
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of metallosupramolecular single-chain polymeric nanoparticles and their characterization by Taylor dispersion†
Laura N.
Neumann
 ,
Dominic A.
Urban
,
Philipp
Lemal
,
Sushila
Ramani
,
Alke
Petri-Fink
,
Sandor
Balog
,
Christoph
Weder
and
Stephen
Schrettl
*
,
Dominic A.
Urban
,
Philipp
Lemal
,
Sushila
Ramani
,
Alke
Petri-Fink
,
Sandor
Balog
,
Christoph
Weder
and
Stephen
Schrettl
*
Adolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: stephen.schrettl@unifr.ch
First published on 19th November 2019
Abstract
Intramolecular cross-linking of polymers can furnish single-chain polymeric nanoparticles (SCPNs), and the use of reversible non-covalent bonds for cross-linking can potentially provide such nanoparticles with stimuli-responsive properties. Here, we report the synthesis of acrylic polymers that carry pendant 2,6-bis(1′-methyl-benzimidazolyl)pyridine ligands, and use these for the preparation of SCPNs through the complex formation with different types of metal ions. The addition of the polymer to solutions containing Fe2+, Zn2+, or Eu3+ ions at low concentrations reliably furnishes the metal–ligand complexes. In order to demonstrate the formation of single-chain polymeric nanoparticles, conventional characterization techniques were complemented by Taylor dispersion analysis, which proved to be particularly useful to accurately measure the hydrodynamic radii of the dispersed particles, in spite of the formation of a small fraction of larger aggregates.
Introduction
Polymers that are equipped with non-covalent binding motifs can assemble through the formation of highly directional interactions, and this approach has been successfully exploited to create a wide range of complex supramolecular structures and materials.1–4 The reversible nature of non-covalent bonds generally provides for a responsive behaviour of the corresponding polymeric materials, especially when the association or dissociation of these bonds can be triggered by a defined (external) stimulus.5–7 Aiming to mimic the intricate folding of biomacromolecules, intramolecular non-covalent bond formation has been employed to trigger the assembly of individual macromolecules into nanoparticles,8–11 and strategies based on hydrogen bonding,12–17 host–guest interactions,18,19 hydrophobic interactions,20–22 or metal–ligand complexes23–26 were reported.The use of metal–ligand (ML) interactions appears to be particularly versatile in this context, since the interaction strength and dynamic nature of the complexes can in principle be tuned by variation of the metal ions, ligands, or counter ions.27–29 Examples of single-chain polymeric nanoparticles (SCPNs) that form through intramolecular ML complexes include the use of a poly(styrene) backbone with pending triarylphosphine ligands that assembles in the presence of Pd2+ ions.24 Further, a poly(N-hydroxyethylacrylamide) with pending 2,2′:6′,2′′-terpyridine ligands was prepared and the intramolecular cross-linking through ML complex formation with Fe2+ ions was employed to obtain SCPNs in water.26 Moreover, SCPNs were also prepared from a copolymer of methyl methacrylate and 2-(acetoacetoxy)ethyl methacrylate.25 The formation of Cu2+-complexes with the β-ketoesters triggered the collapse of the polymer into SCPNs, and the latter were used to catalyse the oxidative coupling reaction of terminal alkynes. Whereas these examples illustrate the successful formation of SCPNs upon ML complex formation,30 a comparison of the processes that yield SCPNs upon addition of metal salts to ligand-functionalized polymers is generally impeded by the varying types of employed ligands, metal salts, and polymers. A modular platform that furnishes SCPNs by intramolecular cross-linking of the same polymer backbone with different metal ions is expected to be of added value, as this could allow for a comparison of the influence of different metal salts and assembly conditions on the particle formation and furnish SCPNs with tuneable characteristics. Notably, interchain ML complex formation thereby constitutes a considerable challenge, even under highly diluted conditions. Thus, there is a finite probability that larger multi-chain aggregates form in addition to the desired SCPNs. Their separation by filtration or centrifugation is difficult, aggregates may reform due to the dynamic nature of the ML interactions, and the presence of larger aggregates can pose difficulties for the in situ experimental characterization of SCPNs.
To address this, we pursued the preparation of an acrylic polymer with tridentate 2,6-bis(1′-methyl-benzimidazolyl)pyridine (Mebip) ligands as pendant groups that are known to reliable form bi- or trifold ML coordination complexes with a range of different transition or lanthanide metal ions (Fig. 1).31,32 The polymer was expected to collapse into SCPNs upon intramolecular ML complex formation in the presence of different metal ions. The characterization by UV-vis spectroscopy, size exclusion chromatography (SEC), dynamic light scattering (DLS), and atomic force microscopy (AFM) was complemented by Taylor dispersion analysis (TDA) as an in situ experimental technique to determine the translational diffusion coefficient of dilute suspensions. The method provides for an improved ‘signal-to-noise’ ratio in favour of single particles, even in the presence of a relatively small number of larger aggregates.33–36 The battery of analytic techniques allowed us to demonstrate that the addition of the acrylic, Mebip-containing polymers to solutions containing Fe2+, Zn2+, or Eu3+ ions at low concentrations reliably furnishes SCPNs with intramolecular metal–ligand complexes, irrespective of the molecular weight of the polymer and the nature of the metal ion.
