
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaking the best of it: nitroxide-mediated polymerization of methacrylates via the copolymerization approach with functional styrenics†
Aaron C.
Schmidt‡
ab,
Hatice
Turgut‡
ab,
Dao
Le
 ab,
Ana
Beloqui
abc and
Guillaume
Delaittre
*abde
ab,
Ana
Beloqui
abc and
Guillaume
Delaittre
*abde
aInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstrasse 15, 76131 Karlsruhe, Germany
cCICnanoGUNE, Avda. Tolosa 76, 20018 San Sebastián, Spain
dInstitute for Applied Polymer Chemistry, University of Applied Sciences Aachen, Heinrich-Mußmann-Strasse 1, 52428 Jülich, Germany. E-mail: delaittre@fh-aachen.de; Web: http://www.twitter.com/GPRDelaittre
eDeutsches Textilforschungszentrum Nord-West (DTNW) gGmbH, Adlerstrasse 1, 47798 Krefeld, Germany
First published on 19th November 2019
Abstract
The SG1-mediated solution polymerization of methyl methacrylate (MMA) and oligo(ethylene glycol) methacrylate (OEGMA, Mn = 300 g mol−1) in the presence of a small amount of functional/reactive styrenic comonomer is investigated. Moieties such as pentafluorophenyl ester, triphenylphosphine, azide, pentafluorophenyl, halide, and pyridine are considered. A comonomer fraction as low as 5 mol% typically results in a controlled/living behavior, at least up to 50% conversion. Chain extensions with styrene for both systems were successfully performed. Variation of physical properties such as refractive index (for MMA) and phase transition temperature (for OEGMA) were evaluated by comparing to 100% pure homopolymers. The introduction of an activated ester styrene derivative in the polymerization of OEGMA allows for the synthesis of reactive and hydrophilic polymer brushes with defined thickness. Finally, using the example of pentafluorostyrene as controlling comonomer, it is demonstrated that functional PMMA-b-PS are able to maintain a phase separation ability, as evidenced by the formation of nanostructured thin films.
Introduction
Nitroxide-mediated polymerization (NMP) was developed in the 80s and as such was historically the first reversible-deactivation radical polymerization (RDRP) technique to appear.1 In the following decades, many new techniques emerged, of which copper-mediated radical polymerization (CuRP)2–4 and reversible addition–fragmentation transfer polymerization (RAFTP)5–7 are the most studied. Undoubtedly, thanks to some decisive features (low temperatures, availability of compounds, intellectual property), atom-transfer radical polymerization (ATRP) and RAFTP have become the most popular methods in the academic field. Nevertheless, NMP remains a very useful companion to these techniques. In some specific cases, it can even be stated that NMP is the method of choice, e.g., chain ends being inert to hydrolysis or nucleophilic attack. If one asks an academic well acquainted with all RDRP methods which technique s/he would choose to synthesize a well-defined polystyrenic, chances are relatively high that NMP will be the answer, based on a simpler setup requiring only the addition of an alkoxyamine to the monomer.8–13Nevertheless, NMP suffers from some drawbacks, a classic issue being the lack of control over the polymerization of methacrylates with readily accessible alkoxyamines/nitroxides.14 Until recently, the specifically designed, yet challenging to synthesize, DPAIO (2,2-diphenyl-3-phenylimino-2,3-dihydroindol-1-yloxyl nitroxide) reported by Guillaneuf et al. was the only nitroxide species leading to a clear control of the homopolymerization of methyl methacrylate (MMA).15,16 Yet its inability to control the NMP of other types of monomers – although some improvement for styrene was achieved later on17 – precludes its use for block copolymer synthesis. Remarkably, researchers at POLYMAT research institute have introduced a readily synthesized range of alkoxyamines, of which the so-called Dispolreg 007 variant (3-(((2-cyanopropan-2-yl)oxy)(cyclohexyl)amino)-2,2-dimethyl-3-phenylpropanenitrile) is able to control the polymerization of both methacrylates and styrene (Sty) to a fair extent.18–20 Before these, the nitroxide SG1 (N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide), usually in the form of its alkoxyamine MAMA-SG1 (also registered trademark BlocBuilder®), has been the most efficient commercially available NMP initiator for styrenics and acrylates/acrylamides.21–27 Yet, it failed to control the polymerization of methacrylic esters due to unfavorable kinetic and thermodynamic parameters, notably a too large activation–deactivation equilibrium constant K leading to a high concentration of propagating macroradicals and consequently a high rate of irreversible self-termination. This materializes in the rapid consumption of active species and cessation of the polymerization, typically below 50% of monomer conversion. β-Hydrogen transfer side reactions, which have been described as the main source of the problem in TEMPO-mediated polymerization, leading to a rapid and complete loss of the nitroxide, are however quasi-absent when using SG1.14,28–30 To circumvent the issue related to the high K, an astute method was developed by Charleux,31 on the basis of an earlier report by Hawker and co-workers.32 Indeed, the latter reported that the nitroxide-mediated copolymerization of MMA and styrene could be controlled up to 85 mol% of MMA. Charleux rationalized this result by establishing a theoretical expression of the average activation–deactivation equilibrium constant for RDRP based on reversible termination. She simultaneously inferred that adding a small amount of a so-called controlling comonomer with a significantly lower K and a relatively lower polymerization rate constant kp to the NMP of MMA should lead to an artificial control. In that case, the overall K for the copolymerization is largely governed by the controlling comonomer characteristics. What about copolymerization kinetics? It was at that point demonstrated experimentally that a ratio as low as [Sty]/([MMA] + [Sty]) = 0.044 leads to a polymerization exhibiting controlled/living features, at least up to 60% conversion. These polymerizations could be performed at temperatures notably lower than classic NMP of styrenics and acrylics, owing to the presence of a MMA-styrene-SG1 sequence at the end of the dormant polymer chains and a penultimate effect on the dissociation constants of the corresponding alkoxyamine structures.33
Subsequently, various methacrylic ester/controlling comonomer pairs were reported in classic bulk and solution experiments, as well as in the more advanced context of polymerization-induced self-assembly.14,34–36 While the effect of the presence of the controlling comonomer on the overall properties of the methacrylic polymers has been rarely considered,37–39 only a handful of examples have shown the incorporation of a functional controlling comonomer in the NMP of methacrylic esters in low amounts (≤10 mol%): fluorescent 9-(4-vinylbenzyl)-9H-carbazole (VBC),39–41 ionizable sodium 4-styrenesulfonate,42 pentafluorostyrene (PFS) for post-polymerization modification with nucleophiles,43 and the cyclic ketene acetal 2-methylene-4-phenyl-1,3-dioxolane (MPDL), allowing chain degradation by incorporation of main-chain ester moieties44 (Scheme 1). We therefore sought to establish the ability of a range of functional styrenics as controlling comonomers, particularly because: (i) many are commercially available, (ii) they usually exhibit a polymerization behavior relatively close to that of styrene, at least in comparison to other monomer classes, and (iii) they additionally and quite obviously may impart new functionality/reactivity to the polymer, thereby “making the best” of the copolymerization approach (Scheme 1). The final copolymers are particularly interesting in the development of reactive nanostructured materials based on phase-separating block copolymers involving low fractions of reactive comonomers.45–47 In brief, the goal of the present article, rather than to provide an in-depth rate constant-based investigation, is to exemplify in a practical manner the opportunity to control the polymerization of methacrylates by SG1-mediated polymerization, while introducing functionality without strongly compromising initial physical properties.
