
Self-assembling morphology-tunable single-component supramolecular antibiotics for enhanced antibacterial manipulation†
Xin
Song‡
a,
Xudong
Deng‡
b,
Qinghua
Wang‡
c,
Jinjin
Tian
a,
Feng-Li
He
b,
Hai-Yu
Hu
 c and
Wei
Tian
*a
c and
Wei
Tian
*a
aMOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions and Shaanxi Key Laboratory of Macromolecular Science and Technology, School of Science, Northwestern Polytechnical University, Xi'an, 710072, Shaanxi, P. R. China. E-mail: happytw_3000@nwpu.edu.cn
bKey Laboratory for Space Bioscience and Biotechnology, School of Life Sciences, Northwestern Polytechnical University, Xi'an 710072, Shaanxi, P. R. China
cState Key Laboratory of Bioactive Substances and Function of Natural Medicine, and Beijing Key Laboratory of Active Substances Discovery and Druggability Evaluation, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100050, P. R. China
First published on 20th November 2019
Abstract
The problem of bacterial resistance to antibiotics has become a major cause of concern in the modern world, due to the occurrence of multidrug-resistant bacterial strains. A single-component supramolecular antibiotic with different self-assembling morphologies that can effectively regulate antibacterial efficacy and present recyclable antimicrobial activity is proposed. This supramolecular antibiotic consists of a cationic polymer-grafted host–guest-conjugated amphiphilic molecule. With sequential ultrasonic and redox stimuli, its self-assemblies presented reversible morphology transitions from spherical micelles to branched aggregates and finally to dot-like assemblies in aqueous solution. Branched aggregates showed the strongest antibacterial ability for E. coli and S. aureus amongst the three self-assemblies, owing to the special enrichment form and high distribution density of the grafted cationic polymer chains on their surfaces. Fluorogen-activating protein imaging was innovatively employed to deeply investigate the supramolecular antibiotic mechanism. It is anticipated that the design of self-assembling, single-component supramolecular antibiotics may be a successful strategy to fight against the increasingly serious problem of drug-resistant bacteria.
Introduction
Antibiotics have been widely used in various fields, including clinical therapy, agricultural utilization, environmental treatment and so on.1 However, following the long-term abuse of antibiotics, the problem of bacterial resistance to antibiotics has become a major cause of concern in the modern world, due to the occurrence of multidrug-resistant bacterial strains.1b,2 Therefore, there is an urgent need for developing novel antibiotic approaches.1b,c,3,4 Recently, a supramolecular strategy has been proposed to prepare antibacterial agents because they possess unique properties and functions.5–7 Thus, the antibacterial activity of supramolecular antibacterial agents can be controlled on demand via regulating the function of their components and noncovalent interactions. As a significant supramolecular interaction, host–guest interaction has been used to construct new supramolecular antibacterial systems.1c,2b,6,7 Zhang and co-workers1c,6a–c reported a supramolecular photosensitizer constructed through host–guest interactions between a porphyrin derivative and cucurbit[7]uril (CB[7]) with enhanced antibacterial activity. Wang and co-workers2b,6d developed a supramolecular antibiotic switch to reversibly turn on and turn off antibacterial activity on demand based on host–guest interactions between CB[7] and cationic polymers. Additionally, other supramolecular host–guest systems were also found to have antibacterial activity.7 Although these systems indeed prevented the accumulation of the active antibiotic in the environment, the presence of two components (host and guest), or even three components (an additional competitive guest), is the essential condition for regulating antibacterial activity, thereby causing possible adverse effects on their practical application. Thus, an effective supramolecular antibacterial system with only a single component that is still multifunctional is urgently needed.5c Furthermore, a single-component self-assembling system means that the self-assemblies are constructed by only one kind of assembly unit, thereby avoiding the mutual interference occurring in multicomponent systems. For example, multifunctional single-component self-assembling systems have shown tremendous advantages in cancer treatment.8 In addition, it is difficult to clarify the interaction between supramolecular antibacterial agents and bacterial cell membranes due to limited methods, so a visible approach is considered to investigate this issue for further understanding the antibacterial mechanism.To address the above challenges, we developed a self-assembling morphology-tunable single-component supramolecular antibiotic for enhanced antibacterial manipulation (Scheme 1). The reported supramolecular antibiotic consists of only a cationic polymer-grafted host–guest-conjugated amphiphilic molecule. The antibacterial activity can be effectively adjusted by regulating the self-assembling morphology of this amphiphilic molecule in aqueous solution. The morphology transitions driven by the synergy between host–guest and hydrophobic interactions can change the enrichment form and distribution density of the grafted cationic polymer chains on the surfaces of supramolecular self-assemblies, resulting in different antibacterial efficiencies. According to other research studies,9 supramolecular self-assemblies driven by hydrogen bonding, hydrophobic and electrostatic interactions represent an emerging and important class of antimicrobial agents. Moreover, the interaction between this supramolecular antibiotic self-assembly and the bacterial cell membrane is observed for the first time by the fluorogen-activating protein imaging method.