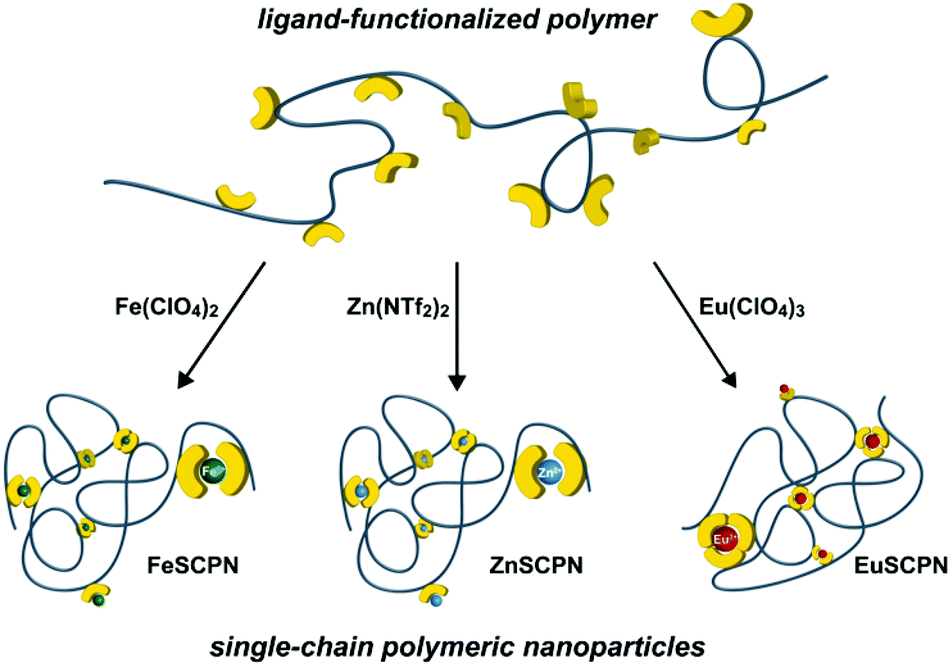 | ||
| Fig. 1 Schematic representation of a polymer chain that carries ligands as pendant groups and its collapse into single-chain polymer nanoparticles (SCPNs) upon formation of intramolecular metal–ligand complexes in the presence of different metal salts with Fe2+, Zn2+, or Eu3+ ions (counter-ions are omitted for clarity). | ||
Results and discussion
For the current study, we chose the tridentate 2,6-bis(1′-methylbenzimidazolyl)pyridine (Mebip) ligand because its coordination with various transition- and lanthanide-metal ions has been demonstrated in many small molecules as well as polymeric systems.37–41 To prepare the desired polymers with pendant Mebip ligands, the statistical copolymerization of butyl acrylate with a functional monomer was pursued. Thus, a Mebip-carrying acrylate derivative was prepared and co-polymerized with n-butyl acrylate by means of reversible addition–fragmentation chain transfer (RAFT) polymerization following a modified literature procedure (Scheme 1).42 The polymer thus obtained was characterized by SEC, which revealed a number-average molar weight (Mn) of 2.4 × 104 g mol−1 and a dispersity (Đ) of 1.04. The analysis of the 1H NMR spectra of the polymer suggests a successful incorporation of ca. 5 mol% of the monomers featuring the Mebip ligand, in accordance with the feed ratio. This concentration was independently confirmed by measurement of the UV-vis absorption spectrum of the polymer that suggested a degree of functionalization of 4.8 mol% on the basis of the extinction coefficient of the ligand (Fig. S1†). The Mn and the fraction of incorporated Mebip repeat units, allow the calculation that each polymer molecule carries on average approximately eight ligands.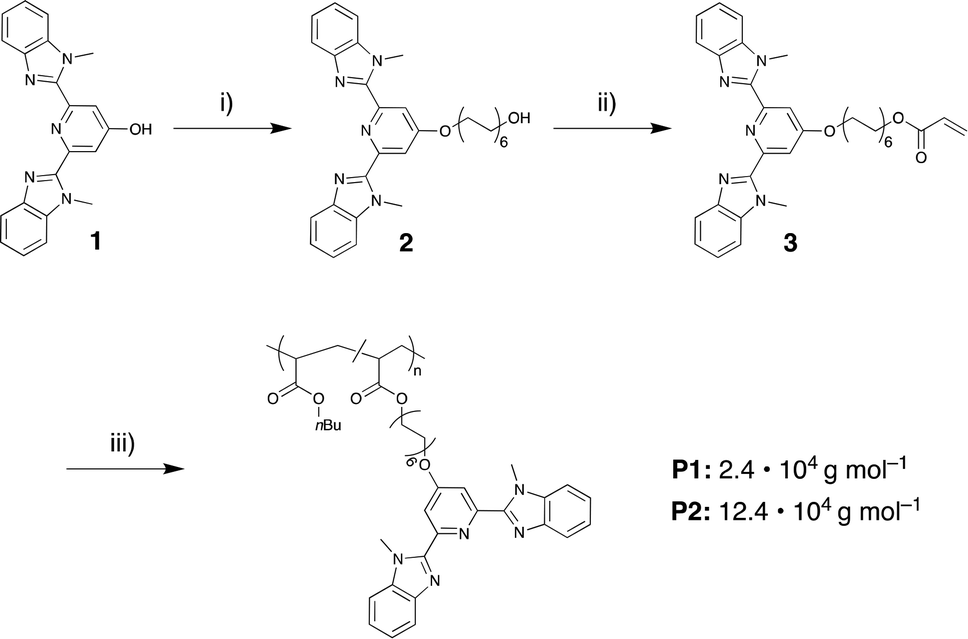 | ||
| Scheme 1 Synthesis of the 2,6-bis(1′-methylbenzimidazolyl)pyridine (Mebip) ligand-functionalized monomer 3 and its co-polymerization with n-butyl acrylate. The resulting poly(n-butyl acrylate-co-Mebip acrylate) P1 and higher-molecular-weight polymer P2 both feature ca. 5 mol% of the ligands as pendant groups. Reagents and conditions: (i) 12-bromo-1-dodecanol, K2CO3, DMF, o/n, 40%; (ii) acryloyl chloride, triethylamine, CHCl3, o/n, 73%; (iii) azobisisobutyronitrile, cyanomethyl dodecyl trithiocarbonate, butyl acrylate, dioxane, 17 h, 60%. | ||
With the ligand-carrying polymer in hand, the preparation of SCPNs by intramolecular chain collapse upon slow addition of a solution of the polymer to solutions of different metal salts was explored. For the current study, three different metal salts were investigated. Since their ML complexes with Mebip are well-studied, the transition metal salts iron perchlorate (Fe(ClO4)2) and zinc bis(triflimide) (Zn(NTf2)2) were selected.39,40,43–45 Both, Fe2+ as well as Zn2+ ions, were expected to give rise to similar bidentate coordination complexes with Mebip ligands, but with different thermodynamic and kinetic binding characteristics.29 Furthermore, europium perchlorate (Eu(ClO4)3) was chosen because of the preference of Eu3+ ions for high coordination numbers,31 which was expected to lead to the formation of trifold coordination complexes with the tridentate Mebip ligands.
To preferentially produce SCPNs and limit the formation of intermolecular cross-links, solutions with low concentrations of the polymer were combined with solutions containing an excess of the metal salts.46,47 Thus, solutions of the polymer (c(THF) = 1 mg mL−1) were added dropwise, using a syringe pump operated at a rate of 1.7 mL h−1, to dilute solutions of the different metal salts in THF (c(Fe(ClO4)2) = 1 mM; c(Zn(NTf2)2) = 1 mM; c(Eu(ClO4)3) = 0.2 mM) in an inert argon atmosphere.26 This approach furnished solutions with dispersed FeSCPN-P1, ZnSCPN-P1, and EuSCPN-P1 featuring polymer P1. In the case of the FeSCPNs, the ML complex formation was indicated by the purple colour that became immediately visible upon addition of the colourless polymer solution to the colourless iron perchlorate solution. A comparison of the UV-vis spectra recorded before and after addition of the polymer solution to the metal salt solution shows that the ligand-to-metal charge-transfer band characteristic for the complex is present at ca. 340 nm in the spectrum of FeSCPN-P1 (Fig. 2a). Moreover, a comparison of the spectrum of FeSCPN-P1 with spectrophotometric titrations that were carried out with solutions of a molecular Mebip model derivative and the same iron perchlorate salt corroborate that full ML complex formation has occurred (Fig. S2†). While the mixtures with zinc and europium salts did not display a comparable distinct colour change upon addition of the polymer solutions, analogous investigations by UV-vis spectroscopy suggest that full complexation of the ligands has occurred in these cases, as well (Fig. 2a and S2†).
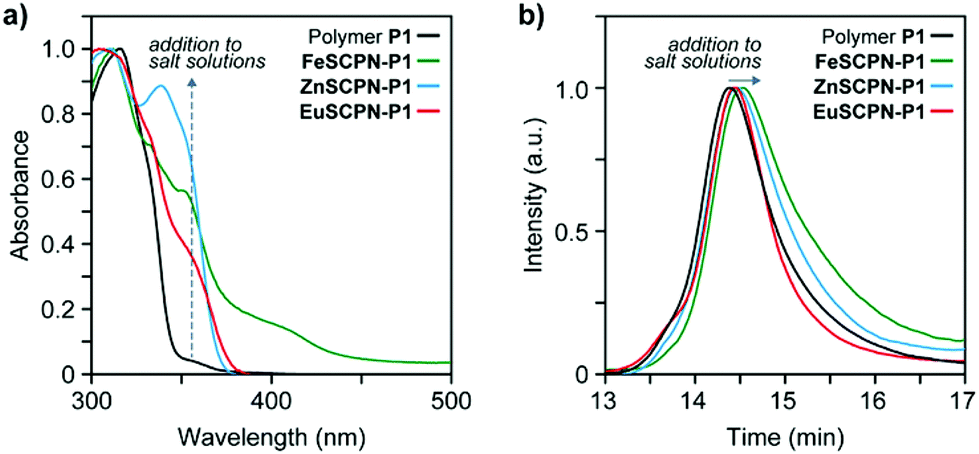 | ||
| Fig. 2 (a) UV-vis spectra of the parent polymer P1 (black line) and the spectra recorded after solutions of the polymer (c(THF) = 1 mg mL−1) were added to dilute solutions of Fe(ClO4)2 (green line), Zn(NTf2)2 (blue line), and Eu(ClO4)3 (red line) in THF as solvent. (b) A comparison of the normalized size exclusion chromatography traces of the parent polymer P1 with those of samples of the polymer after addition to THF solutions of Fe(ClO4)2 (green line, c = 1 mM), Zn(NTf2)2 (blue line, c = 1 mM), and Eu(ClO4)3 (red line, c = 0.2 mM) indicate a reduction of the hydrodynamic radii and suggest formation of SCPNs. | ||
In addition to changes in the absorption spectra, intramolecular cross-linking by ML complex formation should lead to a decrease in the average hydrodynamic volume of the polymers. Indeed, the SEC traces recorded after adding solutions of the polymer P1 to dilute solutions of the three metal salts all show a shift of the elution peak to shorter retention times compared to the parent polymer P1 (Fig. 2b). The relatively small shifts observed for ZnSCPN-P1 and EuSCPN-P1, compared to FeSCPN-P1 may be tentatively attributed to a partial decomplexation due to the dynamic nature of these ML complexes in combination with the significant shear forces and dilution of the sample during SEC measurements.29,48 To probe the hydrodynamic radii of the solvent-dispersed SCPNs, DLS measurements were carried out with samples obtained after addition of polymer P1 to solutions of the different metal salts. In all cases, the measured scattering data reveal two different populations that appear to reflect the presence of the desired SCPNs in addition to much larger aggregates, with dimensions of >100 nm. Indeed, although the formation of SCPNs is a stochastic process that favours the intramolecular collapse of polymer chains under dilute conditions,46,47 the formation of a small number of larger, more strongly scattering aggregates by intermolecular crosslinking of multiple polymer chains can be difficult to exclude.49 Moreover, inter-particle exchange may lead to the formation of larger aggregates over time, particularly in dynamic systems such as the herein investigated SCPNs featuring Zn2+- or Eu3+-based complexes.29,38,44