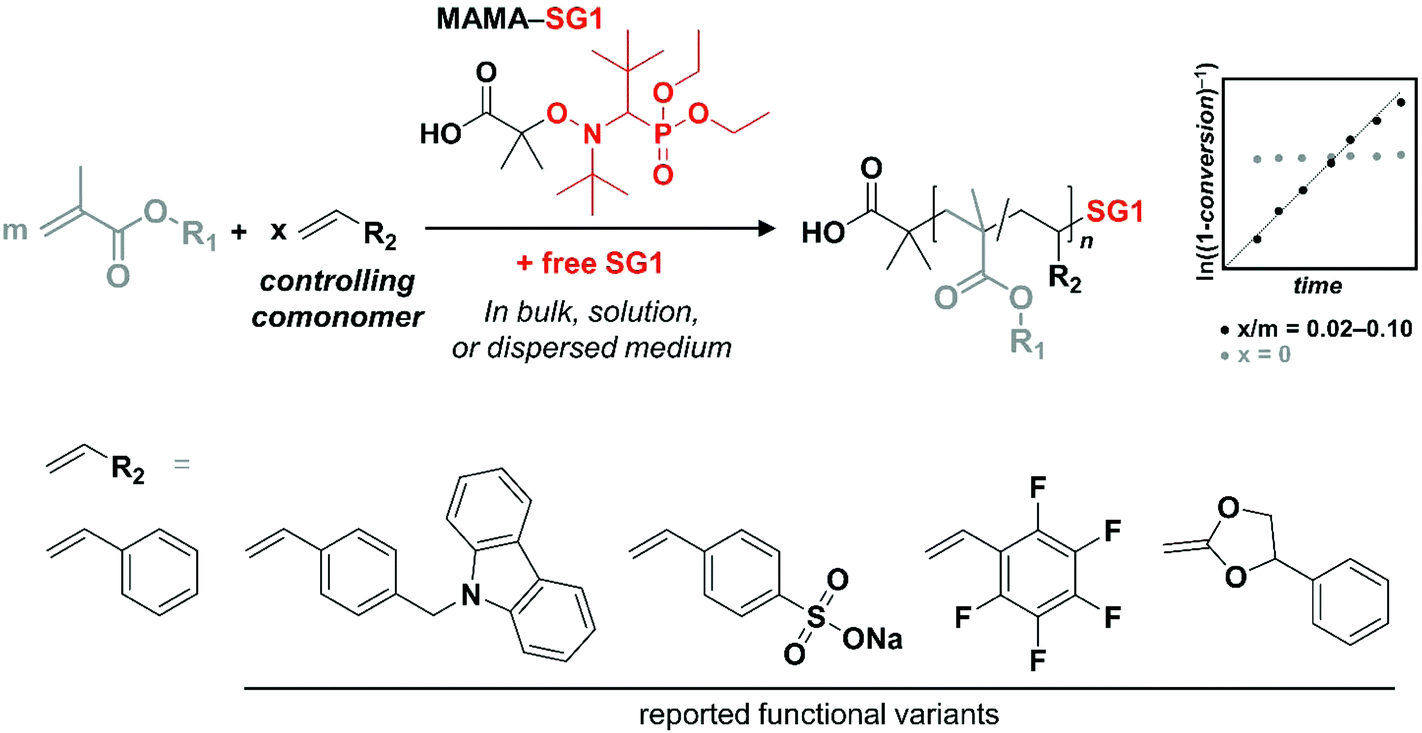 | ||
| Scheme 1 The copolymerization approach to achieve control over the SG1-mediated radical polymerization of methacrylics. On the bottom row, besides styrene, the reported controlling comonomers providing functionality are presented. | ||
Experimental part
Materials
Methyl methacrylate (MMA; 99%, Acros), oligoethylene glycol methyl ether methacrylate (OEGMA; Mn = 300 g mol−1, Sigma Aldrich), styrene (Sty; 99%, Acros), and 2,3,4,5,6-pentafluorostyrene (PFS; 98%, ABCR) were eluted through a basic alumina column (Roth) to remove the inhibitor. 4-Vinylpyridine (4VP; 95%, Aldrich) was distilled prior to use. 4-(Diphenylphosphino)styrene (DPPS; 97%, Aldrich), 4-bromostyrene (BrMS; 98%, ABCR), pentafluorophenyl-4-vinylbenzoate (PFPVB; 99%, Aldrich), 4-chloromethylstyrene (CMS; 97%, Aldrich), 3-aminopropyltriethoxysilane (APTES; 97%, Alfa Aesar), toluene (>95%, Fischer), acetonitrile (99.9%, Acros), acetone (>95%, Fisher), dichloromethane (DCM; ≥99.5%, VWR), ethyl acetate (≥99.5%, Roth), ethanol (96%, Roth), N,N-dimethylacetamide (DMAc; ≥99.0%, VWR), concentrated HCl (conc. HCl; 37%, Roth), hydrogen peroxide (H2O2; 35%, Roth) were used as received. N-tert-butyl-N-[1-diethylphosphono-(2,2-dimethylpropyl)] nitroxide (SG1),48N-(2-methyl-2-propyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-O-(2-carboxyprop-2-yl) hydroxylamine (MAMA-SG1),49 NHS-MAMA-SG1,50 and 4-azidomethylstyrene (AzMS)51 were synthesized according to literature procedures. Following our published protocols, homopolymer PMMAATRP for refractive index determination as well as hydroxy end-functional random copolymer P(MMA-co-Sty) for neutralization before spin-coating were obtained by ATRP46 and homopolymer POEGMARAFT for dynamic light scattering investigations was synthesized by RAFT polymerization.52Silicon wafers ([100], p-doped with boron) were obtained from International Wafer Source, Inc.
Characterization methods
Experimental procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 g mol−1, Đ = 1.20), styrene (1.197 g, 11.5 mmol, 1600 eq.) and toluene (1.8 mL) were introduced in a 5 mL round-bottom flask, which was sealed with a rubber septum. The mixture was cooled in an ice-bath while purging with nitrogen for 30 minutes. The flask was then placed in a pre-heated oil bath at 120 °C for 2 hours. After cooling the mixture to room temperature, the conversion was calculated by 1H NMR spectroscopy and the molar mass was determined by size-exclusion chromatography.
700 g mol−1, Đ = 1.47), styrene (400 mg, 3.85 mmol, 1280 eq.) and DMAc (0.4 mL) were introduced in a 5 mL round-bottom flask, which was sealed with a rubber septum. The mixture was cooled in an ice-bath while purging with nitrogen for 30 minutes. The flask was then placed in a pre-heated oil bath at 120 °C for 2 hours. After cooling the mixture to room temperature, the conversion was calculated by 1H NMR spectroscopy and the molar mass was determined by size-exclusion chromatography.
:H2O2, 3:1 v/v) for 1 h at 90 °C. Caution: Piranha solution is an extremely strong oxidant and should be handled very carefully! After being rinsed twice with MilliQ water, the wafers were dried under a N2 stream and immediately immersed in an APTES solution in dry toluene (0.02 mM). The reaction was carried out for 2 h at 50 °C, then overnight at room temperature. Finally, any aggregate or weakly bound silane was washed off by sonication in toluene for 5 min. Wafers were rinsed with toluene, acetone, and MilliQ water solutions, and blow-dried with N2.
900 g mol−1, Đ = 1.20), styrene (1.197 g, 11.5 mmol, 1600 eq.) and toluene (1.8 mL) were introduced in a 5 mL round-bottom flask, which was sealed with a rubber septum. The mixture was cooled in an ice-bath while purging with nitrogen for 30 minutes. The flask was then placed in a pre-heated oil bath at 120 °C for 2 hours. After cooling the mixture to room temperature, the conversion was calculated by 1H NMR spectroscopy and the molar mass was determined by size-exclusion chromatography.
700 g mol−1, Đ = 1.47), styrene (400 mg, 3.85 mmol, 1280 eq.) and DMAc (0.4 mL) were introduced in a 5 mL round-bottom flask, which was sealed with a rubber septum. The mixture was cooled in an ice-bath while purging with nitrogen for 30 minutes. The flask was then placed in a pre-heated oil bath at 120 °C for 2 hours. After cooling the mixture to room temperature, the conversion was calculated by 1H NMR spectroscopy and the molar mass was determined by size-exclusion chromatography.
:H2O2, 3:1 v/v) for 1 h at 90 °C. Caution: Piranha solution is an extremely strong oxidant and should be handled very carefully! After being rinsed twice with MilliQ water, the wafers were dried under a N2 stream and immediately immersed in an APTES solution in dry toluene (0.02 mM). The reaction was carried out for 2 h at 50 °C, then overnight at room temperature. Finally, any aggregate or weakly bound silane was washed off by sonication in toluene for 5 min. Wafers were rinsed with toluene, acetone, and MilliQ water solutions, and blow-dried with N2.
APTES-modified silicon wafers were then introduced into large glass vials with NHS-MAMA-SG1 (100 mg, 0.21 mmol) and triethylamine (200 μl, 1.43 mmol) in dry DCM (20 ml). The reaction mixture was kept at room temperature overnight. Non-bound material was removed by rinsing the surfaces three times with DCM. The wafers were finally dried under a N2 stream.
Results and discussion
The present study focuses on eight styrenics as controlling comonomers, including Sty itself as a point of comparison. These structures span a relatively large scope of functionality and reactivity, as shown in Scheme 2. Only Sty had previously been employed with the sole purpose of gaining control over the NMP of methacrylic esters,31,33,53–61 while PFS had been shown to exert a positive influence as well, yet in a study not focusing on the control aspect.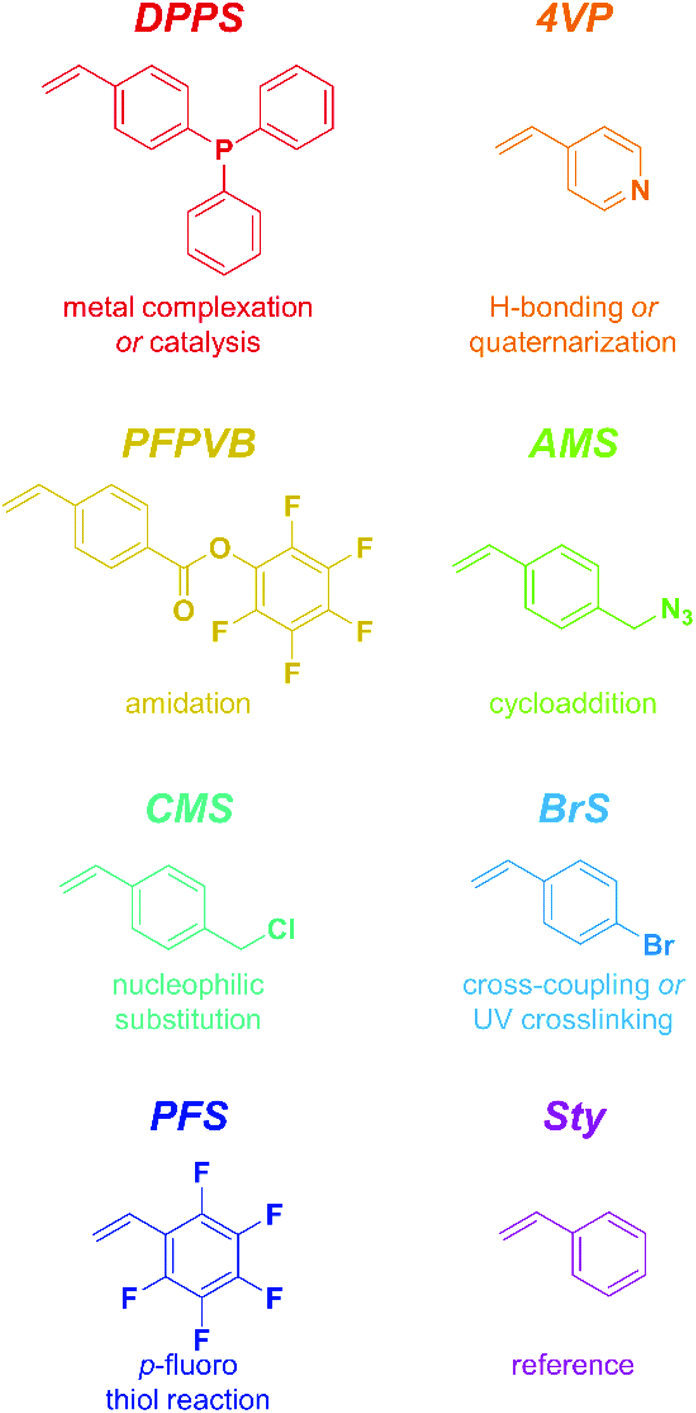 | ||
| Scheme 2 Chemical structures, abbreviations, and potential uses of the functionality of the styrenics introduced as controlling comonomers in the nitroxide-mediated polymerization of methacrylics in the present contribution. | ||
For all experiments, we employed rather commonly encountered values in the field for the following parameters: [comonomers]/[initiator] = 100 and 300 for OEGMA and MMA, respectively; [styrenic]/[comonomers] = 0.05; [MAMA-SG1]/[SG1] = 10; and a polymerization temperature of 80 °C. Although it may not be the optimal ratio, we concentrated our investigations on copolymerizing 5 mol% of styrenics because (i) it was found that a rather decent control could be achieved with 4.4–8.8 mol% styrene31 and (ii) it fits well our general goal of imparting side-chain reactivity to (block) copolymers without strongly altering their physical properties.45–47
Kinetics and macromolecular control in solution
The first methacrylic ester we investigated was logically methyl methacrylate, which is probably the most ubiquitously used one, with applications spanning from glass replacement62 or contact lenses63 to microdevices.64The simplest way to perform NMP is to mix an alkoxyamine with a given monomer and to raise the temperature in the absence of oxygen. This is a so-called bulk polymerization. When the present investigation was commenced, bulk polymerization was therefore the first approach. However, it was observed that with many styrene derivatives, e.g., 4VP, AMS, CMS, molar masses seemed to plateau after a certain conversion was reached and sampling became complicated by a large increase in viscosity of the medium. In addition, some comonomers exhibited poor solubility in pure MMA. Therefore, the decision was made to carry out further experiments in solution. Toluene was employed since it has a convenient boiling point at ca. 110 °C, which is significantly higher than the temperature required for the SG1-mediated polymerization of methacrylates, yet low enough for easy removal by rotary evaporation.