 | ||
| Scheme 1 Regulating antibacterial activity by tunable morphology transition via a single-component supramolecular antibiotic under ultrasonic and redox stimuli. (A and B) Fc-(CD-g-(QA-PDMAEA))2 as single-component assembly units to form the initial spherical micelles with relatively moderate antibacterial efficiency in aqueous solution. (B and C) Morphology transition of spherical micelles into branched aggregates with enhanced antibacterial activity induced by ultrasonic vibration. (C and D) Dissociation of branched aggregates by peroxide to form dot-like assemblies with the weakest antibacterial activity. (D and B) Recovery of host–guest interaction by adding GSH to form spherical micelles again. | ||
Herein, a cationic polymer-grafted host–guest-conjugated amphiphilic molecule Fc-(CD-g-(QA-PDMAEA))2 containing a single ferrocene (Fc) moiety and two β-cyclodextrin (β-CD) units carrying two quaternized P(N,N-dimethylaminoethyl acrylate) (QA-PDMAEA) chains was first synthesized (Schemes S1–S3†). Fc-(CD-g-(QA-PDMAEA))2 self-assembled to form spherical micelles with relatively moderate antibacterial efficiency in aqueous solution (Scheme 1A and B). When ultrasonic vibration was imposed on the spherical micelles, their self-assembling morphologies were transformed into branched aggregates with enhanced antibacterial efficiency (Scheme 1B and C). The branched aggregates were dissociated by the addition of peroxide and consequently formed dot-like assemblies with the weakest bactericidal efficiency of the morphologies tested (Scheme 1C and D). The self-assembling morphology and antibacterial efficiency could be recovered by adding L-glutathione (GSH) (Scheme 1D and B). Thus, the antibacterial activity of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies as a single-component supramolecular antibiotic was effectively adjusted by regulating their morphologies under the action of external stimuli in aqueous solution. Furthermore, the destruction of the cell membrane was confirmed by zeta potentials and SEM. Specifically, the effect of supramolecular antibiotics on bacterial cell membrane permeability was visually examined by confocal laser scanning microscopy (CLSM) using fluorogen-activating protein (FAP) imaging technology.
Results and discussion
The Fc-(CD-N3)2 precursor with one Fc moiety and two β-CD units carrying two azido groups was first synthesized (Scheme S1†). The PDMAEA-≡ chain was prepared by reversible addition–fragmentation chain transfer (RAFT) polymerization (Scheme S2†). Finally, Fc-(CD-g-(QA-PDMAEA))2 was synthesized through the azide–alkyne click reaction between Fc-(CD-N3)2 and PDMAEA-≡ and the subsequent quaternization of two PDMAEA chains as antibacterial active sites (Scheme S3†). The detailed characterization data, including 1H NMR, 13C NMR, ESI-MS, MALDI-TOF-MS and GPC results, are shown in the ESI (Fig. S1–S11†), jointly confirming the successful synthesis of Fc-(CD-g-(QA-PDMAEA))2.The morphology transitions of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies were conducted by imposing an ultrasonic or a redox stimulus. Spherical micelles were first obtained by directly dissolving Fc-(CD-g-(QA-PDMAEA))2 in aqueous solution without an external stimulus. TEM and AFM images revealed that these spherical micelles had average diameters of 38 ± 3 nm (Dav, TEM, Fig. 1A(a)) and 41 ± 2 nm (Dav,AFM, Fig. 1B(a)), respectively, which were similar to the hydrodynamic diameter (Dh) of 42 nm, as determined by DLS (Fig. 1C). The central structures of the spherical self-assemblies did not collapse according to the AFM image, demonstrating that the self-assembly is a solid micellar structure rather than a hollow vesicular structure.10 Furthermore, the inner structures of the spherical micelles were quantitatively confirmed by the Rg/Rh value of 0.80, which was consistent with the theoretical value of 0.774, indicating a solid micellar structure.11 After imposing ultrasonic vibration on the micellar structure for 15 min, typical branched aggregates were observed by TEM and AFM (Fig. 1A(b) and B(b)). The DLS results showed that the Dh of branched aggregates increased to 200 ± 30 nm (Fig. 1C) compared with spherical micelles. Subsequently, with the addition of peroxide, branched aggregates dissociated into random dot-like assemblies (Fig. 1A(c) and B(c)) with a size of approximately 10 nm, accompanied by a decrease in the Dh value of 15 nm (Fig. 1C). After adding excess GSH to the dot-like assembly solution, the morphology of the assemblies returned to the initial spherical micelles with Dav, TEM, Dav,AFM, Dh, and Rg/Rh values of 35 ± 3 nm, 39 ± 4 nm, 43 nm, and 0.82 (Fig. S12†), respectively. In addition, the Dh value maintained a cyclic change corresponding to the alternating ultrasonic and redox stimuli at least three times, as shown in Fig. 1D. These results implied that ultrasonic vibration and redox stimuli could induce the reversible morphology transitions of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies from spherical micelles to branched aggregates and then to dot-like assemblies.
