
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate-independent Cu(0)-mediated controlled radical polymerization: grafting of block copolymer brushes from poly(dopamine) modified surfaces†
Daniel
Hafner
and
Rainer
Jordan
 *
*
Professur für Makromolekulare Chemie, Technische Universität Dresden, Mommsenstr. 4, 01062 Dresden, Germany. E-mail: rainer.jordan@tu-dresden.de
First published on 16th January 2020
Abstract
A method is presented allowing the preparation of polymer brushes from numerous monomers and on a variety of substrates. Poly(dopamine) (PDA) is used to cover a series of different surfaces (SiO2, Au, Cu, Al/Al2O3, Teflon) and for the subsequent preparation of bromine containing initiator layers by conversion with 2-bromoisobutyryl bromide (BiBB). Surface-initiated Cu(0)-mediated controlled radical polymerization (SI-CuCRP) enables a rapid polymerization of methacrylates and styrene on the PDA/BiBB layers with minimal effort. When the substrates are faced with a copper plate and submerged into reaction solution containing monomer, solvent and ligand, thick polymer brushes up to hundreds of nanometers can be achieved within 1 h. Despite the simplicity of the method and the fast polymerization rates at room temperature, an outstanding endgroup fidelity, which is unmatched for surface polymerization, can be observed. This fact is demonstrated by the preparation of pentablock copolymer brushes representing the highest block number ever reported for surface-bound polymer.
Introduction
Surface-initiated polymerization (SIP) has become a popular approach for the synthesis of surface-bound polymers allowing the introduction of chemical functionality, adjustment of wettability and surface energy or control of bioadhesion.1,2 As advantageous polymer brush characteristics are ensured by the high grafting densities achieved with SIP, all types of polymerization have been adapted to this method.Most attention was drawn to surface-initiated atom transfer radical polymerization (SI-ATRP).3 Since its development in 1995, ATRP has been labeled a useful tool for the synthesis of complex architecture due to excellent control of structure under mild reaction conditions and applicability to various monomers.4–6 However, sufficiently high concentrations of the catalyst, namely Cu(I), had to be used and addition of Cu(II) salts was necessary to ensure good control of polymerization.3 Therefore, several techniques have been developed to reduce the amount of Cu catalyst. All of them rely on the reduction of excess Cu(II) back to the activating species, be it via use of chemical agents,6 electrical current7 or photochemistry.5 Additionally, these approaches improved the oxygen tolerance of the polymerization. Still, only limited preservation of endgroup fidelity is possible, when ATRP is employed on surface, which is shown by the fact that attempts of block copolymerization result in diblocks at maximum.6 Another approach, i.e. the surface-initiated Cu(0)-mediated controlled radical polymerization (SI-CuCRP), was reported by our group.8 Here, a copper plate fitting the geometrical demand of the substrate is used as Cu source and reaction takes place in between the facing surfaces. Polymerization is restricted to the initiator bearing substrate, so that reaction solution and copper plate can be reused and only minimum amounts of solution are needed.8,9 Furthermore, this method not only enables synthesis of polymer brushes with unmatched growth rates, but is also applicable on wafer-scale and under ambient conditions without loss of initiating moieties. Synthesis of tetrablock copolymers demonstrated the excellent endgroup fidelity provided by this SIP approach.9
No matter what kind of SIP is employed the introduction of an initiating layer prior to the polymerization itself is necessary since most substrates only show insufficient reactivity or do not provide the required functionality.10,11 Therefore, a variety of techniques have been developed including layer-by-layer (LbL)12,13 and Langmuir–Blodgett (LB)14 films, spin coating15,16 or chemical vapor deposition (CVD).17 Also, self-assembled monolayers (SAMs) have been widely used as they form dense and uniform layers leading to well defined reaction mechanisms.8,10,11 Unfortunately, surface modification with low molecular substances as well as the use of non-covalent methods like dip or spin coating may struggle with a lack of solvolytical or thermal stability.18,19 Furthermore, these techniques often demand tedious preparation, complex experimental set up and are time consuming procedures. Hence, more straightforward approaches are desirable.
After a publication on poly(dopamine) (PDA) by Lee et al. in 2007, the mussel-inspired coating received a lot of attention.20 Mussels are well-known fouling organisms able to attach to nearly any kind of organic or inorganic surfaces. The key to these excellent adhesive properties was found in the Mytilus edulis foot protein 5 (Mefp-5), which contains high amounts of 3,4-dihydroxyphenylalanine (DOPA) and lysine. Lee et al. came up with the small molecule compound dopamine (DA) to mimic the two main functions of Mefp-5, i.e. the catechol group of DOPA and the amino group of lysine. It was found that DA undergoes spontaneous self-polymerization/self-cross linking in slightly alkaline water (pH = 8.5) and ambient conditions. If a substrate is immersed into solution, PDA layers can be controllably obtained on surface reaching up to 50 nm. As speculated, such deposition was performable on a wide range of materials independent from their original surface nature.
Its self-polymerizing/self-cross linking nature allows the formation of PDA layers with minimum experimental set-up, offering new strategies for simple modification and functionalization of substrates.20–22 Although the actual polymerization mechanism as well as its structure are still a topic of discussion, it seems certain that PDA provides many functional groups including carboxylic units, primary and secondary amines and hydroxyl groups.23–26 A recent report by Maier et al. suggests that amine as well as hydroxyl groups synergistically contribute to the robust adhesion of PDA.27 Especially, these outstanding adhesive properties make poly(dopamine) an universal mediating agent for surface polymerization as deposition is possible on nearly any kind of surface, including (noble) metals and metal oxides,20,28,29 steel,23 carbon nanotubes (CNTs)30,31 or graphene,32 nanoparticles33,34 and poly(tetrafluoroethylene) (PTFE).17,19,25 Furthermore, the quantity and variety of functions enable the additional modification with polymers via Michael/Schiffbase reaction,35 self-initiated photografting and photopolymerization (SIPGP),36,37 different SI-ATRP methods22,28,38–40 or surface-initiated reversible addition fragmentation chain transfer (SI-RAFT).41,42
In this work we present the combination of PDA deposition and SI-CuCRP for the controlled grafting of homogeneous polymer brushes on a series of different substrates within short time frames (Fig. 1). Moreover, it is demonstrated that this method allows the preparation of block copolymer brushes on poly(dopamine) layers. Thus, a universal method for fast, controlled and substrate-independent grafting is attained.