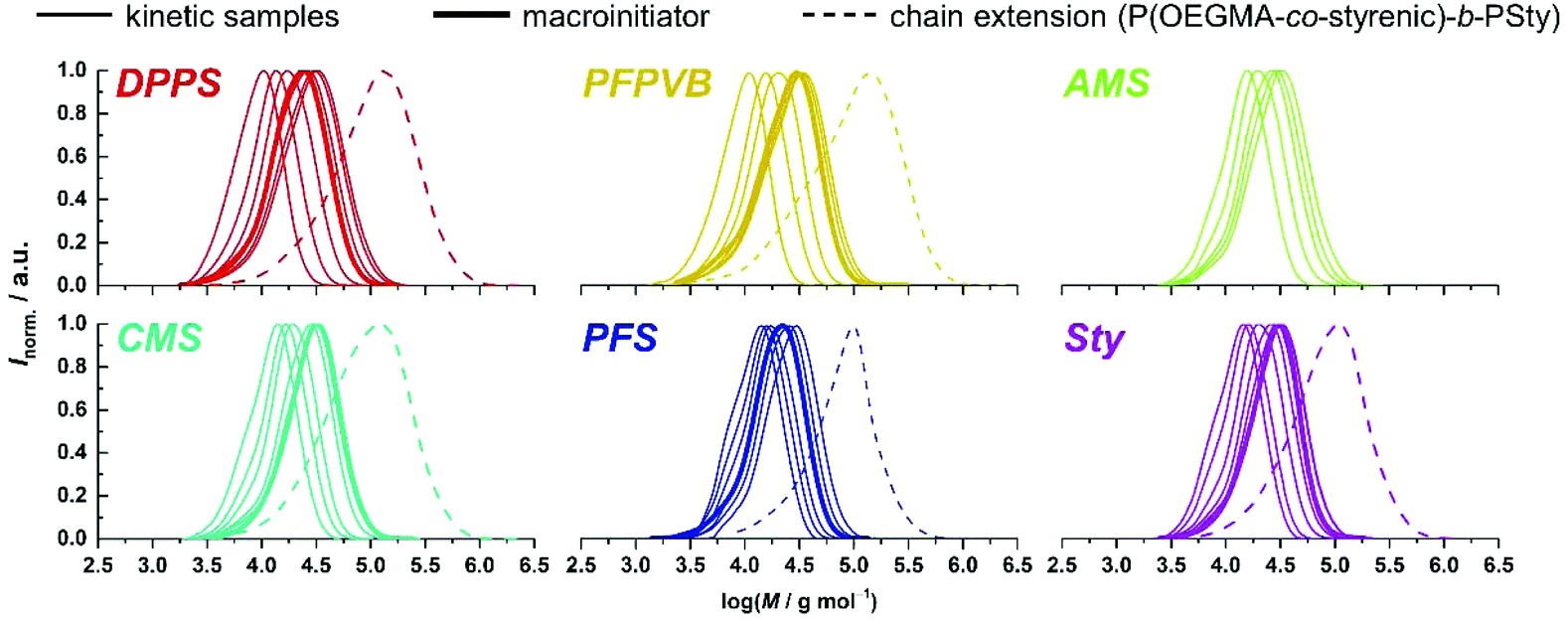
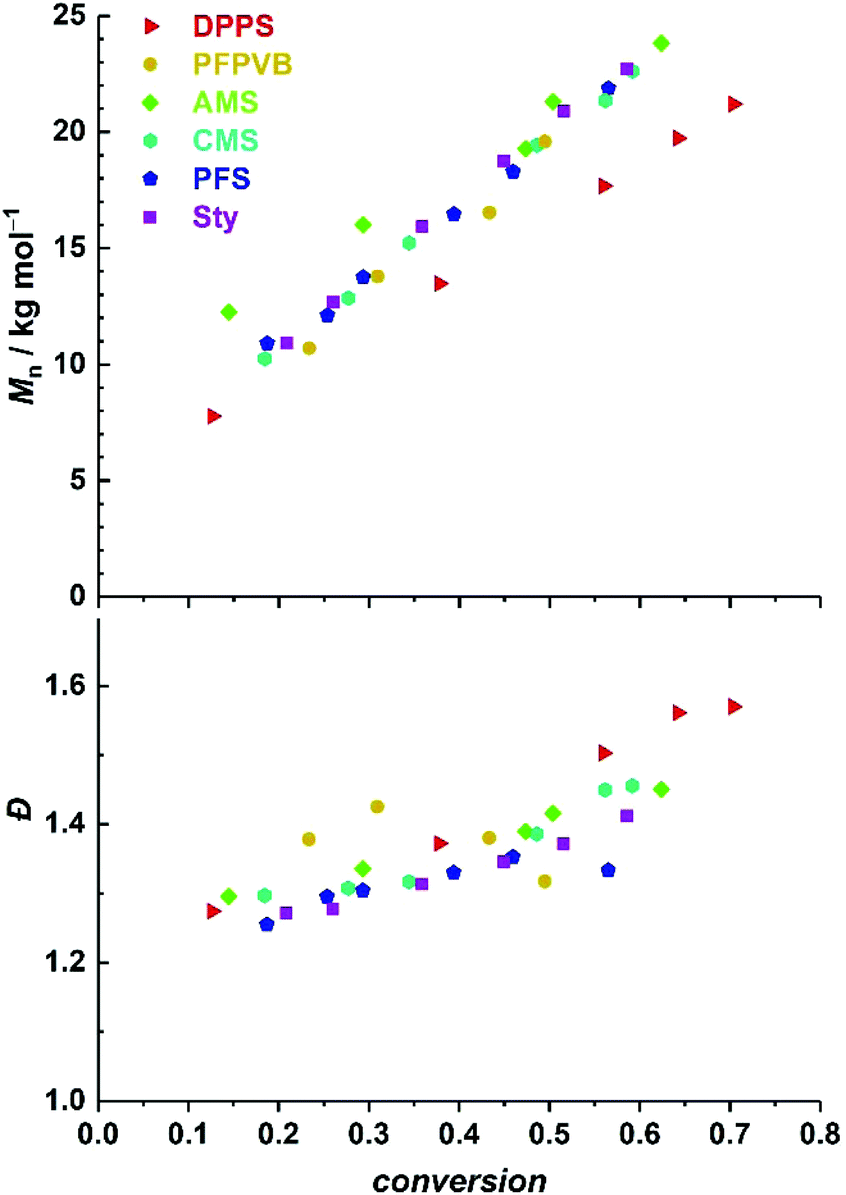
Fig. 1 compiles the molar mass distribution data obtained for the copolymerization of MMA with the 8 styrenics with MMA/toluene 4:1 w/w. Clear shifts of rather symmetrical molar mass distributions are observed for all comonomers. Fig. 2 provides the numerical data extracted from the chromatograms of Fig. 1. The entire ensemble of kinetic points represents a cloud with a clear increase of the number-average molar mass with conversion. Importantly, all individual series display a linear increase in Mn. Differences between all series are obvious and may be related to the respective molar mass of each particular comonomer. Nevertheless, and although it is our aim to not strongly alter the physical attributes of the polymethacrylics obtained via this methodology, it should not be excluded that the solution behavior may be affected (vide infra). Therefore, while the SEC data may suggest distinct deviations in the degree of polymerization between experiments, it is clear that the SEC calibration based on pure PMMA does not provide absolute values. Furthermore, for essentially all styrene derivatives, the dispersity remains stable or even decreases with conversion. High values observed for azidomethylstyrene (AMS) and 4-vinylpyridine (4VP) may originate from two distinct phenomena. For the former, the presence of side reactions (e.g., crosslinking) cannot be discarded due to the instability of the azide group. Nevertheless, the SEC traces displayed a symmetrical shape throughout the reaction along clean shifts to higher molar masses with increasing conversion (Fig. 1). However, two unknown peaks appeared in the 1H NMR spectrum in the 4–5 ppm region (Fig. S1†) during the course of the reaction and can be found in the precipitated polymer as well (Fig. S6†). The latter data allowed us to calculate a fraction of incorporated AMS corresponding to 4.9 mol%, while only 62% of the azidomethyl substituents seemed to be intact. For the 4VP series, interaction with the SEC columns are possibly responsible for such values.