 | ||
| Fig. 1 Reversible morphology transitions of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies from spherical micelles to branched aggregates and then to dot-like assemblies. (A) Typical TEM images (freeze-dried samples); (B) AFM images; (C) DH distributions; (D) cyclic changes of DH values corresponding to the alternating ultrasonic and redox stimuli. For A–D: [Fc-(CD-g-(QA-PDMAEA))2] = 1 mg mL−1. | ||
The above-mentioned reversible morphology transitions of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies under ultrasonic and redox triggers were attributed to the synergy of host–guest and hydrophobic interactions. This viewpoint was further confirmed by 2D NOESY (Fig. 2A), I1/I3 values of the pyrene-loaded self-assemblies in aqueous solution12 (Fig. S13 and Table S1†) and 1H NMR analysis of the molecules in DMSO or D2O (Fig. 2B). First, the formation of initial spherical micelles was mainly driven by hydrophobic interactions. As seen from the 2D NOESY spectra of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies in D2O without any external stimulus (Fig. 2A(a)), the proton peak of H3 and H5 (δ = 3.6–3.9) in the β-CD cavity and Fc moiety (Ha-c, δ = 4.1–4.6) only formed a slight cross-peak, indicating a weak host–guest interaction between β-CD and Fc.13 The I1/I3 value (0.712) of the spherical micelles decreased dramatically compared with pure pyrene aqueous micelles (1.025) (Fig. S13 and Table S1†), confirming the formation of hydrophobic domains. Moreover, compared with the 1H NMR spectra of the molecules dissolved in DMSO (Fig. 2B(a)), the 1H NMR signals corresponding to the hydrophobic Fc moiety were not observed when the spherical micelles were dissolved in D2O directly (Fig. 2B(b)). This result verified that the spherical micelles driven by hydrophobic interactions were composed of a hydrophobic Fc moiety in the inner core and a surrounding hydrophilic β-CD and QA salt outer shell in aqueous solution. When ultrasonic vibration was applied, the spherical micelles transformed into branched aggregates, accompanied by enhanced cross-peaks in 2D NOESY spectra (Fig. 2A(b)), indicating that ultrasonic vibration could promote host–guest interaction between β-CD and Fc. Additionally, the I1/I3 value of branched aggregates increased to 0.736 (Fig. S13 and Table S1†), probably because the hydrophobic guest moieties were encapsulated into the β-CD cavity, resulting in a shield of the hydrophobic microdomain.14 In the 1H NMR spectra (Fig. 2B(c)), the peak intensity of protons in the Fc moiety increased with ultrasonic vibration application, implying that hydrophobic Fc moieties could again contact the solution directly. These results revealed that the reinforced host–guest interactions between β-CD and Fc became the main driving force and further induced the dissociation of original spherical micelles and the formation of branched aggregates. After adding H2O2 to the solution, the original hydrophobic Fc moiety transformed into hydrophilic oxidized Fc (Fc+), and the host–guest interaction between β-CD and Fc+ discontinued.15 Thus, the branched aggregates dissociated and then formed dot-like assemblies. This conclusion was verified by 2D NOESY spectra, as shown in Fig. 2A(c); the cross-peaks disappeared after the addition of H2O2. The decreased I1/I3 value of 0.704 (Fig. S13 and Table S1†) further indicated that the dot-like assemblies possessed hydrophobic domains in the interior. The proton signal peaks of Fc drastically decreased after the addition of peroxide into the solution (Fig. 2B(d)) because the electron paramagnetic resonance was triggered by the oxidized Fc (Fc+) moiety.16 Finally, when treating the solution with GSH, the self-assembling morphology was again restored to spherical micelles. The slight cross-peak in the 2D NOESY spectra appeared again (Fig. 2A(d)), demonstrating that Fc+ was reduced to Fc and induced a weak host–guest interaction between β-CD and Fc; the I1/I3 value recovered to 0.717, which was similar to the initial state. Moreover, the proton signal peaks of Fc recovered to their initial states (Fig. 2B(e)) because the hydrophobic interaction was rebuilt. Additionally, during the morphological transition process, the protons in quaternary ammonium salt (δ = 0.7–1.3) maintained a high signal intensity and no obvious change was observed, which confirmed that the hydrophilic quaternary ammonium salt moieties were always located on the surface of these self-assemblies. These results jointly verified that ultrasonic vibration and redox stimuli could regulate the balance between hydrophobic and host–guest interactions of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies, thereby achieving reversible transitions amongst three morphologies from spherical micelles to branched aggregates, and finally forming dot-like assemblies.
 | ||
| Fig. 2 The investigation of the driving force during the assembly process. (A) 2D NOESY spectra of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies in D2O at 298 K. (a) Without stimulus; (b) with ultrasonic vibration for 15 min; (c) adding peroxide to the solution; (d) further treating with GSH. (B) 1H NMR spectra of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies under different conditions at 298 K. The picture on the right-hand side is a magnified view of the Fc moiety; the solid arrow represents the enhancement of the signal and the dotted arrow represents the weakening of the signal. | ||
The antibacterial activities of the above-mentioned different Fc-(CD-g-(QA-PDMAEA))2 self-assemblies were further investigated. E. coli (typical Gram-negative bacterium) and S. aureus (typical Gram-positive bacterium) were chosen as representative strains. The minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) values of different self-assemblies against E. coli and S. aureus (Table 1) were recorded by the routine method.17 The branched aggregates showed the lowest MIC value amongst the three self-assemblies, indicating the strongest antibacterial efficiency. Upon comparison, dot-like assemblies did not show distinct bactericidal activity (MIC > 1024 μg mL−1). Furthermore, the MIC values of the three self-assemblies were beyond their critical aggregation concentration (CAC) values (Table 1 and Fig. S14†), demonstrating that the antibacterial behavior was produced by the self-assemblies themselves rather than the unassembled Fc-(CD-g-(QA-PDMAEA))2 units.7a,b The relationships between antibacterial efficiency and the concentration of self-assemblies for E. coli are shown in Fig. 3A, which shows that the antibacterial behaviors of the three self-assemblies were completely different near specific concentration ranges (64–256 μg mL−1). Compared with the spherical micelles, the branched aggregates showed enhanced antibacterial efficiency, whereas the dot-like assemblies only showed slight antibacterial activity. Furthermore, the antibacterial activities of the three self-assemblies were demonstrated by colony forming unit (CFU) statistics after co-cultivation with bacteria for 12 h, as shown in Fig. 3B and Fig. S17A.† The branched aggregates showed a higher killing efficiency (90%) than the spherical micelles and dot-like assemblies, which showed killing efficiencies of 75% and less than 30% towards E. coli, respectively.