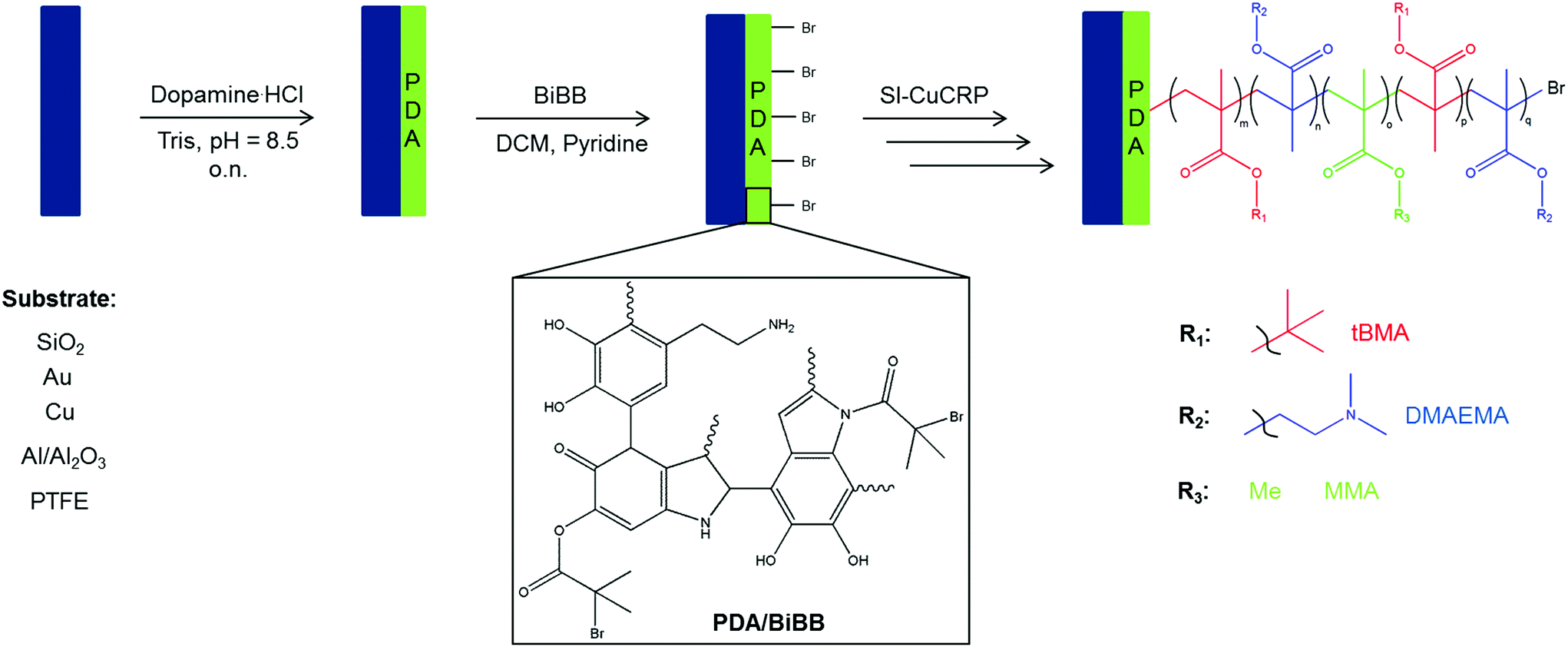 | ||
| Fig. 1 Schematic illustration of the synthetic pathway to pentablock copolymer brushes via SI-CuCRP on PDA surface. | ||
Results and discussion
As SI-CuCRP was never before performed on PDA surface, a series of experiments were dedicated to establish the combination of both as a versatile method for surface modification. In that context, it had to be proven that PDA does not limit control of SI-CuCRP due to its own partly radical character.43,44 Starting from PDA deposition, substrates were prepared for further polymerization steps by subsequent binding of initiator. Then, SI-CuCRP was performed to graft polymer brushes on SiO2 and several other substrates to exploit PDA chemistry. Furthermore, grafting of block copolymer brushes was accomplished to demonstrate the method's robustness.SI-CuCRP on PDA/BiBB
First, a series of monomers were grafted on an initiator bearing PDA surface. PDA was deposited on a silicon wafer (SiO2, 300 nm) in a previously reported manner by placing the substrate into a freshly prepared solution of dopamine (1 g l−1) in 10 mM trishydroxymethyl aminomethane/HCl buffer (Tris/HCl, pH = 8.5).37 The deposition was allowed to proceed overnight. Then, 2-bromoisobutyryl bromide (BiBB), a typical ATRP initiator, was reacted with the PDA layer to give PDA/BiBB. Finally, the wafer was covered with a copper plate at a distance of 0.5 mm and immersed into a degassed solution of monomer, solvent and ligand (1,1,4,7,7-pentamethyldiethylendiamine, PMDETA) at room temperature. In a similar fashion polymer brushes on SAM-bound initiator (APTES-BiBB) were synthesized for comparison.Successful conversion of PDA and APTES surface with BiBB was confirmed by the change of static water contact angle θs from 36° to 68° for PDA/BiBB and from 54° to 68° for APTES-BiBB. Furthermore, X-ray photoelectron spectroscopy (XPS, Fig. S1†) show the appearance of a Br 3d signal at ∼70 eV after conversion with BiBB proving the binding of initiator to the respective surface.