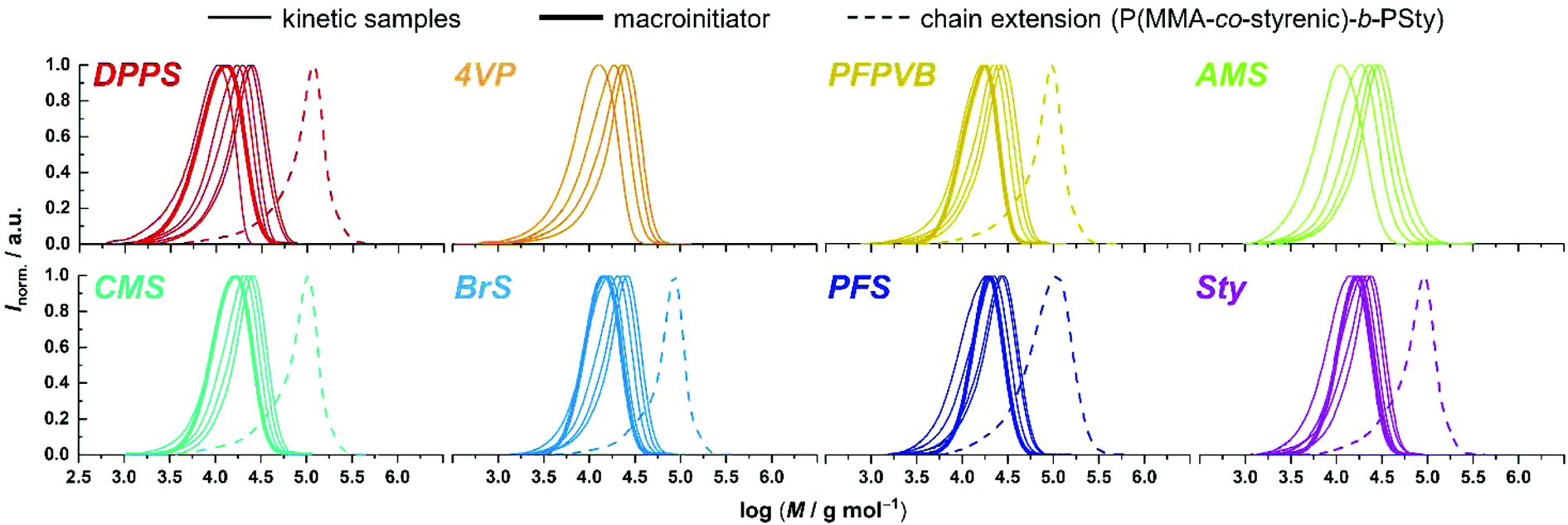 | ||
| Fig. 1 Size-exclusion chromatograms obtained at various conversions during the SG1-mediated polymerization of MMA in the presence of 5 mol% of various styrenics (thin lines) at 80 °C in toluene, as well as after chain extension with styrene (dashed lines) at 120 °C in toluene from a corresponding macroinitiator (thick lines). | ||
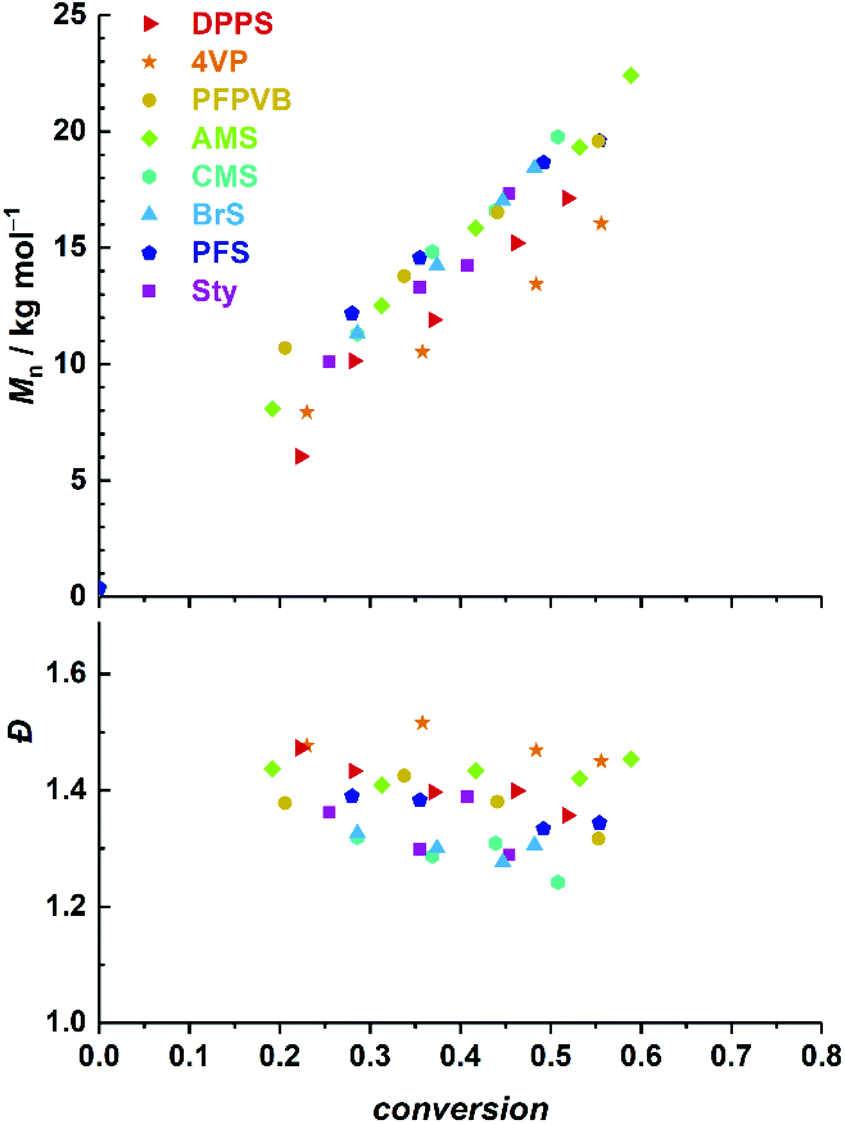 | ||
| Fig. 2 Evolution of number-average molar mass (top) and dispersity (bottom) with MMA conversion for the SG1-mediated polymerization of MMA in toluene at 80 °C, in the presence of 5 mol% of various styrenics. | ||
Time-lapse monitoring of the MMA monomer conversions provided data series with overall linear trends, up to 55–65% (Fig. 3A). Note that kinetic experiments were stopped around this MMA conversion, as it is commonly observed that loss of linearity, hence of control, occurs beyond. This can be explained by the generally higher consumption rate of the controlling comonomer compared to MMA (see Fig. S2†), which leads to constantly decreasing styrenic-to-MMA ratios during the polymerization, until complete depletion, eventually resulting in pure uncontrolled homopolymerization. In a rather disparate way, 50% conversion (i.e., −ln(1 − conversion) ≈ 0.7) was achieved between three and six hours, depending on the styrenic comonomer. Linear regressions could be well fitted and converted to apparent rate constants of polymerization (Fig. 3B). As expected, the latter span a relatively broad range and provide a ranking in terms of polymerization rate as follows: DPPS > 4VP > PFPVB > AMS > CMS > BrS > PFS > Sty. The influence of the aromatic ring substituent(s) can clearly be observed and may have various origins. By electronic effects, the reactivity of the styrenic double bond is obviously directly impacted. In addition, steric effects cannot be excluded. These two parameters – together with the reactivity of the chain-end radical formed by incorporation of a comonomer unit – in fact directly influence the reactivity ratios.65 The dissociation rate of the alkoxyamine bond is certainly impacted as well. Interestingly, higher apparent polymerization rates correlate rather well with increasing electron-withdrawing characteristics of the substituents. Assuming that, irrespective of the exact controlling comonomer, dormant chain ends have the structure MMA-styrenic-SG1, as previously demonstrated for styrene,33 a higher electron-withdrawing nature of the styrenic substituent would result in a stronger propensity of the alkoxyamine for hemolytic cleavage and a lower recombination rate. One exception is that of AMS and CMS, which should be reversed since the azido moiety is less electron-withdrawing than the chloride group. This could possibly originate from side-reactions of the azido group66,67 – which do occur here (vide infra) – that could result in alteration of the overall electron-withdrawing properties. It should also be noted that the diphenylphosphino substituent of DPPS can act as both an electron-withdrawing (via inductive effect) as well as an electron-donating group (via mesomeric effect). To decipher all these parameters in details, further work will be needed.