 | ||
| Fig. 3 Antibacterial activities of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies towards E. coli. (A) The relationship between antibacterial efficiency and the concentration of the three self-assemblies for 12 h. (B) CFUs of bacteria treated with the three self-assemblies on LB agar plates: (a) spherical micelles, (b) branched aggregates, and (c) dot-like assemblies. (C) CLSM images of antibacterial activities amongst the three self-assemblies using live/dead bacterial viability assays. The blue signal emission by Hoechst 33258 represents all bacteria, while the red signal emission by PI represents dead bacteria. Scale bars: 10 μm. (D) The relationship between antibacterial efficiency and time for the three self-assemblies. (E) Antibacterial activity change trends of the three self-assemblies under periodic cycles of morphology transitions. For B–E: [Fc-(CD-g-(QA-PDMAEA))2] = 128 μg mL−1. | ||
| CACa (μg mL−1) | MICb (μg mL−1) | MBCb (μg mL−1) | |||
|---|---|---|---|---|---|
| E. coli | S. aureus | E. coli | S. aureus | ||
| a CAC values determined by the conductometric method are described in the Experimental section. b The method to obtain MIC and MBC values is described in the section on antibacterial efficiency tests in the ESI.† | |||||
| Spherical micelles | 23.7 | 256 | 256 | 256 | 512 |
| Branched aggregates | 28.2 | 128 | 128 | 256 | 256 |
| Dot-like assemblies | 27.8 | >1024 | >1024 | >1024 | >1024 |
Next, the live/dead cell staining method by CLSM (Hoechst 33258/propidium iodide (PI))17,18 was used to further investigate the antibacterial efficiency of the self-assemblies, which were first co-cultured with bacteria and then stained with live/dead cell stains. As shown in Fig. 3C, approximately half of the bacteria were dead after their co-cultivation with spherical micelles, while the signal of dead bacteria increased obviously for branched aggregate group samples. By contrast, less than 30% of bacteria died when treated with the dot-like assemblies. The relationship between antibacterial activity and time is shown in Fig. 3D. The time-dependent antibacterial experiments indicated that these self-assemblies presented relatively long-term antibacterial activity. Moreover, these self-assemblies also showed a recyclable antibacterial ability, as shown in Fig. 3E. After three cycles of morphology transitions, the branched aggregates still showed more than 65% antibacterial efficiency, and spherical micelles also maintained good antibacterial activity. Similar antibacterial results of these self-assemblies were also found in the S. aureus system (Fig. S15–S18†). Therefore, the antibacterial activity of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies as supramolecular antibiotics has been successfully regulated through reversible morphology transitions. In addition, these self-assemblies did not show obvious cytotoxicity on human red blood cells under antibacterial conditions in a hemolysis assay (Fig. 4), thus indicating good biocompatibility.
 | ||
| Fig. 4 Hemolytic activity of different morphologies: (A) spherical micelles, (B) branched aggregates, and (C) dot-like assemblies on human RBC. The inset images show the photographs of the corresponding experimental results. (i) and (ii) are the positive and negative controls, and (iii)–(x) are the concentrations ranging from 50 to 1000 μg mL−1. | ||
The antibacterial activity of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies has been effectively regulated, but the antibacterial mechanism still needs to be more deeply elucidated. First, the binding ability of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies of various morphologies to bacteria was confirmed via zeta potentials. E. coli (−37.2 ± 0.2 mV, Fig. 5A and S19†) showed negative zeta potential values, since the bacterial cell membrane is negatively charged.19 When the bacteria were co-cultured with the three self-assemblies for 15 min, the zeta potential values of the bacteria underwent a reversal from a negative potential to a positive potential for spherical micelles (6.5 ± 0.6 mV) and branched aggregates (12.9 ± 0.4 mV) due to the electrostatic binding between the cationic chain QA-PDMAEA in the self-assemblies and the bacterial cell membrane.20 However, the zeta potential value of the bacteria co-cultured with dot-like assemblies (−13.9 ± 0.3 mV) was much lower, indicating a weak binding affinity for the cell membrane. Thus, branched aggregates possessed the strongest binding affinity to the cell membrane. In addition, the zeta potential values of the bacteria co-cultured with the initial and secondary spherical micelles (2.9 ± 0.3 mV) were similar, indicating a recyclable antibacterial ability of the self-assemblies. Second, SEM was employed to confirm the degree of damage on the cell membrane surface. As shown in Fig. 5B, compared with the smooth surface of the control group, the surfaces of E. coli cell membranes co-cultured with the three self-assemblies suffered from different degrees of collapse. Furthermore, branched aggregates presented the strongest membrane damage, as shown in Fig. 5B(c). Similar zeta potential and SEM results were found in the S. aureus system (Fig. 5A, S19 and S20†). In addition, Fc-(CD)2 self-assemblies without the QA-PDMAEA chain did not show any evident antibacterial activity compared with the initial spherical micelles with QA-PDMAEA chains (Fig. S21 and S22†). Therefore, Fc-(CD-g-(QA-PDMAEA))2 self-assemblies with QA-PDMAEA chains first interacted with the cell membrane, then accumulated on its surface, and finally destroyed its integrity, resulting in cell death.