After SI-CuCRP the resulting polymer brushes were analyzed with ellipsometry and contact angle measurements. All polymer brushes exhibit a typical contact angle (Table 1) showing the successful grafting of each monomer on both APTES-BiBB and PDA/BiBB. Furthermore, remarkable brush thicknesses (d) were reached with every monomer considering that reaction time did not exceed 1 h (Table 1). Especially hydrophilic monomers reacted with high rates reaching up to 250 nm for poly(2-hydroxyethyl methacrylate) (PHEMA).
| Monomer | Solvent | t R [min] | APTES | PDA | ||
|---|---|---|---|---|---|---|
| d [nm] | θ s [°] | d [nm] | θ s [°] | |||
| HEMA | H2O | 60 | 238 ± 15 | 52 ± 3 | 250 ± 20 | 56 ± 2 |
| DMAEMA | 10%(v/v)-H2O in iPrOH | 60 | 58 ± 6 | 58 ± 5 | 45 ± 5 | 59 ± 5 |
| MMA | DMSO | 60 | 127 ± 10 | 69 ± 2 | 79 ± 5 | 68 ± 4 |
| St | DMSO | 60 | 25 ± 5 | 90 ± 1 | 29 ± 6 | 87 ± 2 |
| tBMA | DMSO | 20 | 41 ± 8 | 95 ± 6 | 51 ± 5 | 101 ± 2 |
Even with only small amounts of water in an otherwise unsuited solvent as isopropanol (iPrOH) thicknesses of 58 nm and 45 nm were achieved for poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA).
Hydrophobic monomers display slower growth of polymer brush. These results are not very surprising, as radical polymerizations in the presence of Cu(0) are known to proceed faster in H2O than in dimethyl sulfoxide (DMSO).45,46 However, styrene was still polymerizable in DMSO and reached a polymer brush height of around 25–30 nm after 60 minutes. Furthermore, it is remarkable that for tert-butyl methacrylate (tBMA) thicknesses of 41 nm and 51 nm were reached even after reducing reaction time to 20 minutes.
It is of particular interest that the results of SI-CuCRP are similar for both surface-bound initiator systems making PDA/BiBB an alternative to the common APTES-BiBB without restriction of polymerization and providing new options for surface modification.
Polymer brush synthesis on different substrates
As PDA is well known for its strong adhesion on any kind of substrates, SI-CuCRP can be exploited to graft polymer brushes on different surfaces in a facile, fast and controlled way. Therefore, polymerization was carried out on several substrates including metal (Au, Cu), oxides (Al/Al2O3, SiO2) and repellent organic surface (PTFE). The experimental procedure remained as described above. However, reaction time was decreased to 15–30 minutes (Table S1†), since the used monomer (HEMA) showed high polymerization rates.The grafting of PHEMA on the different substrates was confirmed by Fourier transform infrared spectroscopy (FT-IR) and contact angle measurements. While static contact angles (Fig. 2) ranged from very hydrophilic (SiO2, θs < 10°) to very hydrophobic (PTFE, θs = 112°) for the pure substrate, it changed strongly upon deposition of PDA (θs = 29–42°) and subsequent conversion with BiBB (θs = 68–74°) indicating successful modification on all substrates. After SI-CuCRP typical θs for PHEMA was observed on every surface exhibiting similar values of 54° and 57° on Al/Al2O3 and Cu, respectively (Fig. 2). SiO2 and gold both had a contact angle of 51° after SI-CuCRP. Interestingly, while polymerization worked well on every surface, conversion of BiBB on PDA grafted PTFE led to a successive destruction of the PDA layer within 1 hour. After all, it seems that adhesion of PDA on PTFE is not as robust as expected, so that reaction time had to be reduced to 20 minutes. Still, subsequent polymerization of HEMA decreased the contact angle to 58° due to the hydrophilicity of the resulting polymer brush on the PTFE substrate.