As attempts to remove the larger particles by multiple filtrations proved unsuccessful in the present study, the DLS data were evaluated with a bimodal linear combination of stretched exponentials (Kohlrausch functions) to account for the presence of larger aggregates and good fits were obtained. Based on this evaluation, average hydrodynamic radii of 22 nm for FeSCPN-P1, 6 nm for ZnSCPN-P1, and 8 nm for the EuSCPN-P1 were determined (Fig. 3a–c). The hydrodynamic radius determined for FeSCPN-P1 is significantly higher than that of both ZnSCPN-P1 and EuSCPN-P1, which may be an artefact attributed to the absorption band of the iron complexes at the wavelength of the laser (660 nm) used for excitation in the DLS setup (Fig. S3†). The DLS experiments indicate that P1 barely undergoes compaction upon complex formation in THF as a good solvent,50 as was previously also observed for a range of different SCPNs under good solvent conditions that appear to primarily adopt conformations close to the random coil.47,51–54
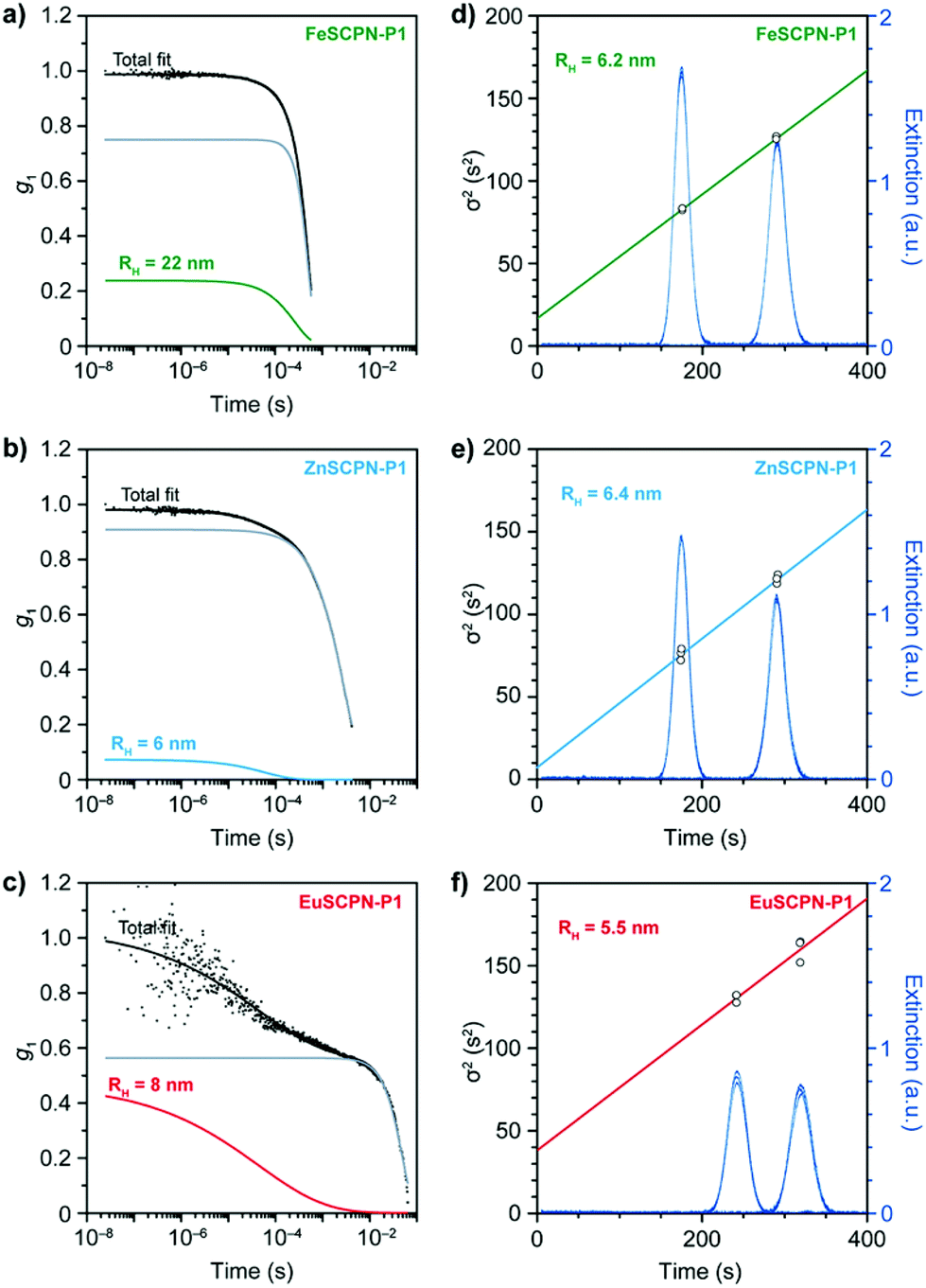 | ||
| Fig. 3 (a–c) Dynamic light scattering (DLS) data of dispersions of (a) FeSCPN-P1 (c(THF) = 24 μmol L−1), (b) ZnSCPN-P1 (c(THF) = 29 μmol L−1), and (c) EuSCPN-P1 (c(THF) = 38 μmol L−1) showing nanometre-sized particles in all samples (raw data (black squares); fits for the SCPNs (coloured), aggregates (grey), and the total fit with a bimodal decay function (black)). (d–f) Taylor dispersion analysis for samples of (d) FeSCPN-P1 (c(THF) = 24 μmol L−1), (e) ZnSCPN-P1 (c(THF) = 29 μmol L−1), and (f) EuSCPN-P1 (c(THF) = 38 μmol L−1) showing the time-evolved concentration profiles (navy) as well as the corresponding fits for FeSCPN-P1 (green), ZnSCPN-P1 (blue), and EuSCPN-P1 (red). | ||
A minor compaction renders the unambiguous characterization of dispersed SCPNs difficult. Moreover, the quality of the DLS measurements – i.e. the accuracy and precision of the determined particle sizes – is dictated by sample homogeneity. Given the small size and their relatively low optical refractive index, the optical contrast and polarizability of SCPNs is small, and scattering of light is therefore weak. At the same time, the polarizability is proportional to the squared volume, leading to an increased light scattering intensity from larger aggregates.55 Even if the number of multi-chain aggregates is small compared to the SCPNs, their presence contributes unpredictable and anomalous intensity fluctuations to the overall scattering intensity that can be significant enough to distort the correlation function, which may severely bias the results of an analysis by means of DLS measurements.51–54,56 Techniques such as viscometry coupled with SEC, small-angle X-ray or neutron scattering measurements, as well as 2D DOSY NMR spectroscopy have been successfully employed in this context,51,52,54 but a reliable characterization of dispersed SCPNs remains challenging.