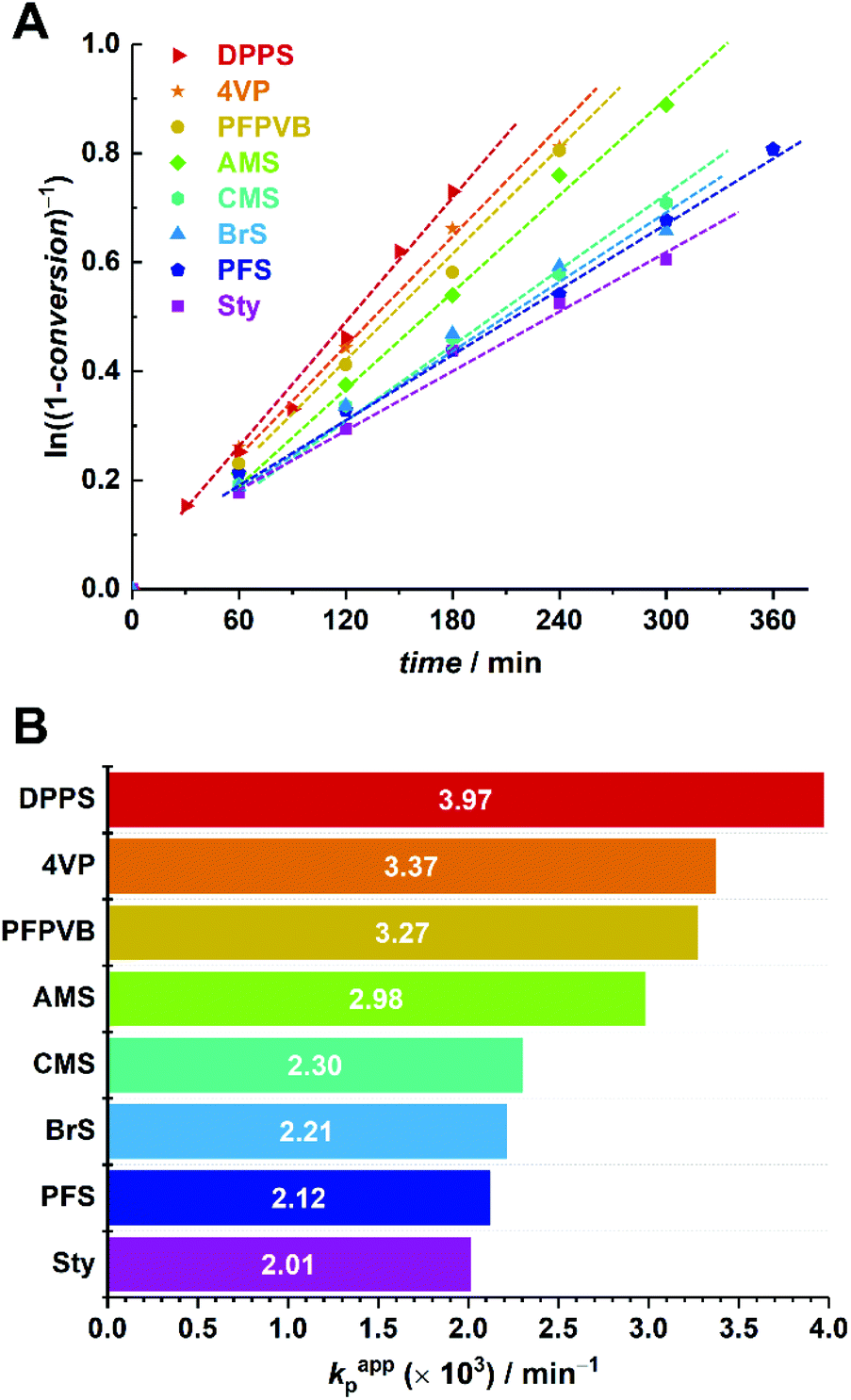 | ||
| Fig. 3 (A) First-order kinetic plots for the SG1-mediated polymerization of MMA in toluene at 80 °C, in the presence of 5 mol% of various styrenics. (B) Apparent rates of polymerization calculated for the kinetic data presented in A. | ||
An important criterion to assess the level of control on the polymerization is the degree of functionalization of the chain ends, particularly the associated ability to synthesize block copolymers by chain extension.68 With high interest for functional phase- separating PMMA-b-PS,46 we therefore performed the polymerization of styrene with the functional PMMAs as macroinitiators, in toluene at 120 °C without free SG1. To this end, macroinitiators were produced by repetition of the aforementioned kinetic experiments, yet by stopping the polymerization between 2 and 3 hours without sampling, to reach between 30 and 40% conversion (Fig. 3A), guaranteeing a good degree of livingness. SEC characterization presented in Fig. 1 satisfactorily revealed clear shifts of the molar mass distributions, with low amounts of unreacted or dead PMMA.
Next, we explored the possibility of regulating the NMP of oligoethylene glycol methyl ether methacrylate (OEGMA) with a similar set of controlling comonomers as it would provide well-defined hydrophilic polymers with additional lateral reactivity. POEGMA is particularly interesting as it is frequently employed in the biomaterials field, due to demonstrated biocompatibility and ease of synthesis.37,69 It was already polymerized by NMP following the copolymerization approach, using Sty,54,57 acrylonitrile,37,70 PFS,43 and MPDL.44 The same type of kinetic experiments as for MMA was therefore undertaken. Again, a continuous shift of the molar mass distribution was observed with increasing conversion for all comonomer series (Fig. 4).
 | ||
| Fig. 4 Size-exclusion chromatograms obtained at various conversions during the SG1-mediated polymerization of OEGMA in the presence of 5 mol% of various styrenics (thin lines) at 80 °C in acetonitrile, as well as after chain extension with styrene (dashed lines) at 120 °C in DMAc from a corresponding macroinitiator (thick lines). | ||
In a similar manner, number-average molar masses increased linearly with conversion (Fig. 5, top). As opposed to MMA, however, dispersity values were rather low in the first instants and continuously increased with conversion (Fig. 5, bottom), revealing a lower degree of control with time. This may be due to a relatively faster consumption of the controlling comonomer in comparison to the methacrylate monomer – OEGMA polymerizes slower than MMA in the current setup – which results in a progressively lower fraction of the styrenic (vide supra), a shift of the activation–deactivation equilibrium towards higher values, hence a loss of control.31
 | ||
| Fig. 5 Evolution of number-average molar mass (top) and dispersity (bottom) with OEGMA conversion for the SG1-mediated polymerization of OEGMA in acetonitrile at 80 °C, in the presence of 5 mol% of various styrenics. | ||
Observation of the kinetics data reveals overall similar trends as for MMA, with the following order of apparent rate of polymerization: DPPS > PFPVB ≈ AMS > CMS > Sty ≈ PFS (Fig. 6). The small differences may arise from the differences in the polarity of the solvent, which is known to influence NMP.71 As previously demonstrated in several publications for MMA, the absence of controlling comonomer led to fast and incomplete polymerization for OEGMA as well (Fig. 6A, black dots). As with the PMMA system, chain extension with styrene as single monomer was performed in similar conditions with most of the functional POEGMAs. Clean molar mass distribution shifts were observed, with again a broadening of the molar mass distribution, slightly more pronounced when macroinitiators were obtained in the upper end of the conversion range (Fig. 4). It was thus possible to produce amphiphilic block copolymers, which may be used as self-assembling blocks towards core–shell nanoparticles with a shell that is essentially biocompatible, yet functionalizable.
