
Hyperbranched multiple polythioamides made from elemental sulfur for mercury adsorption†
 a
Yagang
Zhang
*abc
a
Yagang
Zhang
*abc
Abstract
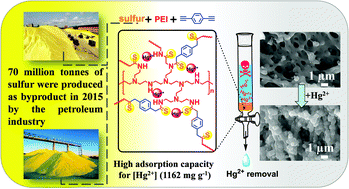
Different from traditional polyethylenimine (PEI) modified Hg(II) adsorbent materials, a novel hyperbranched polythioamide adsorbent (SPD) was prepared by using sulfur, PEI and 1,4-diethynylbenzene (DEB) as monomers. There are tens and thousands of tons of sulfur produced from oil refinery processes as a major by-product each year, and its utilization becomes a concern due to its limited consumption, environmental problems and safety. Herein, a SPD was fabricated through a one-pot, catalyst-free polymerization which even can be solvent-free via using sulfur to solidify the liquid PEI. A kinetic study showed that the adsorption of Hg(II) ions followed the pseudo-second-order model, indicating chemical adsorption between Hg(II) ions and SPD. The SPD had a maximum adsorption capacity of 1161.99 mg g−1. FT-IR and XPS measurements indicated that the removal of Hg(II) by SPD was mainly controlled by the coordination interaction between Hg(II) ions and nitrogen groups (i.e., amine and imine groups), and also C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds of SPD. By using elemental sulfur, PEI becomes the backbone of the polymer system by forming –C(S)NH– and –C(S)N< groups unlike the grafted part of traditional PEI modified materials. TG, DTG, and DSC measurements showed that the main degradations occurred at around 160 °C and 360 °C.
S bonds of SPD. By using elemental sulfur, PEI becomes the backbone of the polymer system by forming –C(S)NH– and –C(S)N< groups unlike the grafted part of traditional PEI modified materials. TG, DTG, and DSC measurements showed that the main degradations occurred at around 160 °C and 360 °C.
 Please wait while we load your content...
Please wait while we load your content...