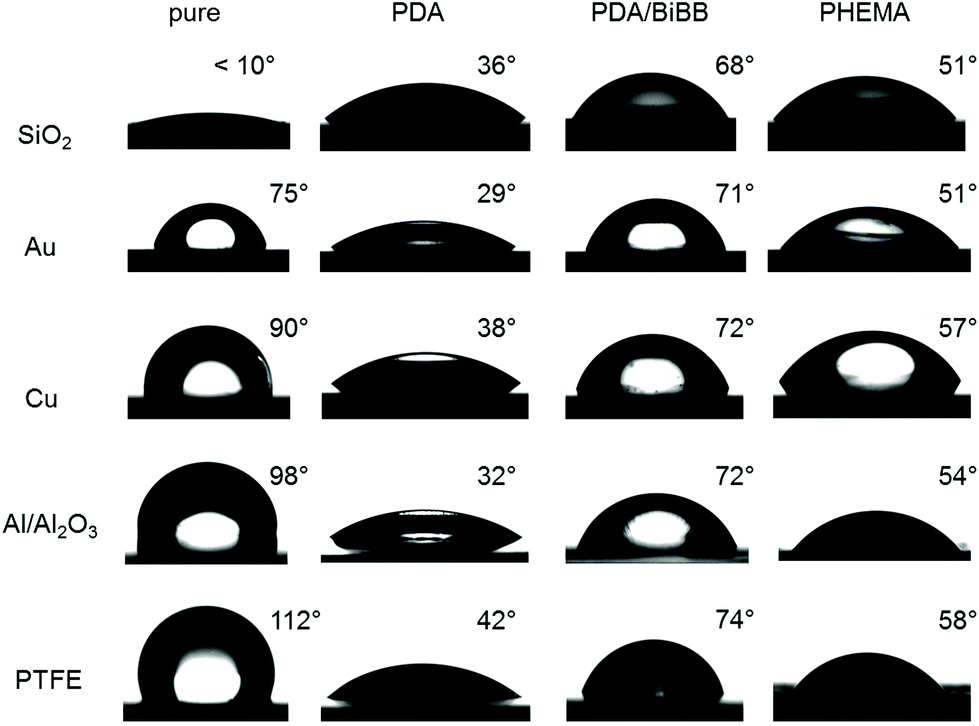 | ||
| Fig. 2 Photographs of static water contact angle on pure substrate and substrates grafted with PDA, PDA/BiBB and PHEMA. | ||
It is necessary to point out that the functionalization of PDA with initiator on PTFE is the only occasion, where destruction of PDA was observed. This was not the case on any other substrate. Furthermore, no indication of PDA weakness was observed after polymerization even after grafting multiple blocks of polymers up to 270 nm (see below). A partial destruction of PDA would lead to a sudden decrease of thickness after grafting due to rupture of polymer brushes. However, such behavior was never detected. As reported by Jordan et al., even small amount of polymer chain detachment could be otherwise seen in the AFM.9
Moreover, FT-IR spectra taken after surface polymerization clearly display successful functionalization of PTFE. Although signals of C–F stretching (1100–1300 cm−1) originating from the substrate are dominating the spectrum (Fig. 3e), a broad signal of O–H stretching from PHEMA can be observed between 3680 cm−1 and 3050 cm−1. Same signals prove the grafting of PHEMA on the other substrates (Fig. 3a–d). Furthermore, additional signals of OH deformation (1276–1248 cm−1), twisting and rocking of CH2 (1389–1365 cm−1) and several stretching vibrations of C–O bonds (Table S2†) were detected. Stretching of Si–O–Si is very prominent in the spectrum of PHEMA on SiO2 at 1122 cm−1 (Fig. 3d) even though the same kind of surface was used as background. However, all signals mentioned above can still be distinguished. Therefore, it is evident that grafting of PHEMA was accomplished on all used substrates.
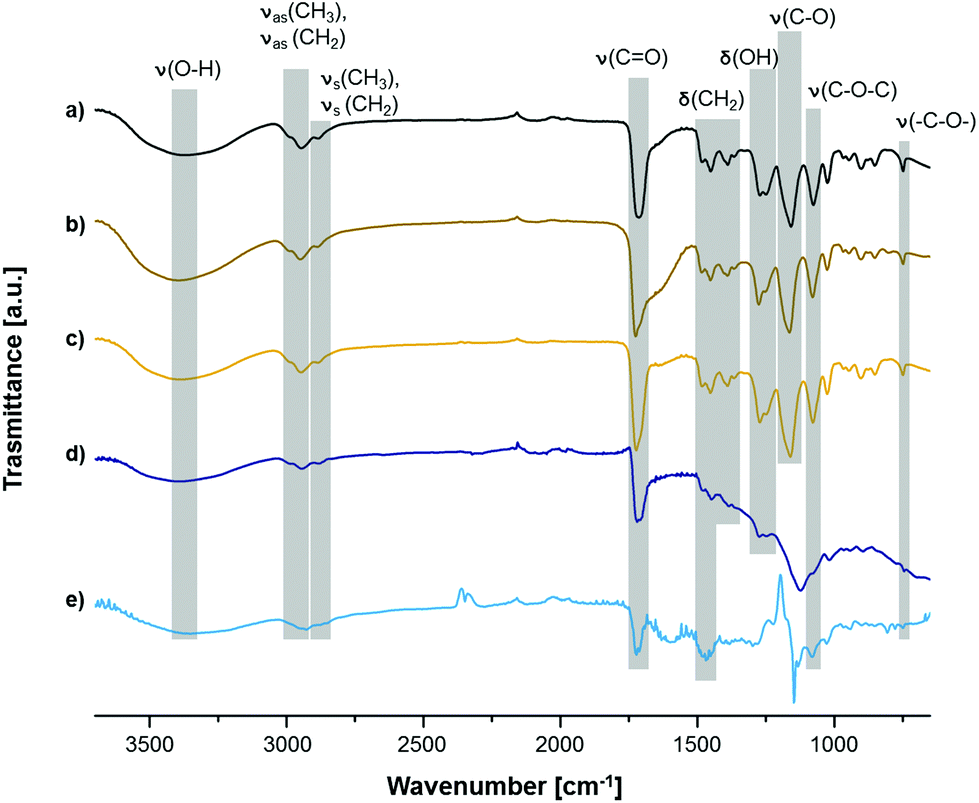 | ||
| Fig. 3 FT-IR spectra of PHEMA on Al/Al2O3 (a), Cu (b), Au (c), SiO2 (d) and PTFE (e). | ||
Grafting block copolymer on surface
To demonstrate the high end group fidelity of SI-CuCRP on PDA modified surface grafting of block copolymer brushes was carried out. A silicon surface was chosen as model substrate and functionalized with PDA/BiBB as described above. Then, the polymerization procedure was performed and the resulting polymer brush was washed and dried. Subsequently, another layer of polymer was grafted via SI-CuCRP on the same sample. The whole process was repeated several times without further functionalization with initiator between polymerizations.As can be seen in Table 2 a five block copolymer brush was synthesized with alternating hydrophilic and hydrophobic blocks. The first block of tBMA gave a comparable brush thickness as in previous experiments (Table 1) and a contact angle of 96° owing to its hydrophobic nature. Before polymerizations a double scratch was made on the PDA/BiBB layer so that atomic force microscopy (AFM) could always be measured at the same spot. In that way a height increase of 11 nm was detected after the following polymerization of DMAEMA (Fig. 4). The static contact angle θs dropped to 52° (Fig. 4). Analogously PMMA and further layers of PtBMA and PDMAEMA were grafted resulting in consecutive brush growth and exhibiting 61°, 100° and 76° for θs, respectively. Since the grafting of the second block (PDMAEMA) resulted in a thickness gain of only 11 nm, the polymerization time of following polymerizations was prolonged for 5 min or 10 min. This resulted in sufficiently higher thickness increases of 61 nm for PMMA, 52 nm for PtBMA and 86 nm for another layer of PDMAEMA (Fig. 4).