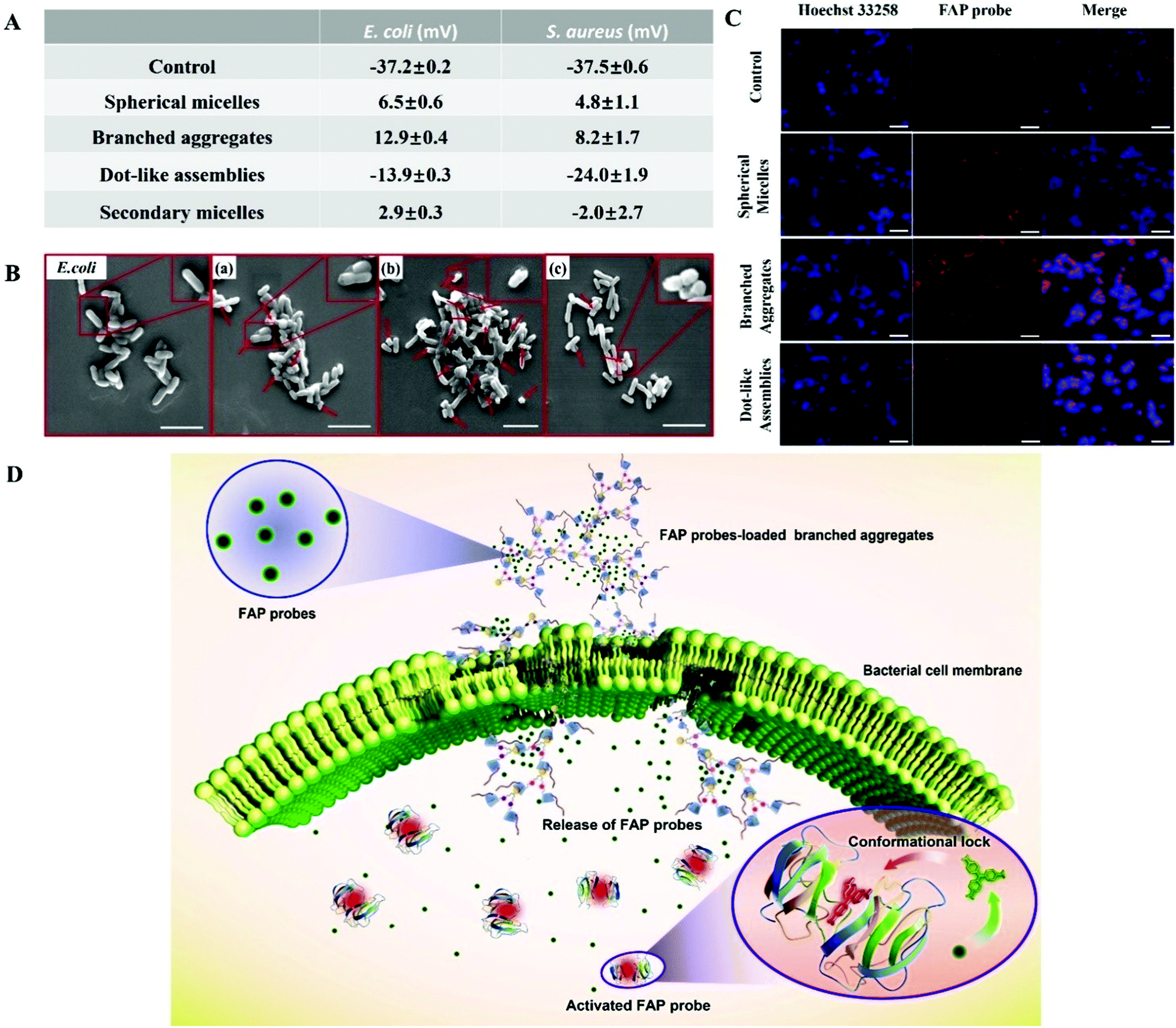 | ||
| Fig. 5 Antibacterial mechanism of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies. (A) Zeta potential values of the three self-assemblies co-cultured with E. coli for 30 min at 25 °C. (B) SEM images of E. coli without treatment and treated with (a) spherical micelles, (b) branched aggregates, and (c) dot-like assemblies. The red arrows indicate the destruction and collapse of cell membranes. Scale bars: 5 μm. (c) CLSM images of the three self-assemblies by physical loading of FAP probes co-cultivated with E. coli, setting the pure probe as the control group. Scale bars: 10 μm; [Fc-(CD-g-(QA-PDMAEA))2] = 128 μg mL−1. (D) Proposed interaction modes of the self-assemblies to destroy the cell membrane by a physically loaded membrane permeability study (taking branched aggregates as a typical example). | ||
Although the antibacterial mechanism of Fc-(CD-g-(QA-PDMAEA))2 self-assemblies has been preliminarily proved, the effect of supramolecular antibiotics on bacterial cell membrane permeability, which is important for a deeper understanding of the antibacterial mechanism, has not yet been observed. The above process was further investigated using a fluorescent dye, malachite green (MG, Fig. S23†). Although MG itself is not fluorescent, it can hinder the rotation of the aromatic rings upon binding to the fluorogen-activating protein (FAP), thereby strongly enhancing the fluorescence.21 Therefore, FAP probes were physically pre-loaded into the three different self-assemblies and then co-cultured with E. coli specifically. The fluorescence signal intensity of the FAP probe (red signal) observed by CLSM showed an increasing transformation from dot-like assemblies into spherical micelles, and then to branched aggregates (Fig. 5C). Thus, branched aggregates showed the strongest cell membrane permeability. In general, the electro-positive quaternary ammonium moiety can damage the cell membrane integrity after combining with the cell membrane compactly by electrostatic attraction, causing the cell membrane to lose its original selective permeability.9e,22 Therefore, FAP-loaded self-assemblies were likely to enter the interior of the bacteria, and then released FAP molecules (Fig. S24†), thereby binding with fluorogen-activating proteins inside E. coli to produce a fluorescence signal (Fig. 5D).