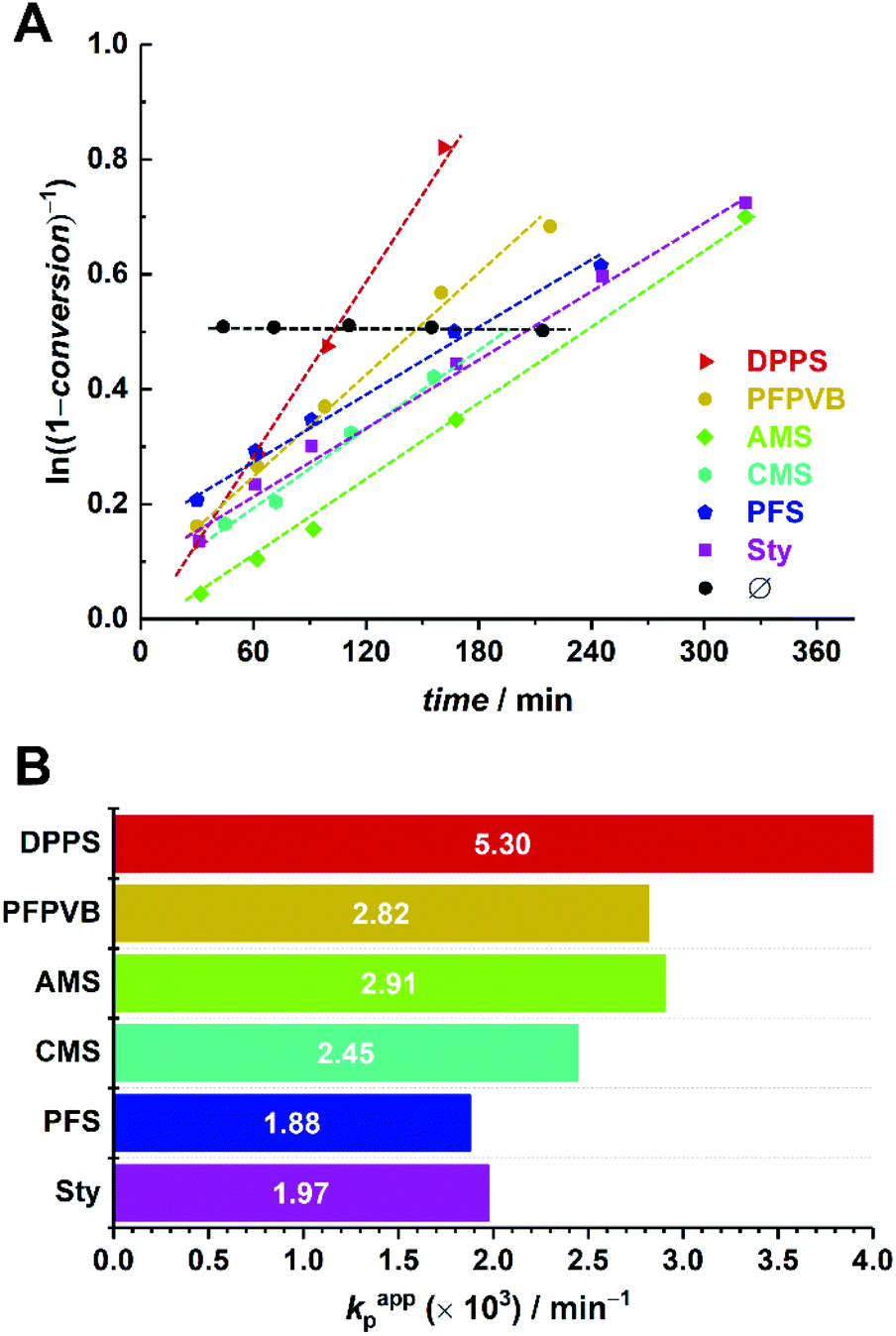 | ||
| Fig. 6 (A) First-order kinetic plots for the SG1-mediated polymerization of OEGMA in acetonitrile at 80 °C, in the presence of 5 mol% of various styrenics, in the linear regime. (B) Apparent rates of polymerization calculated for the kinetic data presented in A. | ||
Characterization of the polymers
Importantly, at the exception of AMS which suffers from the aforementioned loss of functionality, all comonomer units incorporated within the polymethacrylic scaffold remained intact, as observed by 1H NMR spectroscopy (Fig. S3–10†).Concerning PMMA, being a material used for many optical applications63,64 or simply when transparence in the final object is required along its particular properties,62,72–74 it is important to ascertain that its optical properties are not strongly impacted by the presence of the controlling comonomer. As a relevant reference sample, we synthesized a PMMA sample by ATRP (PMMAATRP), which does not necessitate the introduction of a controlling comonomer. Its refractive index nD was determined to be 1.513 by ellipsometry at 589 nm, which is somewhat higher than the reported values of about 1.490 obtained using an Abbe refractometer. The presence of a few percentages of each of the styrenic comonomers induces only a slight increase, i.e., 0.001–0.030 (Table 1). The highest increases (+0.030) is found in the case of DPPS, which is not surprising considering the presence of 3 aromatic rings for each comonomer unit. The second largest increase is found for BrS, which is typical for brominated polymers (see Table S1†). The presence of the functional comonomer should therefore not drastically impact the optical properties of the PMMA material and even may provide additional opportunities for corresponding applications such as crosslinking for reinforcement or covalent integration of dyes.

|
POEGMA typically exhibits a temperature-dependent solubility in aqueous solutions, which is related to the length of OEG sidechains. For the present POEGMA, lower critical solution temperatures around 64 °C were reported. Here, as a reference, we took a POEGMA obtained by RAFT polymerization POEGMARAFT with a molar mass in a similar range as our POEGMAs obtained by NMP in the presence of a styrenic controlling comonomer. We evaluated the thermoresponsive behavior by following the diameter of scattering objects present in a 3 mg mL−1 solution of POEGMA by dynamic light scattering (DLS). The results are collated in Fig. 7. While we found a phase transition temperature (PTT) at around 70 °C for our reference, the presence of the controlling comonomer units systematically led to a decrease of the PTT. This was particularly pronounced for PFS (PTT ≈ 52 °C), which is not surprising considering the hydrophobic properties of fluorinated compounds. This was followed by the two comonomers with more than one aromatic ring in the sidechains, DPPS and PFPVB (PTT ≈ 55 °C), and the chlorinated CMS (PTT ≈ 59 °C). AMS and Sty led to the same PTT of about 63–64 °C, which could be explained by the polarity of the azide offsetting the hydrophobicity of the additional methylene group. All in all, the presence of the controlling comonomers induces a hydrophobization of POEGMA, yet does not a priori hamper potential biological and biotechnological applications, for which the classic usage temperature spans the 4–40 °C range.
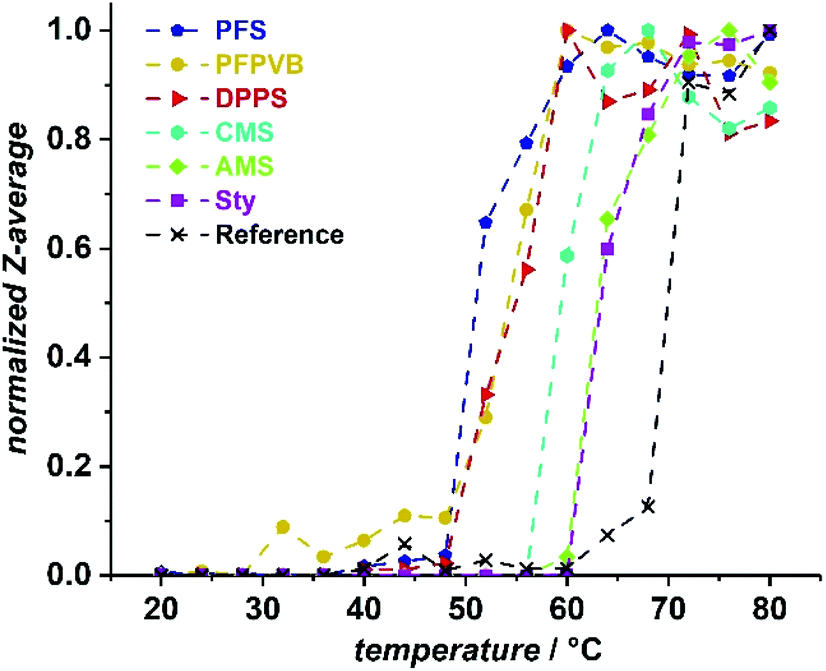 | ||
| Fig. 7 Normalized evolution of the hydrodynamic diameter (Z-average) with temperature in deionized water for POEGMAs obtained by NMP in the presence of styrenics as controlling monomers, compared to a well-defined POEGMA homopolymer obtained by RAFT polymerization. Polymer concentration is 3 mg mL−1. | ||
Application to the synthesis of functional/reactive nanostructured materials
After having demonstrated the possibility of imparting simultaneously control over the polymerization and functionality to the polymer chains, we sought to showcase the utility of the method in various contexts. While we will not present any functionalization in this contribution – because the reactions in which the functional groups present on the styrenic derivatives can be involved are well known and widely reported – we wish to show that their presence facilitates (polymer brushes) or at least may not hamper (block copolymer films) the synthesis of functional nanomaterials.Functional/reactive polymer brushes are a robust avenue to homogeneous reactive coatings of tunable thicknesses.75–78 Polymer brushes are mostly obtained by surface-initiated (SI) RAFT polymerization or ATRP and examples of SI-NMP are relatively scarce.79 There is in fact no example of brushes based on (near-)pure methacrylics obtained by SI-NMP in a controlled manner. The sole examples of SI-NMP involving methacrylates are based on unsuitable nitroxides (e.g., TEMPO),80–83 random copolymers with relatively high amounts of non-methacrylic comonomers (min. 33 mol%),84,85 or no unequivocal demonstration of controlled growth.86–88 Here, silicon wafers were modified in two steps by silanization with 3-aminopropyltriethoxysilane and subsequent amidation with a N-hydroxysuccinimidyl ester of MAMA-SG1 (Fig. 8A). The SI-NMP of OEGMA was then performed by simply immersing functionalized wafers in a classic NMP mixture. The presence of free MAMA-SG1 as sacrificial initiator guarantees a limited impact of possible traces of deactivators (e.g., oxygen) towards the low amount of initiating sites on the flat 2D surface. As a particular example, PFPVB was chosen as the controlling comonomer because activated ester-functionalized polymer brushes are highly meaningful for the immobilization of (bio)molecules on/in polymer brushes.89–93 Monomer conversion was monitored by 1H NMR spectroscopy, along with the thickness of the growing coating by ellipsometry. As observed in Fig. 8B, it was found that the latter significantly increases during the polymerization, demonstrating the pseudo-living character of the polymerization within this conversion range and showcasing the possibility to reliably obtain coatings of specific thicknesses. It must also be noted that these results are obtained after stopping the polymerization for each data point by cooling down the flask, opening to remove one wafer, closing, deoxygenating anew, and reheating, which demonstrates the robustness of the process.