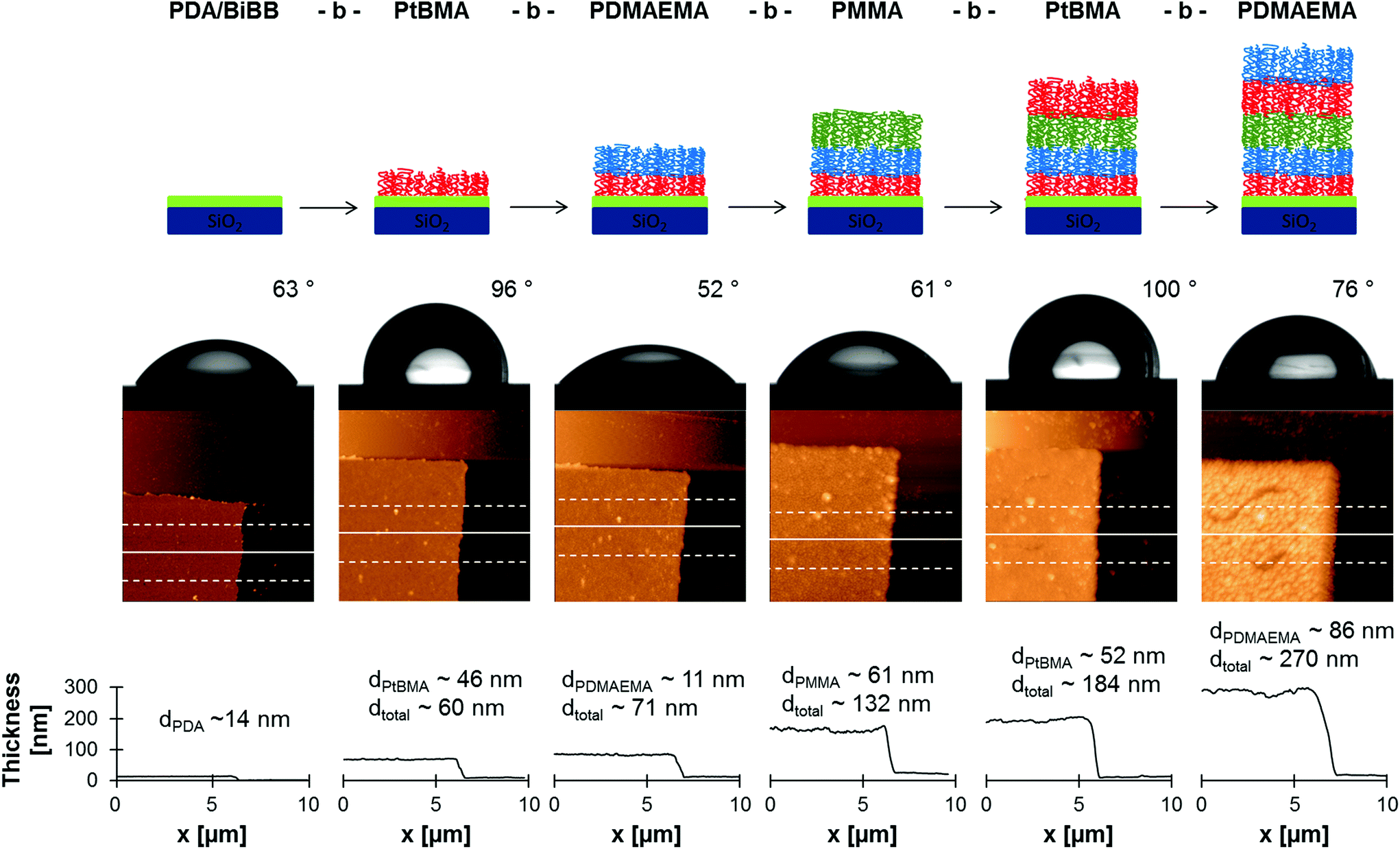 | ||
| Fig. 4 Block copolymerization of different monomers: development of contact angle depending on polymer brush (top) and the corresponding AFM image (below) with respective profile (bottom). AFM images are 10 × 10 μm2. | ||
| Block | Monomer | Solvent | t R [min] | d Block [nm] | θ s [°] |
|---|---|---|---|---|---|
| 1 | tBMA | DMSO | 15 | 46 ± 1 | 96 ± 4 |
| 2 | DMAEMA | 10% (v/v) H2O in iPrOH | 15 | 11 ± 2 | 52 ± 5 |
| 3 | MMA | DMSO | 20 | 61 ± 4 | 61 ± 7 |
| 4 | tBMA | DMSO | 25 | 52 ± 7 | 100 ± 5 |
| 5 | DMAEMA | 10% (v/v) H2O in iPrOH | 25 | 86 ± 12 | 76 ± 8 |
All these values confirm successful polymerization of the used monomers. As all contact angles match to the respective polymer brush, it can be concluded that each polymerization step results in an extra layer of polymer rather than in a system with mixed polymer brushes on surface. In case of mixed (not block-like) brushes a value resembling both brushes would be expected. For a hydrophobic/hydrophilic brush mixture, the hydrophobic brush will collapse and the contact angle will represent the hydrophilic layer. This is not the case in our results. The slightly higher θs for the last PDMAEMA block (θs = 76°) can be explained by enhanced surface roughness, as can be seen from the higher deviation from the mean thickness (Table 2). Therefore, it can be assumed that a block-like structure is attained. Furthermore, this explains the stepwise increase of the polymer brush from 60 nm after the first SI-CuCRP to 270 nm after the last step. Such a gain in thickness as well as height increase after each polymerization (52–86 nm, Fig. 4) is unlikely to be caused by only higher grafting density. Especially, it has to be taken into account that grafting density was calculated to be very high for SI-CuCRP already after one polymerization.9 Therefore, a further growth will occur on top of the previously grafted brush. Still, a certain interpenetration depth cannot be completely excluded.