To circumvent these issues, Taylor dispersion analysis (TDA) was carried out as a reliable method capable of accurately determining particle sizes across several orders of magnitude,57 while offering outstanding precision.33–36,58 This technique relies on the UV-light absorption of the particles (Fig. 2a and 3d–f), which scales linearly with their hydrodynamic radius, and thus allows for a reliable determination of particle sizes, even in the presence of minor amounts of larger aggregates.35,36 In TDA measurements, the translational self-diffusion and sheer-enhanced dispersion of SCPNs that are subjected to a steady laminar flow through a microfluidic channel is monitored by measuring the absorption profile (λ = 280 ± 10 nm) at fixed positions.57 The width of the concentration band of the absorbing particles changes at the different positions in the channel due to the combined effects of convection and diffusion, which, after fitting to the Stokes–Einstein equation, yields the hydrodynamic radii (see the ESI† for a detailed description). Through TDA measurements with the dispersions of the different SCPNs, average hydrodynamic radii of 6.2 ± 0.1, 6.4 ± 0.3, and 5.5 ± 3.5 nm were determined for the samples of FeSCPN-P1, ZnSCPN-P1, and EuSCPN-P1, respectively (Fig. 3d–f), whereas the parent polymer P1 displayed a hydrodynamic radius of 5.6 ± 1.1 nm (Fig. S4†). In TDA measurements, multi-chain aggregates are expected to appear at the tails of the Gaussian-like absorption profile as a wider second mode (Fig. S5†). However, no significant contribution was observed in the measurements, corroborating that the small number of larger aggregates does not interfere critically with the TDA analysis. Indeed, the data suggest that this experimental technique is much more reliable for the evaluation of the sizes of SCPNs in the presence of a fraction of larger aggregates than DLS. A comparison of the determined values suggests that the formation of intramolecular ML complexes between the Mebip ligands and the Fe2+ and Zn2+ ions results in an insignificant compaction of the polymer in the case of FeSCPN-P1 and ZnSCPN-P1. Samples of EuSCPN-P1 that feature the lanthanoid complex similarly do not show a substantial decrease in the hydrodynamic radius, but a relatively large deviation that may be tentatively attributed to the more dynamic nature of these complexes in the employed dilute conditions.29,48
In order to explore the influence of molecular weight, an additional polymer P2 with a higher Mn of 12.4 × 104 g mol−1 (Đ = 1.6, bimodal distribution) was prepared by copolymerization of n-butyl acrylate with 5 mol% of the Mebip-carrying acrylate 3 (Fig. S6†). As in the case of P1, the evaluation of the 1H NMR spectra confirms that the incorporation of 3 in the polymer mirrors the feed ratio, while the analysis of the UV-vis spectra reflects a degree of functionalization of 5.8 mol% (Fig. S1†), which amounts to approximately forty ligands per polymer chain. SCPNs were then prepared in analogy to P1 with Fe(ClO4)2 and Eu(ClO4)3 in order to form FeSCPN-P2 with the least dynamic Fe2+-complexes and EuSCPN-P2 with the more dynamic Eu3+-complexes. TDA measurements reveal a pronounced reduction of the hydrodynamic radius from 14.3 ± 2.1 nm for the parent polymer P2 to 8.2 ± 0.6 nm for samples of FeSCPN-P2 (Fig. S7–S8†). By contrast, samples of EuSCPN-P2 were found to again display an increase in the radius to 18.8 ± 1.3 nm, possibly on account of the more dynamic nature of the europium complexes, which in turn may limit intramolecular cross-linking under dilute conditions. At the same time, previous investigations of the size reduction upon SCPN formation through reversible47 or irreversible cross-links53 by Pomposo and co-workers suggest that a higher cross-linking density does not necessarily lead to an increased compaction, and an analysis of the relation between the molecular weight of a precursor polymer and resulting particle sizes by Blasco et al. found that very low densities can be observed for dispersed SCPNs.54 As such, increased intramolecular cross-linking of the same polymer precursor, as could be observed in SCPNs with complexes of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 metal–ligand ratio (Eu3+) compared to a 1:2 ratio (Fe2+), would not necessarily lead to an increased compaction, but instead to highly polydisperse topologies.
:3 metal–ligand ratio (Eu3+) compared to a 1:2 ratio (Fe2+), would not necessarily lead to an increased compaction, but instead to highly polydisperse topologies.
In addition to the different analyses performed under dilute conditions, the SCPNs made with P1 and P2 were also investigated in the dry state by means of AFM. Thus, dilute dispersions (c = 0.5 mg L−1) of the SCPNs were drop-cast onto silicon wafers and AFM images were recorded after drying under ambient conditions (Fig. 4a–c). As can be seen from the micrographs of the different particles as well as the corresponding height profiles, the AFM measurements corroborate the formation of SCPNs, as particles with sizes of ca. 3–8 nm, 4–8 nm, and 1–4 nm were observed for FeSCPN-P1, ZnSCPN-P1, and EuSCPN-P1, respectively; and similar particle sizes were also observed for SCPNs obtained from P2 (Fig. S9†). Given that the AFM experiments were conducted with dried particles, while the DLS and TDA measurements were performed in a dispersed state, it is clear that the data cannot be directly compared. Indeed, the data reported should be considered indicative for relative sizes and not necessarily as representative absolute values, because, as frequently observed in SCPN analysis,54 different types of measurement techniques yield different average particle sizes. Overall, however, the results confirm the successful formation of SCPNs by intramolecular ML complex formation between the ligand functionalized polymer and the different metal ions.
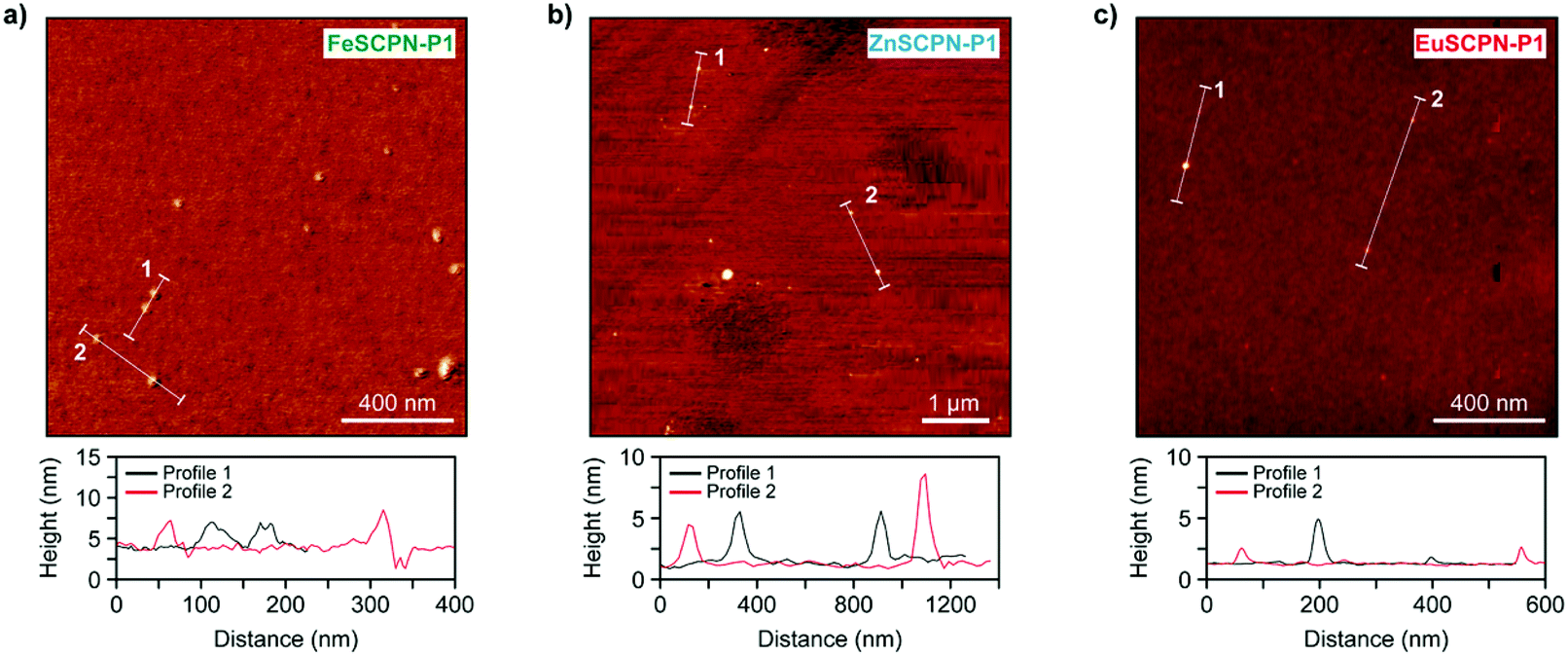 | ||
| Fig. 4 (a–c) AFM height images (top) and corresponding line scans (bottom) for drop-cast samples of (a) FeSCPN-P1 showing values of 3–8 nm in the height profile, (b) ZnSCPN-P1 with particle sizes of 4–8 nm according to the height profile, as well as (c) EuSCPN-P1 showing particles with sizes of 1–4 nm in the height profile. | ||
Conclusions
In summary, a versatile route for the preparation of metallosupramolecular single-chain polymer nanoparticles is presented. The employed modular approach allows for the straightforward preparation of SCPNs featuring different metal ions. To this end, polymers with ca. 5 mol% of pendant, tridentate Mebip ligands were prepared by the statistical copolymerization of n-butyl acrylate with a ligand carrying monomer. The addition of solutions of the polymer to THF solutions containing Fe2+, Zn2+, or Eu3+ ions led to the formation of ML-complexes, which according to the analysis by DLS, TDA, and AFM measurements promoted the formation of predominantly the desired SCPNs. While the formation of a small fraction of larger, multi-chain aggregates hampered the unambiguous determination of the hydrodynamic radii by DLS, measurements by TDA were found to be reliable and confirmed the presence of nanometre-sized SCPNs for all the different metal-ions. Recently developed methods to analyse TDA results promise to allow for a quantification of size polydispersity,59,60 and this technique hence appears to be a valuable (complementary) characterization method of SCPNs and related polymer particles, as it does not suffer from the typical problems the presence of larger aggregates causes. Moreover, the developed approach toward metallosupramolecular SCPNs is expected to provide a viable scaffold for a comparative investigation of the rupture forces of ML complexes on the single-molecule level, and further work is anticipated to allow for a detailed investigation of the different binding strengths and kinetic parameters of different ML complexes. Establishing these parameters in a reliable and comparative fashion is expected to greatly aid in the development of novel biomimetic and mechanoresponsive materials.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors gratefully acknowledge Franziska Marx for help with elemental analysis and mass spectrometry measurements. L. N. N., A. P. F., S. B., C. W., and S. S. gratefully acknowledge financial support through the National Center of Competence in Research Bio-Inspired Materials, a research instrument of the Swiss National Science Foundation (SNF) as well as the Adolphe Merkle Foundation. P. L. and A. P. F. acknowledge additional financial support from the SNF (project 200021_15980).References
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 CrossRef CAS.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2015, 37, 29–46 CrossRef.
- M. Gonzalez-Burgos, A. Latorre-Sánchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS.
- E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2010, 49, 118–126 CrossRef.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS.
- P. J. M. Stals, M. A. J. Gillissen, R. Nicolaÿ, A. R. A. Palmans and E. W. Meijer, Polym. Chem., 2013, 4, 2584–2514 RSC.
- T. Mes, R. van der Weegen, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS.
- G. M. ter Huurne, L. N. J. de Windt, Y. Liu, E. W. Meijer, I. K. Voets and A. R. A. Palmans, Macromolecules, 2017, 50, 8562–8569 CrossRef CAS.
- J. Romulus and M. Weck, Macromol. Rapid Commun., 2013, 34, 1518–1523 CrossRef CAS.
- E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS.
- E. A. Appel, J. D. Barrio, J. Dyson, L. Isaacs and O. A. Scherman, Chem. Sci., 2012, 3, 2278–2274 RSC.
- M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2013, 52, 12–20 CrossRef.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- T. Akagi, P. Piyapakorn and M. Akashi, Langmuir, 2012, 28, 5249–5256 CrossRef CAS.
- J. Willenbacher, O. Altintas, P. W. Roesky and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 35, 45–51 CrossRef.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS.
- F. Wang, H. Pu, M. Jin and D. Wan, Macromol. Rapid Commun., 2016, 37, 330–336 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS.
- A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC.
- R. Dobrawa and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981–4995 CrossRef CAS.
- I. Berkovich, V. Kobernik, S. Guidone and N. G. Lemcoff, in Single-Chain Polymer Nanoparticles, ed. J. A. Pomposo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, vol. 126, pp. 217–257 Search PubMed.
- C. Piguet, J. Bunzli, G. Bernardinelli, G. Hopfgartner and A. F. Williams, J. Alloys Compd., 1995, 225, 324–330 CrossRef CAS.
- S. J. Rowan and J. B. Beck, Faraday Discuss., 2005, 128, 43–53 RSC.
- U. Sharma, N. J. Gleason and J. D. Carbeck, Anal. Chem., 2005, 77, 806–813 CrossRef CAS.
- J. Chamieh and H. Cottet, J. Chromatogr. A, 2012, 1241, 123–127 CrossRef CAS.
- S. Balog, D. A. Urban, A. M. Milosevic, F. Crippa, B. Rothen-Rutishauser and A. Petri-Fink, J. Nanopart. Res., 2017, 19, 67 CrossRef.
- D. A. Urban, A. M. Milosevic, D. Bossert, F. Crippa, T. L. Moore, C. Geers, S. Balog, B. Rothen-Rutishauser and A. Petri-Fink, Colloid Interface Sci. Commun., 2018, 22, 29–33 CrossRef CAS.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS.
- A. Escande, L. Guénée, K.-L. Buchwalder and C. Piguet, Inorg. Chem., 2009, 48, 1132–1147 CrossRef CAS PubMed.
- J. B. Beck, J. M. Ineman and S. J. Rowan, Macromolecules, 2005, 38, 5060–5068 CrossRef CAS.
- D. W. R. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS PubMed.
- L. N. Neumann, C. Calvino, Y. C. Simon, S. Schrettl and C. Weder, Dalton Trans., 2018, 47, 14184–14188 RSC.
- A. C. Jackson, F. L. Beyer, S. C. Price, B. C. Rinderspacher and R. H. Lambeth, Macromolecules, 2013, 46, 5416–5422 CrossRef CAS.
- R. H. Holyer, C. D. Hubbard, S. F. A. Kettle and R. G. Wilkins, Inorg. Chem., 1966, 5, 622–625 CrossRef CAS.
- R. Dobrawa, P. Ballester, C. R. Saha-Möller and F. Würthner, in Metal-Containing and Metallosupramolecular Polymers and Materials, ed. U. S. Schubert, G. R. Newkome and I. Manners, American Chemical Society, Washington, DC, 2009, vol. 928, pp. 43–62 Search PubMed.
- S. Coulibaly, C. Heinzmann, F. L. Beyer, S. Balog, C. Weder and G. L. Fiore, Macromolecules, 2014, 47, 8487–8496 CrossRef CAS.
- A. M. Hanlon, R. Chen, K. J. Rodriguez, C. Willis, J. G. Dickinson, M. Cashman and E. B. Berda, Macromolecules, 2017, 50, 2996–3003 CrossRef CAS.
- J. A. Pomposo, J. Rubio-Cervilla, A. J. Moreno, F. Lo Verso, P. Bacova, A. Arbe and J. Colmenero, Macromolecules, 2017, 50, 1732–1739 CrossRef CAS.
- Y. Liu, Z. Wang and X. Zhang, Chem. Soc. Rev., 2012, 41, 5922–5911 RSC.
- O. Altintas, T. S. Fischer and C. Barner-Kowollik, in Single-Chain Polymer Nanoparticles, ed. J. A. Pomposo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, vol. 181, pp. 1–45 Search PubMed.
- Physical Properties of Polymers Handbook, ed. J. E. Mark, Springer New York, New York, NY, 2007 Search PubMed.
- A. Arbe and J. Colmenero, in Single-Chain Polymer Nanoparticles, ed. J. A. Pomposo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, vol. 44, pp. 129–181 Search PubMed.
- A. M. Hanlon, K. J. Rodriguez, R. Chen, E. Bright and E. B. Berda, in Single-Chain Polymer Nanoparticles, ed. J. A. Pomposo, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, vol. 2, pp. 91–138 Search PubMed.
- J. A. Pomposo, I. Perez-Baena, F. Lo Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767–772 CrossRef CAS.
- E. Blasco, B. T. Tuten, H. Frisch, A. Lederer and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5845–5851 RSC.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 1–19 Search PubMed.
- D. Bossert, F. Crippa, A. Petri-Fink and S. Balog, Anal. Chem., 2018, 90, 3656–3660 CrossRef CAS.
- P. Lemal, A. Petri-Fink and S. Balog, Anal. Chem., 2018, 91, 1217–1221 CrossRef.
- P. Taladriz-Blanco, B. Rothen-Rutishauser, A. Petri-Fink and S. Balog, Anal. Chem., 2019, 91, 9946–9951 CrossRef CAS PubMed.
- L. Cipelletti, J.-P. Biron, M. Martin and H. Cottet, Anal. Chem., 2014, 86, 6471–6478 CrossRef CAS.
- S. Balog, Anal. Chem., 2018, 90, 4258–4262 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and a comprehensive account of all experimental details, including synthetic procedures, analytical data, and NMR spectra. See DOI: 10.1039/c9py01264h |
| This journal is © The Royal Society of Chemistry 2020 |