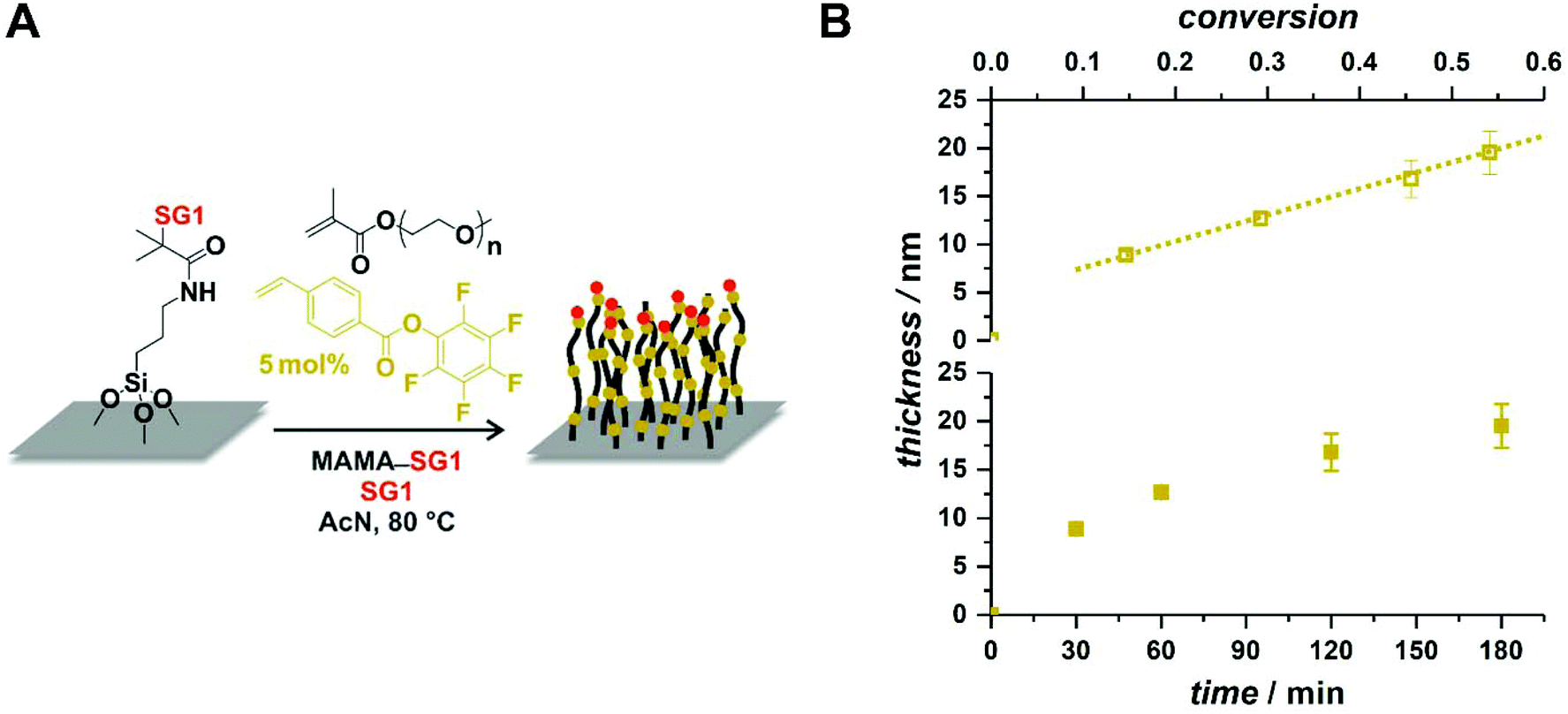 | ||
| Fig. 8 (A) Schematic depiction of the preparation of POEGMA brushes by NMP on a silicon substrate with the help of PFPVB as controlling comonomer. (B) Evolution with polymerization time of the thickness of dry POEGMA brushes obtained by surface-initiated NMP of OEGMA in the presence of 5 mol% of PFPVB as controlling comonomer in acetonitrile at 80 °C. | ||
We recently showed that a range of functionalities could be introduced in block copolymers without necessarily impacting or at least hampering phase separation in the solid state and the formation of nanoscaled patterns. In those cases, the functional comonomers were generally introduced in segments on the same nature, i.e., methacrylics in PMMA segments and styrenics in polystyrene segments, by using ATRP or RAFT polymerization. Here, in contrast, styrenics are introduced in polymethacrylic segments. We therefore were wondering whether it was still possible to obtain phase separation and most importantly polymer films with nanoscale patterns, potentially exhibiting surface reactivity. To showcase this possibility in one example, we chose P(MMA-co-PFS)-b-PSty. The coating process consists of two stages: (i) neutralization of a Si wafer surface by covalent grafting of an OH-end functional random P(MMA-co-Sty) copolymer and (ii) spin-coating of a toluene solution of the block copolymer and annealing (Fig. 9). The atomic force micrograph presented in the same figure reveals fingerprint-like patterns characteristics of either parallel cylinders or perpendicular lamellae. Considering the molar mass (41.7 kg mol−1) and the ratio between the two blocks (fPS = 0.53), the latter morphology is the most likely. Notably, the domain spacing L0 was 45 nm, which is consistent with previous observations with PMMA-b-PS of similar characteristics.46 The present library of functional PMMA-b-PSty block copolymers, obtained by NMP, is therefore promising for nanostructured immobilization.
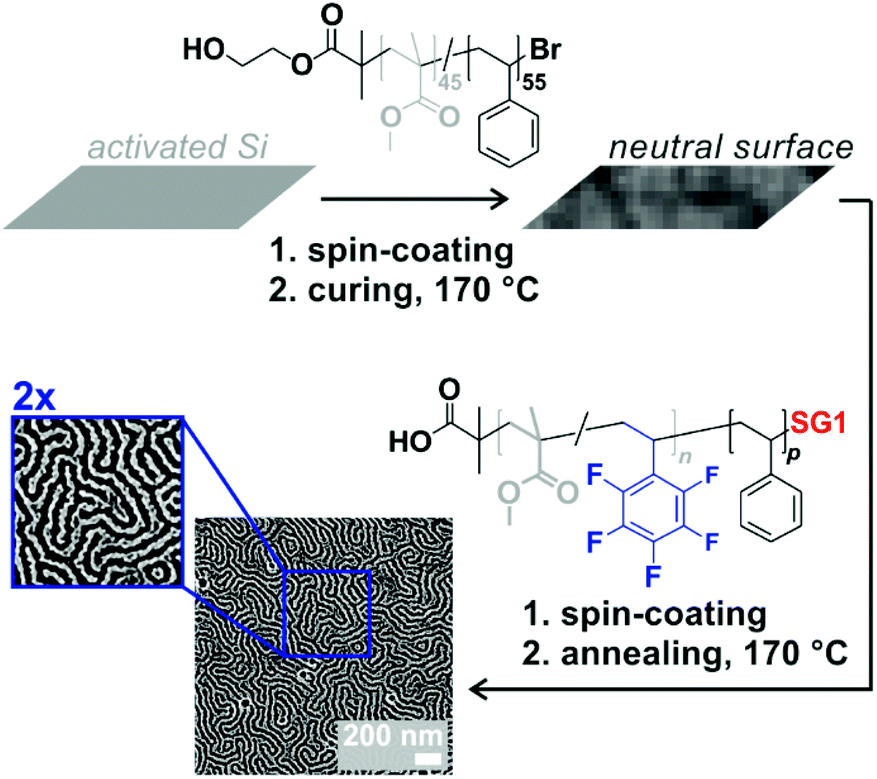 | ||
| Fig. 9 Schematic depiction of the two-step preparation of nanostructured thin films of P(MMA-co-PFS)-b-PS and corresponding phase-based atomic force micrographs with two magnifications. | ||
Conclusions
For a deeper understanding of the relative ability of a given styrenic derivative to impart control to the SG1-mediated polymerization of a given methacrylate, several parameters would need to be determined: (i) the kp of each styrenic, (ii) the reactivity ratios of each comonomer pair, (iii) the nature of the chain-end alkoxyamine sequence (MMA-comonomer-SG1?), and (iv) the kd of such alkoxyamines. Nevertheless, even without these numerical values, the practical impact of just 5 mol% of various styrenics on the SG1-mediated polymerization of MMA and OEGMA is clearly evidenced by the present elementary kinetic and macromolecular data. Primary assessment reveals that an increasing electron-withdrawing ability of the styrenic substituent may lead to faster polymerization in a controlled/living manner. Most importantly, while providing reactive handles along the polymer backbone, the controlling comonomers only weakly alter the optical properties of PMMA and lead to a decrease of the solubility of POEGMA in water, yet to an extent that is irrelevant for envisioned uses. The present study demonstrates the applicability of NMP for the synthesis of (nanostructured) methacrylic-based materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GD thanks the German Federal Ministry of Education and Research (BMBF) for funding in the frame of the Molecular Interaction Engineering program (Biotechnologie 2020+). The Soft Matter Synthesis laboratory (KIT, Helmholtz BIFTM program) is acknowledged for instrumentation support. The authors are grateful to Dr Meike Koenig (IFG, KIT) for her help on ellipsometry. Let the Gliemann (IFG, KIT) and Levkin (ITG, KIT) groups be thanked for access to AFM and DLS, respectively.Notes and references
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, 2008 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- K. Van De Wetering, C. Brochon, C. Ngov and G. Hadziioannou, Macromolecules, 2006, 39, 4289–4297 CrossRef CAS.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- A. S. Lang and M. Thelakkat, Polym. Chem., 2011, 2, 2213 RSC.
- M. Zamfir, P. Theato and J.-F. Lutz, Polym. Chem., 2012, 3, 1796–1802 RSC.
- K. Satoh, J. E. Poelma, L. M. Campos, B. Stahl and C. J. Hawker, Polym. Chem., 2012, 3, 1890–1898 RSC.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092–8101 CrossRef CAS.
- E. Guégain, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2015, 36, 1227–1247 CrossRef.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108–3114 CrossRef CAS.
- M. Edeleva, S. R. A. Marque, D. Bertin, D. Gigmes, Y. Guillaneuf, S. V. Morozov and E. G. Bagryanskaya, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6828–6842 CrossRef CAS.
- P. Astolfi, L. Greci, P. Stipa, C. Rizzoli, C. Ysacco, M. Rollet, L. Autissier, A. Tardy, Y. Guillaneuf and D. Gigmes, Polym. Chem., 2013, 4, 3694–3704 RSC.
- N. Ballard, M. Aguirre, A. Simula, A. Agirre, J. R. Leiza, J. M. Asua and S. van Es, ACS Macro Lett., 2016, 5, 1019–1022 CrossRef CAS.