Only after grafting of the fifth block no further growth of polymer brush was possible. It is suspected that initiating moieties are lost to a certain amount during the experimental procedure. Furthermore, a drop in accessibility of Br-groups due to surface reconstruction or mismatch in wettability between polymer brush and used monomer–solvent mixture might be a reason. Still, the synthesized pentablock represents the highest block number reached for copolymer brushes on surface up to date and demonstrates the high level of control and robustness of the SI-CuCRP. A possibility to further increase block thickness as well as re-initiation efficiency could be the addition of small amounts of Cu(II) salts. As reported by Benetti et al., Cu(II) amplifies dissolution of catalytic species and increases control over reaction in SI-CuCRP, when performed in DMSO. However, in case of PDA the additional amount of Cu(II) might lead to a strong incorporation into the PDA layer. Hence, the quantity of copper within the PDA layer and its effect on the grafting would have to be carefully evaluated in future studies.
Conclusion
In conclusion, we combined PDA surface chemistry with the powerful tool of surface-initiated Cu(0)-mediated controlled radical polymerization (SI-CuCRP). It was shown that PDA does not affect SI-CuCRP in any way compared to a typical initiator (APTES-BiBB). Additionally, it enables controlled polymerization on different substrates including noble metal (Au) as well as repellent, unreactive surface (PTFE). It is possible to reach polymer brush thicknesses of hundreds of nanometers within 1 h for hydrophilic monomers in aqueous system. Furthermore, hydrophobic methacrylates and styrene were polymerizable with comparably fast grafting rates in DMSO. Despite fast polymerization an outstanding end group preservation is observed. This was demonstrated by consecutive polymerization steps on previously grafted polymer brush. In that way a pentablock copolymer brush was synthesized on PDA/BiBB.All in all, our study presents a general method for manifold, controlled surface functionalization starting from a PDA/BiBB initiating layer. Due to the nature of PDA deposition this approach is applicable on all kind of surfaces and does not require complex experimental set up.
Experimental
Materials
Methyl methacrylate (MMA), 2-(dimethylamino)ethyl methacrylate (DMAEMA), 2-hydroxyethyl methacrylate (HEMA), 2-ethylhexyl methacrylate (EHMA), tert-butyl methacrylate (tBMA) and styrene (St) were purchased from Sigma-Aldrich (Weinheim, Germany), purified before use by passing through a basic alumina column to remove the inhibitor. 2-Bromoisobutryl bromide (BiBB), 3-aminopropyltriethoxysilane (APTES), 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA, 99%), pyridine (99%), tris(hydroxymethyl)aminomethane (Tris, >99.8%), dichloromethane (DCM, dry), dimethyl sulfoxide (DMSO) and isopropanol (iPrOH) (all from Sigma-Aldrich) were used as received. Dopamine as HCl salt (DA·HCl) was purchased from Acros Organics and used as received. Water for reaction solutions and contact angle measurements was deionized water.4-Inch silicon wafers with a 300 nm oxide layer were obtained from Wacker AG (Burghausen, Germany). 4-Inch copper wafers were from MicroChemicals GmbH, Germany: Prime CZ-Si wafer 4 inch, 1-side polished, p-type (boron) TTV < 10 μm, 1–10 Ohm cm; 10 nm Ti adhesion layer; 200 nm Cu (purity > 99.9%), RMS < 10 nm. The copper coated side of the wafer was consecutively washed with portions of water and ethanol under ultrasonication (5 min). The cleaned Cu plate was immediately used.
Methods
Atomic force microscopy (AFM) was performed on a customized Ntegra Aura/Spectra from NT-MDT (Moscow, Russia) with a SMENA head in semicontact mode. The probes have a typical curvature radius of 6 nm, a resonant frequency of 47–150 kHz, and a force constant of 0.35–6.10 N m−1. Editing, height determination and calculation of the surface roughness was performed with the software Nova Px 3.2.5 from NT-MDT.Ellipsometry measurements of the optical thickness, d, were performed with a SE800 ellipsometer from (SENTECH Instruments GmbH) equipped with a He–Ne laser source (λ = 632.8 nm) and a fixed angle of incidence of 60° at ambient conditions. The accumulated spectra were modeled using SpectraRay 3 software. The d-value was determined from three individual series of measurements.
X-ray photoelectron spectroscopy (XPS) was performed on an Omicron Multiprobe spectrometer using monochromatic aluminum Kα radiation. The spectra were calibrated by setting the Si 2p signal to 102.0 eV. Spectra were fitted by symmetric Voigt functions with a Shirley background correction.
Attenuated total reflection Fourier transform infrared spectroscopy (ATR FT-IR) was measured with a Nicolet 5700 (Thermo) IR-spectrometer with MCT detector. For the measurement a GladiATR setup from PIKE Technologies was used and operated under OMNIC software. Before collection of sample spectra the device was flushed with nitrogen for 30 min and the respective substrate was scanned to obtain background. Then IR of the polymer brush was obtained by summing up 256 scans for each sample.
Static water contact angle measurements were carried out with the Drop Shape Analysis System DSA 10 from Kruss to characterize the wettability of the polymer layers. For each sample, individual measurements at three different spots were performed and averaged. The measurements were performed at RT with bidistilled water. The contact angles (θs) were obtained using the tangent method fitting.
Self-assembled monolayer (SAM) of APTES-BiBB initiator on Si wafer
An oxygen plasma cleaning system (PDC-002, 200 W) from Harrick (USA) was used to clean the surface of the silicon wafer. The oxygen source of the chamber was supplied by a flow of air of 10 mL min−1 in 5 min for each wafer.The clean substrates were functionalized by immersion of the wafers into a 5%-(v/v) 3-aminopropyltrimethoxysilane (APTES) solution in dry acetone and ultrasonicated during the SAM formation for 30 min at RT. After SAM formation, the samples were extensively rinsed with dry acetone and dried under argon atmosphere. This procedure results in a highly reproducible and uniform APTES layer. The substrate was then immersed in dry DCM (3 mL) under nitrogen. Pyridine (51 μL) was added, followed by addition of 100 μL 2-bromoisobutyryl bromide (BiBB) in 3 mL DCM, and the reaction was allowed to complete within 3 h under stirring at RT. The substrate was removed, washed with portions of DCM, water and ethanol and then dried by a jet of nitrogen. The successful surface modification was confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. S1†) and water contact angle measurements. The resulting APTES-BiBB SAM layer thickness d ≈ 2.0 nm was determined by ellipsometry.
Synthesis of PDA/BiBB on different substrates
Poly(tetrafluoroethylene) (PTFE), Au, Cu and Al/Al2O3 were extensively rinsed with deionized water and ethanol and ultrasonicated for 5 min each. SiO2 was cleaned as described above.DA·HCl (12.4 mg) was dissolved in 10 mL of Tris/HCl-buffer (10 mM, pH = 8.5) and the substrates were immersed into the freshly prepared solution. Deposition was allowed to proceed for approximately 24 h. Then, the samples were washed with deionized water and ethanol and ultrasonicated for 5 min each. Reaction with BiBB was carried out in the same way as for APTES. For PTFE reaction time was reduced to 20 min. The successful surface modification was confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. S1†) and water contact angle measurements. The resulting PDA/BiBB layer thickness was determined by ellipsometry and lay within the range of d ≈ 30–35 nm.
Surface-initiated Cu(0)-mediated controlled radical polymerization (SI-CuCRP)
A silicon wafer piece modified with APTES-BiBB or a substrate modified with PDA/BiBB was sandwiched with a copper plate at a typical distance of D = 0.5 mm using two spacers. This assembly was put into a degassed solution of monomer (1 mL), solvent (0.5 mL) and PMDETA (20 μL) and closed with a rubber septum. The assembly was left for 1 h at RT. The plates were separated and the substrate immediately washed with a fresh good solvent for the respective monomer and ultrasonicated for 1 min. Finally, the substrates were dried by a jet of dry nitrogen and analyzed.Grafting of pentablock copolymer brushes on PDA/BiBB
First, SI-CuCRP was performed with tBMA on PDA/BiBB as described above. The polymer brush was washed with DMSO and ethanol und ultrasonicated for 1 min. Then, a layer of PDMAEMA was grafted, followed by a third layer of PMMA. Both were rinsed with ethanol and ultrasonicated for 1 min each. The whole process was repeated for another block of PtBMA and PDMAEMA. Each resulting polymer brush was analyzed with AFM and contact angle measurement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. J. acknowledges financial support by the Cluster of Excellence “Center for Advancing Electronics Dresden” (cfAED). Dr Enrico Langer is acknowledged for measuring XPS spectra.References
- R. C. Advincula, W. J. Brittain, K. C. Caster and J. Rühe, Polymer Brushes: Synthesis, Characterization and Applications, Wiley-VCH, 2004 Search PubMed.
- W. Senaratne, L. Andruzzi, C. K. Ober, S. Monolayers, P. Brushes, W. Senaratne, L. Andruzzi, C. K. Ober, S. Monolayers and P. Brushes, Biomacromolecules, 2005, 6, 2427–2448 CrossRef CAS PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS.
- D. Gieseler and R. Jordan, Polym. Chem., 2015, 6, 4678–4689 RSC.
- T. Zhang, T. Chen, I. Amin and R. Jordan, Polym. Chem., 2014, 5, 4790–4796 RSC.
- K. Matyjaszewski, D. Hongchen, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef CAS PubMed.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS.
- T. Zhang, Y. Du, F. Müller, I. Amin and R. Jordan, Polym. Chem., 2015, 6, 2726–2733 RSC.
- T. Zhang, Y. Du, J. Kalbakova, R. Schubel, R. D. Rodriguez, T. Chen, D. Zahn and R. Jordan, Polym. Chem., 2015, 6, 8176–8183 RSC.
- M. Steenackers, R. Jordan, A. Küller and M. Grunze, Adv. Mater., 2009, 21, 2921–2925 CrossRef CAS.
- M. Steenackers, A. Küller and S. Stoycheva, Langmuir, 2009, 25, 2225–2231 CrossRef CAS PubMed.
- M. E. Buck and D. M. Lynn, Langmuir, 2010, 26, 16134–16140 CrossRef CAS.
- N. C. Estillore and R. C. Advincula, Macromol. Chem. Phys., 2011, 212, 1552–1566 CrossRef CAS.
- K. B. Blodgett, J. Am. Chem. Soc., 1935, 57, 1007–1022 CrossRef CAS.
- N. Meyerbröker and M. Zharnikov, Adv. Mater., 2014, 26, 3328–3332 CrossRef.
- Y. Okamura, K. Kabata, M. Kinoshita, H. Miyazaki, A. Saito, T. Fujie, S. Ohtsubo, D. Saitoh and S. Takeoka, Adv. Mater., 2013, 25, 545–551 CrossRef CAS.
- G. Dearnaley and J. H. Arps, Surf. Coat. Technol., 2005, 200, 2518–2524 CrossRef CAS.
- A. Wang, H. Tang, T. Cao, S. O. Salley and K. Y. S. Ng, J. Colloid Interface Sci., 2005, 291, 438–447 CrossRef CAS PubMed.
- J. Schlenoff, M. Li and H. Ly, J. Am. Chem. Soc., 1995, 117, 12528–12536 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS.
- R. Zangmeister, T. Morris and M. Tarlov, Langmuir, 2013, 29, 8619–8628 CrossRef CAS.
- M. Kohri, Y. Shinoda, H. Kohma, Y. Nannichi, M. Yamauchi, S. Yagai, T. Kojima, T. Taniguchi and K. Kishikawa, Macromol. Rapid Commun., 2013, 34, 1220–1224 CrossRef CAS.
- F. Yu, S. Chen, Y. Chen, H. Li, L. Yang, Y. Chen and Y. Yin, J. Mol. Struct., 2010, 982, 152–161 CrossRef CAS.
- J. Liebscher, R. Mrówczyński and H. Scheidt, Langmuir, 2013, 29, 10539–10548 CrossRef CAS.
- N. F. Della Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS.
- D. Dreyer, D. Miller and B. Freeman, Langmuir, 2012, 28, 6428–6435 CrossRef CAS PubMed.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Isrealachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- H. Watanabe, A. Fujimoto, R. Yamamoto, J. Nishida, M. Kobayashi and A. Takahara, ACS Appl. Mater. Interfaces, 2014, 6, 3648–3653 CrossRef CAS PubMed.
- S. Wang, J. Song, Y. Li, X. Zhao, L. Chen, G. Li, L. Wang, Z. Jia and X. Ge, React. Funct. Polym., 2019, 140, 48–55 CrossRef CAS.
- J. Tian, D. Xu, M. Liu, F. Deng, Q. Wan, Z. Li, K. Wang, X. He, X. Zhang and Y. Wei, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1872–1879 CrossRef.
- Q. Wan, M. Liu, J. Tian, F. Deng, G. Zeng, Z. Li, K. Wang, Q. Zhang, X. Zhang and Y. Wei, Polym. Chem., 2015, 6, 1786–1792 RSC.
- S. Wang, H. Meng, Y. Li, D. Sun, Y. Zhan, X. Ge and L. Chen, J. Polym. Sci., Part A: Polym. Chem., 2018, 1–10 Search PubMed.
- W. Wang, X. Ji, H. B. Na, M. Safi, A. Smith, G. Palui, J. M. Perez and H. Mattoussi, Langmuir, 2014, 30, 6197–6208 CrossRef CAS PubMed.
- A. Lak, J. Dieckhoff, F. Ludwig, J. M. Scholtyssek, O. Goldmann, H. Lünsdorf, D. Eberbeck, A. Kornowski, M. Kraken, F. J. Litterst, K. Fiege, P. Mischnick and M. Schilling, Nanoscale, 2013, 5, 11447–11455 RSC.
- X. Zhang, J. Ji, X. Zhang, B. Yang, M. Liu, W. Liu, L. Tao, Y. Chen and Y. Wei, RSC Adv., 2013, 3, 21817 RSC.
- W. Sheng, B. Li, X. Wang, B. Dai, B. Yu, X. Jia and F. Zhou, Chem. Sci., 2015, 6, 2068–2073 RSC.
- D. Hafner, L. Ziegler, M. Ichwan, T. Zhang, M. Schneider, M. Schiffmann, C. Thomas, K. Hinrichs, R. Jordan and I. Amin, Adv. Mater., 2015, 28, 1489–1494 CrossRef PubMed.
- N. Y. Kostina, O. Pop-georgievski, M. Bachmann, M. Bastmeyer, N. Neykova, M. Bruns and C. Rodriguez-emmenegger, Macromol. Biosci., 2016, 16, 83–94 CrossRef CAS.
- O. Pop-Georgievski, C. Rodriguez-Emmenegger, A. d. l. S. Pereira, V. Proks, E. Brynda and F. Rypáček, J. Mater. Chem. B, 2013, 1, 2859–2867 RSC.
- C. Rodriguez-Emmenegger, C. M. Preuss, B. Yameen, O. Pop-Georgievski, M. Bachmann, J. O. Mueller, M. Bruns, A. S. Goldmann, M. Bastmeyer and C. Barner-Kowollik, Adv. Mater., 2013, 25, 6123–6127 CrossRef CAS.
- R. Gu, W. Z. Xu and P. A. Charpentier, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3941–3949 CrossRef CAS.
- S. P. Le-Masurier, G. Gody, S. Perrier and A. M. Granville, Polym. Chem., 2014, 5, 2816 RSC.
- O. Z. Fisher, B. L. Larson, P. S. Hill, D. Graupner, M. T. Nguyen-Kim, N. S. Kehr, L. De Cola, R. Langer and D. G. Anderson, Adv. Mater., 2012, 24, 3032–3036 CrossRef CAS PubMed.
- K. Y. Ju, Y. Lee, S. Lee, S. B. Park and J. K. Lee, Biomacromolecules, 2011, 12, 625–632 CrossRef CAS.
- N. H. Nguyen, J. Kulis, H.-J. Sun, Z. Jia, B. van Beusekom, M. E. Levere, D. A. Wilson, M. J. Monteiro and V. Percec, Polym. Chem., 2013, 4, 144–155 RSC.
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4419 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01343a |
| This journal is © The Royal Society of Chemistry 2020 |