Based on the above results, it can be concluded that the branched aggregates show the strongest antibacterial ability among the three self-assemblies due to the following three possible reasons. First, the branched aggregates possess the highest zeta potential value amongst the three self-assemblies, which showed the strongest binding ability to the bacterial cell membrane. Second, the relatively large self-assembled size of the branched aggregates can increase their local concentration and accelerate the accumulation process on the surfaces of bacterial cell membranes.7b Finally, the branched aggregates also possess the highest specific surface area, which might increase the QA-PDMAEA chain density due to their special configuration.23 Although the spherical micelles also have a relatively strong membrane binding ability, their specific surface area is smaller than that of the branched aggregates due to their spherical structure, resulting in a slightly lower antibacterial ability. Owing to the lowest binding force with bacteria and local concentration, it is difficult for the dot-like assemblies to completely destroy the cell membrane, leading to their lowest antibacterial activity.
Conclusions
In summary, we have successfully designed and constructed a single-component supramolecular antibiotic on the basis of a cationic polymer-grafted host–guest-conjugated amphiphilic molecule. This supramolecular antibiotic could undergo reversibly self-assembling morphology transitions from spherical micelles to branched aggregates and finally to dot-like assemblies in aqueous solution under sequential ultrasonic and redox stimuli due to the synergy between host–guest and hydrophobic interactions. The biocompatible supramolecular antibiotic self-assemblies with different morphologies showed effective antibacterial regulation and a recyclable antibacterial ability against E. coli and S. aureus. Branched aggregates showed the strongest antibacterial activity compared with spherical micelles and dot-like assemblies. The fluorogen-activating protein imaging method provided a new way to observe and deeply investigate the supramolecular antibiotic mechanism. This work provides an effective model for the design of self-assembled supramolecular antibiotics to fight against the increasingly serious problem of drug-resistant bacteria.Experimental section
Materials, characterization methods and synthesis of Fc-(CD-g-(QA-PDMAEA))2
These sections are described in the ESI.†Preparation of various self-assemblies under different conditions
Characterization of the assembly behaviour
Biological properties of various self-assemblies
Antibacterial efficiency = 1 − (A1 − A2)/(A0 − A3) × 100%where A1 represents the absorbance values of the LB medium containing bacteria and assemblies, A2 represents the absorbance values of the LB medium containing only assemblies, A0 represents the absorbance values of the LB medium containing only bacteria and A3 represents the absorbance values of the LB medium alone. Then, 10 μL of the culture solution was extracted, diluted and spread on LB agar plates, and colonies were observed after incubation at 37 °C for 12 h for CFU counting. All samples were collected as three biological replicates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21674086, 21778077, and 21805229), the Natural Science Basic Research Plan in Shaanxi Province of China (2018JZ2003), the Fundamental Research Funds for the Central Universities (3102019PY003 and 3102017OQD039), and the CAMS Innovation Fund for Medical Sciences (CIFMS 2017-I2M-2-004). W. T. thanks Dr Jiangfei Xu (Tsinghua University) for the useful discussion on the manuscript. We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University.Notes and references
- (a) J. Carlet, P. Collignon, D. Goldmann, H. Goossens, I. C. Gyssens, S. Harbarth, V. Jarlier, S. B. Levy, B. NÏDoye, D. Pittet, R. Richtmann, W. H. Seto, J. W. M. van der Meer and A. Voss, Lancet, 2011, 378, 369–371 CrossRef PubMed; (b) W. A. Velema, J. P. van der Berg, M. J. Hansen, W. Szymanski, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924–928 CrossRef CAS PubMed; (c) Y. Yang, P. He, Y. Wang, H. Bai, S. Wang, J. F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16239–16242 CrossRef CAS; (d) M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS.
- (a) Q. Zhang, G. Lambert, D. Liao, H. Kim, K. Robin, C.-K. Tung, N. Pourmand and R. H. Austin, Science, 2011, 333, 1764–1767 CrossRef CAS; (b) H. Bai, H. Yuan, C. Nie, B. Wang, F. Lv, L. Liu and S. Wang, Angew. Chem., Int. Ed., 2015, 54, 13208–13213 CrossRef CAS PubMed.
- (a) L.-L. Li, J.-H. Xu, G.-B. Qi, X. Zhao, F. Yu and H. Wang, ACS Nano, 2014, 8, 4975–4983 CrossRef CAS PubMed; (b) T. Wei, Q. Yu and H. Chen, Adv. Healthcare Mater., 2019, 8, 1801381 CrossRef CAS PubMed; (c) Y. Hong, Y. Xi, J. Zhang, D. Wang, H. Zhang, N. Yan, S. He and J. Du, J. Mater. Chem. B, 2018, 6, 6311–6321 RSC.
- W. Lee, Z.-H. Li, S. Vakulenko and S. Mobashery, J. Med. Chem., 2000, 43, 128–132 CrossRef CAS.
- (a) H. Bai, F. Lv, L. Liu and S. Wang, Chem. – Eur. J., 2016, 22, 11114–11121 CrossRef CAS; (b) W. Zhan, T. Wei, Q. Yu and H. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 36585–36601 CrossRef CAS; (c) X. Li, H. Bai, Y. Yang, J. Yoon, S. Wang and X. Zhang, Adv. Mater., 2019, 31, 1805092 Search PubMed.
- (a) K. Liu, Y. Liu, Y. Yao, H. Yuan, S. Wang, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 8285–8289 CrossRef CAS PubMed; (b) L. Chen, H. Bai, J. F. Xu, S. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 13950–13957 CrossRef CAS; (c) Z. Huang, H. Zhang, H. Bai, Y. Bai, S. Wang and X. Zhang, ACS Macro Lett., 2016, 5, 1109–1113 CrossRef CAS; (d) H. Bai, H. Zhang, R. Hu, H. Chen, F. Lv, L. Liu and S. Wang, Langmuir, 2017, 33, 1116–1120 CrossRef CAS.
- (a) C. Zhou, D. Wang, M. Cao, Y. Chen, Z. Liu, C. Wu, H. Xu, S. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 30811–30823 CrossRef CAS; (b) C. Zhou, H. Wang, H. Bai, P. Zhang, L. Liu, S. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 31657–31666 CrossRef CAS; (c) T. Wei, W. Zhan, L. Cao, C. Hu, Y. Qu, Q. Yu and H. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 30048–30057 CrossRef CAS; (d) S. Li, N. Jiang, W. Zhao, Y.-F. Ding, Y. Zheng, L.-H. Wang, J. Zheng and R. Wang, Chem. Commun., 2017, 53, 5870–5873 RSC; (e) Q. Li, Y. Wu, H. Lu, X. Wu, S. Chen, N. Song, Y.-W. Yang and H. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 10180–10189 CrossRef CAS; (f) T. Wei, W. Zhan, Q. Yu and H. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 25767–25774 CrossRef CAS; (g) A. Dawn, H. Chandra, C. Ade-Browne, J. Yadav and H. Kumari, Chem. – Eur. J., 2017, 23, 18171–18179 CrossRef CAS; (h) T. Wang, C. Wang, S. Zhou, J. Xu, W. Jiang, L. Tan and J. Fu, Chem. Mater., 2017, 29, 8325–8337 CrossRef CAS; (i) J. Robinson-Duggon, F. Pérez-Mora, L. Valverde-Vásquez, D. Cortés-Arriagada, J. De la Fuente, G. Günther and D. Fuentealba, J. Phys. Chem. C, 2017, 121, 21782–21789 CrossRef CAS.
- (a) X. Li, S. Yu, D. Lee, G. Kim, B. Lee, Y. Cho, B.-Y. Zheng, M.-R. Ke, J.-D. Huang, K. T. Nam, X. Chen and J. Yoon, ACS Nano, 2018, 12, 681–688 CrossRef CAS; (b) X. Li, C.-y. Kim, S. Lee, D. Lee, H. M. Chung, G. Kim, S.-H. Heo, C. Kim, K.-S. Hong and J. Yoon, J. Am. Chem. Soc., 2017, 139, 10880–10886 CrossRef CAS; (c) X. Li, D. Lee, J.-D. Huang and J. Yoon, Angew. Chem., Int. Ed., 2018, 57, 9885–9890 CrossRef CAS; (d) Q. Zou, M. Abbas, L. Zhao, S. Li, G. Shen and X. Yan, J. Am. Chem. Soc., 2017, 139, 1921–1927 CrossRef CAS.