- A. Simula, M. Aguirre, N. Ballard, A. Veloso, J. R. Leiza, S. van Es and J. M. Asua, Polym. Chem., 2017, 8, 1728–1736 RSC.
- A. Simula, F. Ruipérez, N. Ballard, J. R. Leiza, S. van Es and J. M. Asua, Polym. Chem., 2019, 10, 106–113 RSC.
- S. Grimaldi, J. P. Finet, F. Le Moigne, A. Zeghdaoui, P. Tordo, D. Benoit, M. Fontanille and Y. Gnanou, Macromolecules, 2000, 33, 1141–1147 CrossRef CAS.
- D. Benoit, S. Grimaldi, S. Robin, J. P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- L. Couvreur, C. Lefay, J. Belleney, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2003, 36, 8260–8267 CrossRef CAS.
- K. Bian and M. F. Cunningham, Macromolecules, 2005, 38, 695–701 CrossRef CAS.
- K. Bian and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 414–426 CrossRef CAS.
- R. Hoogenboom, D. Popescu, W. Steinhauer, H. Keul and M. Möller, Macromol. Rapid Commun., 2009, 30, 2042–2048 CrossRef CAS.
- G. Delaittre, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 462–470 CrossRef CAS.
- D. Bertin, P. E. Dufils, I. Durand, D. Gigmes, B. Giovanetti, Y. Guillaneuf, S. R. A. Marque, T. Phan and P. Tordo, Macromol. Chem. Phys., 2008, 209, 220–224 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333–6345 CrossRef CAS.
- M. Edeleva, S. R. A. Marque, K. Kabytaev, Y. Guillaneuf, D. Gigmes and E. Bagryanskaya, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1323–1336 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485–5492 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. A. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2018, 1800551 Search PubMed.
- M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291–9303 CrossRef CAS.
- B. Lessard and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2574–2588 CrossRef CAS.
- B. H. Lessard, E. J. Y. Ling and M. Marić, Macromolecules, 2012, 45, 1879–1891 CrossRef CAS.
- B. Lessard, E. J. Y. Ling, M. S. T. Morin and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1033–1045 CrossRef CAS.
- B. H. Lessard, Y. Guillaneuf, M. Mathew, K. Liang, J. L. Clement, D. Gigmes, R. A. Hutchinson and M. Marić, Macromolecules, 2013, 46, 805–813 CrossRef CAS.
- S. Brusseau, J. Belleney, S. Magnet, L. Couvreur and B. Charleux, Polym. Chem., 2010, 1, 720 RSC.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278–1286 CrossRef CAS.
- V. Delplace, E. Guégain, S. Harrisson, D. Gigmes, Y. Guillaneuf and J. Nicolas, Chem. Commun., 2015, 51, 12847–12850 RSC.
- D. Varadharajan, H. Turgut, J. Lahann, H. Yabu and G. Delaittre, Adv. Funct. Mater., 2018, 28, 1800846 CrossRef.
- H. Turgut, D. Varadharajan, N. Dingenouts and G. Delaittre, Macromol. Rapid Commun., 2018, 39, 1800231 CrossRef.
- H. Turgut, N. Dingenouts, V. Trouillet, P. Krolla-Sidenstein, H. Gliemann and G. Delaittre, Polym. Chem., 2019, 10, 1344–1356 RSC.
- L. Hlalele and B. Klumperman, Macromolecules, 2011, 44, 6683–6690 CrossRef CAS.
- S. Harrisson, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2, 1859–1865 RSC.
- J. Vinas, N. Chagneux, D. Gigmes, T. Trimaille, A. Favier and D. Bertin, Polymer, 2008, 49, 3639–3647 CrossRef CAS.
- J.-P. O'Shea, V. Solovyeva, X. Guo, J. Zhao, N. Hadjichristidis and V. O. Rodionov, Polym. Chem., 2014, 5, 698–701 RSC.
- J. Morgenstern, G. Gil Alvaradejo, N. Bluthardt, A. Beloqui, G. Delaittre and J. Hubbuch, Biomacromolecules, 2018, 19, 4250–4262 CrossRef CAS PubMed.
- M. E. Thomson, J. S. Ness, S. C. Schmidt and M. F. Cunningham, Macromolecules, 2011, 44, 1460–1470 CrossRef CAS.
- J. Nicolas, P. Couvreur and B. Charleux, Macromolecules, 2008, 41, 3758–3761 CrossRef CAS.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 CrossRef CAS.
- X. G. Qiao, P.-Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Macromolecules, 2015, 48, 545–556 CrossRef CAS.
- D. Keller, A. Beloqui, M. Martínez-Martínez, M. Ferrer and G. Delaittre, Biomacromolecules, 2017, 18, 2777–2788 CrossRef CAS.
- C. Dire, S. Magnet, L. Couvreur and B. Charleux, Macromolecules, 2009, 42, 95–103 CrossRef CAS.
- S. R. S. Ting, E. H. Min, P. Escalé, M. Save, L. Billon and M. H. Stenzel, Macromolecules, 2009, 42, 9422–9434 CrossRef CAS.
- C. Zhang, B. Lessard and M. Maric, Macromol. React. Eng., 2010, 4, 415–423 CrossRef CAS.
- S. Brusseau, O. Boyron, C. Schikaneder, C. C. Santini and B. Charleux, Macromolecules, 2011, 44, 215–220 CrossRef CAS.
- PLEXIGLAS® – Das Evonik Geschichtsportal – Die Geschichte von Evonik Industries, https://history.evonik.com/sites/geschichte/de/erfindungen/plexiglas/, (accessed 25 September 2019).
- F. Findik, ISRN Mech. Eng., 2011, 2011, 1–4 CrossRef.
- U. Bog, F. Brinkmann, S. F. Wondimu, T. Wienhold, S. Kraemmer, C. Koos, H. Kalt, M. Hirtz, H. Fuchs, S. Koeber and T. Mappes, Adv. Sci., 2015, 2, 1500066 CrossRef.
- F. R. Mayo, F. M. Lewis and C. Walling, J. Am. Chem. Soc., 1948, 70, 1529–1533 CrossRef CAS.
- V. Ladmiral, T. M. Legge, Y. Zhao and S. Perrier, Macromolecules, 2008, 41, 6728–6732 CrossRef CAS.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, W. Van Camp, O. Coulembier, P. Degée, R. Flammang, F. Du Prez and P. Dubois, React. Funct. Polym., 2008, 68, 990–1003 CrossRef CAS.
- M. H. Stenzel and C. Barner-Kowollik, Mater. Horiz., 2016, 3, 471–477 RSC.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- M. Chenal, C. Boursier, Y. Guillaneuf, M. Taverna, P. Couvreur and J. Nicolas, Polym. Chem., 2011, 2, 1523–1530 RSC.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805–810 CrossRef CAS.
- R. Palkovits, H. Althues, A. Rumplecker, B. Tesche, A. Dreier, U. Holle, G. Fink, C. H. Cheng, D. F. Shantz and S. Kaskel, Langmuir, 2005, 21, 6048–6053 CrossRef CAS.
- J. Martín-de León, V. Bernardo and M. Á. Rodríguez-Pérez, Macromol. Mater. Eng., 2017, 302, 1700343 CrossRef.
- F. Kotz, K. Arnold, S. Wagner, W. Bauer, N. Keller, T. M. Nargang, D. Helmer and B. E. Rapp, Adv. Eng. Mater., 2018, 20, 1700699 CrossRef.
- J. Rühe and W. Knoll, J. Macromol. Sci., Polym. Rev., 2002, 42, 91–138 CrossRef.
- A. Mizutani, A. Kikuchi, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2008, 29, 2073–2081 CrossRef CAS.
- R. M. Arnold, D. L. Patton, V. V. Popik and J. Locklin, Acc. Chem. Res., 2014, 47, 2999–3008 CrossRef CAS.
- S. Lamping, C. Buten and B. J. Ravoo, Acc. Chem. Res., 2019, 52, 1336–1346 CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef CAS.
- K. Sill and T. Emrick, Chem. Mater., 2004, 16, 1240–1243 CrossRef CAS.
- F. J. Xu, Y. Song, Z. P. Cheng, X. L. Zhu, C. X. Zhu, E. T. Kang and K. G. Neoh, Macromolecules, 2005, 38, 6254–6258 CrossRef CAS.
- K. Bian and M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2145–2154 CrossRef CAS.
- M. Mičušík, A. Bonnefond, M. Paulis and J. R. Leiza, Eur. Polym. J., 2012, 48, 896–905 CrossRef.
- N. Cherifi, A. Benaboura, M. Save and L. Billon, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3976–3985 CrossRef CAS.
- V. Mittal, J. Colloid Interface Sci., 2007, 314, 141–151 CrossRef CAS PubMed.
- U. Meyer, F. Svec, J. M. J. Fréchet, C. J. Hawker and K. Irgum, Macromolecules, 2000, 33, 7769–7775 CrossRef CAS.
- A. C. Courbaron Gilbert, C. Derail, N. E. El Bounia and L. Billon, Polym. Chem., 2012, 3, 415–420 RSC.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- M. Badoux, M. Billing and H. A. Klok, Polym. Chem., 2019, 10, 2925–2951 RSC.
- K. A. Günay, N. Schüwer and H. A. Klok, Polym. Chem., 2012, 3, 2186–2192 RSC.
- K. Takasu, K. Kushiro, K. Hayashi, Y. Iwasaki, S. Inoue, E. Tamechika and M. Takai, Sens. Actuators, B, 2015, 216, 428–433 CrossRef CAS.
- H. Son, J. Ku, Y. Kim, S. Li and K. Char, Biomacromolecules, 2018, 19, 951–961 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01458f |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |