archer, Nat. Commun., 2015, 6, 10101 CrossRef CAS PubMed.
- C. Yi, S. Zhang, K. T. Webb and Z. Nie, Acc. Chem. Res., 2017, 50, 12–21 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- Q. Ying and B. Chu, Macromolecules, 1987, 20, 362–366 CrossRef CAS.
- S. J. Dalsin, M. A. Hillmyer and F. S. Bates, Macromolecules, 2015, 48, 4680–4691 CrossRef CAS.
- J. R. Dorgan, J. S. Williams and D. N. Lewis, J. Rheol., 1999, 43, 1141–1155 CrossRef CAS.
- F. Khabaz and R. Khare, J. Chem. Phys., 2014, 141, 214904 CrossRef PubMed.
- A. Jabbarzadeh, J. D. Atkinson and R. I. Tanner, Macromolecules, 2003, 36, 5020–5031 CrossRef CAS.
- B. M. Erwin, M. Cloitre, M. Gauthier and D. Vlassopoulos, Soft Matter, 2010, 6, 2825–2833 RSC.
- Y. Takahashi, F. Suzuki, M. Miyachi, I. Noda and M. Nagasawa, Polym. J., 1986, 18, 89–94 CrossRef CAS.
- S. Arietaleaniz, P. Malgaretti, I. Pagonabarraga and R. C. Hidalgo, Phys. Rev. E, 2018, 98, 042603 CrossRef CAS.
- I. Cohen, T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 2004, 93, 046001 CrossRef PubMed.
- X. Cheng, J. H. McCoy, J. N. Israelachvili and I. Cohen, Science, 2011, 333, 1276–1279 CrossRef CAS PubMed.
- E. Zaccarelli, J. Phys.: Condens. Matter, 2007, 19, 323101 CrossRef.
- J. F. Morris, Rheol. Acta, 2009, 48, 909–923 CrossRef CAS.
- J. Mattsson, H. M. Wyss, A. Fernandez-Nieves, K. Miyazaki, Z. Hu, D. R. Reichman and D. A. Weitz, Nature, 2009, 462, 83–86 CrossRef CAS PubMed.
- F. Khabaz, T. Liu, M. Cloitre and R. T. Bonnecaze, Phys. Rev. Fluids, 2017, 2, 093301 CrossRef.
- M. E. A. Zakhari, M. Hütter and P. D. Anderson, J. Rheol., 2018, 62, 543–557 CrossRef CAS.
- A. Scotti, A. R. Denton, M. Brugnoni, J. E. Houston, R. Schweins, I. I. Potemkin and W. Richtering, Macromolecules, 2019, 52, 3995–4007 CrossRef CAS.
- N. Koumakis, A. Pamvouxoglou, A. S. Poulos and G. Petekidis, Soft Matter, 2012, 8, 4271–4284 RSC.
- P. van der Scheer, T. van de Laar, J. van der Gucht, D. Vlassopoulos and J. Sprakel, ACS Nano, 2017, 11, 6755–6763 CrossRef CAS PubMed.
- E. P. K. Currie, W. Norde and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2003, 100–102, 205–265 CrossRef CAS PubMed.
- R. M. Choueiri, E. Galati, H. Thérien-Aubin, A. Klinkova, E. M. Larin, A. Querejeta-Fernández, L. Han, H. L. Xin, O. Gang, E. B. Zhulina, M. Rubinstein and E. Kumacheva, Nature, 2016, 538, 79–83 CrossRef CAS PubMed.
- P. Akcora, H. Liu, S. K. Kumar, J. Moll, Y. Li, B. C. Benicewicz, L. S. Schadler, D. Acehan, A. Z. Panagiotopoulos, V. Pryamitsyn, V. Ganesan, J. Ilavsky, P. Thiyagarajan, R. H. Colby and J. F. Douglas, Nat. Mater., 2009, 8, 354–359 CrossRef CAS PubMed.
- S. Liu, E. Senses, Y. Jiao, S. Narayanan and P. Akcora, ACS Macro Lett., 2016, 5, 569–573 CrossRef CAS.
- J. G. Ethier and L. M. Hall, Macromolecules, 2018, 51, 9878–9889 CrossRef CAS.
- L.-N. Krishnamurthy, N. J. Wagner and J. Mewis, J. Rheol., 2005, 49, 1347–1360 CrossRef CAS.
- P. Voudouris, J. Choi, H. Dong, M. R. Bockstaller, K. Matyjaszewski and G. Fytas, Macromolecules, 2009, 42, 2721–2728 CrossRef CAS.
- M. Asai, A. Cacciuto and S. K. Kumar, ACS Cent. Sci., 2018, 4, 1179–1184 CrossRef CAS PubMed.
- C. Zhang, S. Yang, V. Padmanabhan and P. Akcora, Macromolecules, 2019, 52, 9594–9603 CrossRef CAS.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- R. A. Ramli, W. A. Laftah and S. Hashim, RSC Adv., 2013, 3, 15543–15565 RSC.
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- B. Zhao, W. J. Brittain, W. Zhou and S. Z. D. Cheng, Macromolecules, 2000, 33, 8821–8827 CrossRef CAS.
- E. A. Men'shikov, A. V. Bol'shakova and I. V. Yaminskii, Prot. Met. Phys. Chem. Surf., 2009, 45, 295–299 CrossRef.
- B. Zhao and W. J. Brittain, Macromolecules, 2000, 33, 8813–8820 CrossRef CAS.
- C. M. Wijmans and E. B. Zhulina, Macromolecules, 1993, 26, 7214–7224 CrossRef CAS.
- S. Alexander, J. Phys., 1977, 38, 983–987 CrossRef CAS.
- P. G. De Gennes, J. Phys., 1976, 37, 1445–1452 CrossRef CAS.
- M. Daoud and J. P. Cotton, J. Phys., 1982, 43, 531–538 CrossRef CAS.
- L. C. H. Moh, M. D. Losego and P. V. Braun, Langmuir, 2011, 27, 3698–3702 CrossRef CAS PubMed.
- M. J. A. Hore, Soft Matter, 2019, 15, 1120–1134 RSC.
- Y. Wei, Y. Xu, A. Faraone and M. J. A. Hore, ACS Macro Lett., 2018, 7, 699–704 CrossRef CAS.
- P. A. M. Steeneken, Carbohydr. Polym., 1989, 11, 23–42 CrossRef CAS.
- L. J. Fetters, D. J. Lohse and W. W. Graessley, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1023–1033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01558b |
| This journal is © The Royal Society of Chemistry 2020 |