- (a) S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2011, 4, 31–36 CrossRef; (b) M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek Jr., C. Yao, L. F. Chen, A. L. Ferguson, G. C. Wong and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13155–13160 CrossRef CAS; (c) L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. DeGrado and E. Gazit, Nat. Commun., 2017, 8, 1365 CrossRef; (d) J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed., 2016, 55, 2592–2595 CrossRef CAS; (e) N. Rodrigues de Almeida, Y. Han, J. Perez, S. Kirkpatrick, Y. Wang and M. Conda-Sheridan, ACS Appl. Mater. Interfaces, 2019, 11, 2790–2801 CrossRef CAS; (f) L. P. Datta, R. Mukherjee, S. Biswas and T. K. Das, Langmuir, 2017, 33, 14195–14208 CrossRef CAS PubMed; (g) T. Muthukumarasamyvel, R. Baskar, S. Chandirasekar, K. Umamaheswari and N. Rajendiran, ACS Appl. Mater. Interfaces, 2016, 8, 25111–25126 CrossRef CAS; (h) C. Zhou, F. Wang, H. Chen, M. Li, F. Qiao, Z. Liu, Y. Hou, C. Wu, Y. Fan, L. Liu, S. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 4242–4249 CrossRef CAS; (i) H. Sun, Y. Hong, Y. Xi, Y. Zou, J. Gao and J. Du, Biomacromolecules, 2018, 19, 1701–1720 CrossRef CAS; (j) J. Gao, M. Wang, F. Wang and J. Du, Biomacromolecules, 2016, 17, 2080–2086 CrossRef CAS; (k) L. Gao, M. Li, S. Ehrmann, Z. Tu and R. Haag, Angew. Chem., 2019, 58, 3645–3649 CrossRef CAS; (l) X. Ding, S. Duan, X. Ding, R. Liu and F.-J. Xu, Adv. Funct. Mater., 2018, 28, 1802140 CrossRef; (m) E. Li, Y. Zhou, R. Zhao, K. Jie and F. Huang, Angew. Chem., 2019, 58, 3981–3985 CrossRef CAS; (n) B. Hua, W. Zhou, Z. Yang, Z. Zhang, L. Shao, H. Zhu and F. Huang, J. Am. Chem. Soc., 2018, 140, 15651–15654 CrossRef CAS; (o) X. Chi, G. Yu, L. Shao, J. Chen and F. Huang, J. Am. Chem. Soc., 2016, 138, 3168–3174 CrossRef CAS PubMed; (p) Q. Xu, T. Huang, S. Li, K. Li, C. Li, Y. Liu, Y. Wang, C. Yu and Y. Zhou, Angew. Chem., Int. Ed., 2018, 57, 8043–8047 CrossRef CAS; (q) S. Xu, X. Zhu, C. Zhang, W. Huang, Y. Zhou and D. Yan, Nat. Commun., 2018, 9, 2053 CrossRef; (r) X. Liao, F. Yang, R. Wang, X. He, H. Li, R. Y. T. Kao, W. Xia and H. Sun, Chem. Sci., 2017, 8, 8061–8066 RSC; (s) K. Yuan, Q. Mei, X. Guo, Y. Xu, D. Yang, B. J. Sánchez, B. Sheng, C. Liu, Z. Hu, G. Yu, H. Ma, H. Gao, C. Haisch, R. Niessner, Z. Jiang and H. Zhou, Chem. Sci., 2018, 9, 8781–8795 RSC.
- J. F. Reuther, D. A. Siriwardane, R. Campos and B. M. Novak, Macromolecules, 2015, 48, 6890–6899 CrossRef CAS.
- (a) C. Wu and S. Zhou, Phys. Rev. Lett., 1996, 77, 3053–3055 CrossRef CAS; (b) H. Zhang, X. Fan, R. Suo, H. Li, Z. Yang, W. Zhang, Y. Bai, H. Yao and W. Tian, Chem. Commun., 2015, 51, 15366–15369 RSC.
- (a) C.-G. Mu, X.-D. Fan, W. Tian, Y. Bai and X. Zhou, Polym. Chem., 2012, 3, 1137–1149 RSC; (b) M. Murugesan, M. Aulice Scibioh and R. Jayakumar, Langmuir, 1999, 15, 5467–5473 CrossRef CAS.
- A. Feng, Q. Yan, H. Zhang, L. Peng and J. Yuan, Chem. Commun., 2014, 50, 4740–4742 RSC.
- (a) Z. Yuan, J. Wang, Y. Wang, Y. Zhong, X. Zhang, L. Li, J. Wang, S. F. Lincoln and X. Guo, Macromolecules, 2019, 52, 1400–1407 CrossRef CAS; (b) Y. Shi, H. Li, J. Cheng, T. Luan, D. Liu, Y. Cao, X. Zhang, H. Wei, Y. Liu and G. Zhao, Chem. Commun., 2017, 53, 12302–12305 RSC; (c) Y. Bai, C. Liu, X. Song, L. Zhuo, H. Bu and W. Tian, Chem. – Asian J., 2018, 13, 3903–3911 CrossRef CAS.
- Y. Kang, Y. Ma, S. Zhang, L.-S. Ding and B.-J. Li, ACS Macro Lett., 2015, 4, 543–547 CrossRef CAS.
- (a) W. Yasen, R. Dong, L. Zhou, J. Wu, C. Cao, A. Aini and X. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 9006–9014 CrossRef CAS PubMed; (b) M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- Y. Zhu, C. Xu, N. Zhang, X. Ding, B. Yu and F.-J. Xu, Adv. Funct. Mater., 2018, 28, 1706709 CrossRef.
- K. E. Furse and S. A. Corcelli, J. Am. Chem. Soc., 2008, 130, 13103–13109 CrossRef CAS.
- B. Luan, K. L. Chen and R. Zhou, J. Phys. Chem. Lett., 2016, 7, 2434–2438 CrossRef CAS.
- (a) S. J. Lam, E. H. H. Wong, C. Boyer and G. G. Qiao, Prog. Polym. Sci., 2018, 76, 40–64 CrossRef CAS; (b) L. Liu, K. Xu, H. Wang, P. K. J. Tan, W. Fan, S. S. Venkatraman, L. Li and Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS.
- (a) S. Xu and H.-Y. Hu, Acta Pharm. Sin. B, 2018, 8, 339–348 CrossRef; (b) K. Ferreira, H.-Y. Hu, V. Fetz, H. Prochnow, B. Rais, P. P. Müller and M. Brönstrup, Angew. Chem., Int. Ed., 2017, 56, 8272–8276 CrossRef CAS.
- J. Salta, R. I. Benhamou, I. M. Herzog and M. Fridman, Chem. – Eur. J., 2017, 23, 12724–12728 CrossRef CAS.
- Y. Huang, X. Ding, Y. Qi, B. Yu and F.-J. Xu, Biomaterials, 2016, 106, 134–143 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9py01440c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |