
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral cyclometalated iridium complexes for asymmetric reduction reactions†
Jennifer
Smith
,
Aysecik
Kacmaz
,
Chao
Wang
 ,
Barbara
Villa-Marcos
and
Jianliang
Xiao
*
,
Barbara
Villa-Marcos
and
Jianliang
Xiao
*
Department of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: jxiao@liv.ac.uk
First published on 23rd November 2020
Abstract
A series of chiral cyclometalated iridium complexes have been synthesised by cyclometalating chiral 2-aryl-oxazoline and imidazoline ligands with [Cp*IrCl2]2. These iridacycles were studied for asymmetric transfer hydrogenation reactions with formic acid as the hydrogen source and were found to display various activities and enantioselectivities, with the most effective ones affording up to 63% ee in the asymmetric reductive amination of ketones and 77% ee in the reduction of pyridinium ions.
1. Introduction
Cyclometalated Cp*-iridium complexes, or iridacycles, are easily accessible via base-assisted C–H activation. A number of such complexes have been documented both within our group1 as well as by others with various applications.2 Examples of their use as catalysts are seen in hydrogenation,3 transfer hydrogenation,4 dehydrogenation,5 reductive amination,6 alkylation,7 hydrosilylation,8 racemisation,9 hydroamination10 and aerobic oxidation.11 However, most of these complexes are achiral, and the number of reported chiral iridacycles and their applications in asymmetric catalysis is much smaller.Chiral iridacycle complexes have been sporadically reported over the past years. Selected examples are shown in Scheme 1. Ikariya et al. reported the use of chiral Cp*Ir(C–N) complexes12 for asymmetric transfer hydrogenation (ATH) of acetophenone, yielding (S)-1-phenylethanol in over 90% yield with up to 66% ee. Pfeffer and de Vries et al. reported cyclometalated amino and imidazoline complexes;13 the latter was used for ATH of acetophenone and N-phenyl-(1-phenylethylidene)amine, showing moderate to high yields with low enantioselectivities up to 19% ee.13a Baya et al. also reported the use of cyclometalated iridium complexes for ATH of ketones, affording low to moderate enantioselectivities (up to 58% ee).14 Davies et al. synthesised iridacycles bearing a chiral oxazoline ligand.15 Leung et al. carried out ATH of acetophenone with chiral iridacycles, providing up to 69% ee at −30 °C.16 In recent years, the group of Richards have reported a series of planar chiral iridacycles bearing ferrocene-type ligands, although their application in asymmetric catalysis has rarely been seen.17 More recently, novel chiral iridacycles have been reported by the groups of de la Torre, Sierra and Cramer.18 Our group reported iridium complexes bearing N,O- and N,C-chelating oxazoline ligands for ATH of aromatic ketones; however, the cyclometalated N,C-complex was much less active and enantioselective (up to 7% ee) than the N,O-chelated complex (up to 99% ee).19 In continuation of our study into iridacycles, we have synthesised a range of chiral iridium catalysts and examined their ability in direct asymmetric reductive amination (DARA) of ketones and reduction of pyridiniums via ATH. Reported herein is the results of these studies.
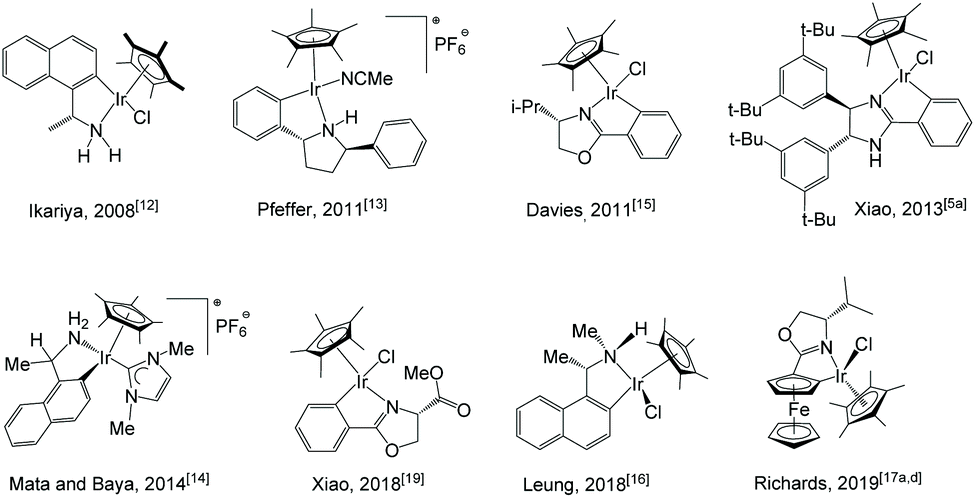 | ||
| Scheme 1 Examples of chiral iridacycles from the literature. | ||
2. Results and discussion
2.1. Synthesis of chiral iridacycles
Base-promoted cyclometallation has been widely studied.15,20 The reaction typically involves C–H bond activation of a chiral ligand with an iridium precursor to form the cyclometalated structure. In 1998, Beck et al.20b reported iridacycles being produced from the reaction of [Cp*IrCl2]2 with substituted 2-phenyl-4-R-5(4H)-oxazolones in the presence of NaOAc. In 2003, Davies20c carried out similar cyclometalation of nitrogen-containing ligands with [Cp*IrCl2]2, [Cp*RhCl2]2 and [RuCl2(p-cymene)]2, revealing the assisting role of the acetate. In most subsequent studies, a similar strategy has been deployed.To access the chiral iridacycles in this study, a range of ligands consisting of chiral oxazoline and imidazoline motifs were prepared. These were easily accessible from the reaction between a benzaldehyde derivative and a chiral amino alcohol or diamine, using reported methods (Scheme 2).21 The subsequent cyclometalation of the iridium precursor [Cp*IrCl2]2 with the chiral oxazoline and imidazoline ligands generally occurred under mild conditions, affording the chiral iridacycles 1–15 (Scheme 2). However, more forcing conditions were required for some iridacycles, e.g.4, likely due to the increased steric hindrance in the ligands.
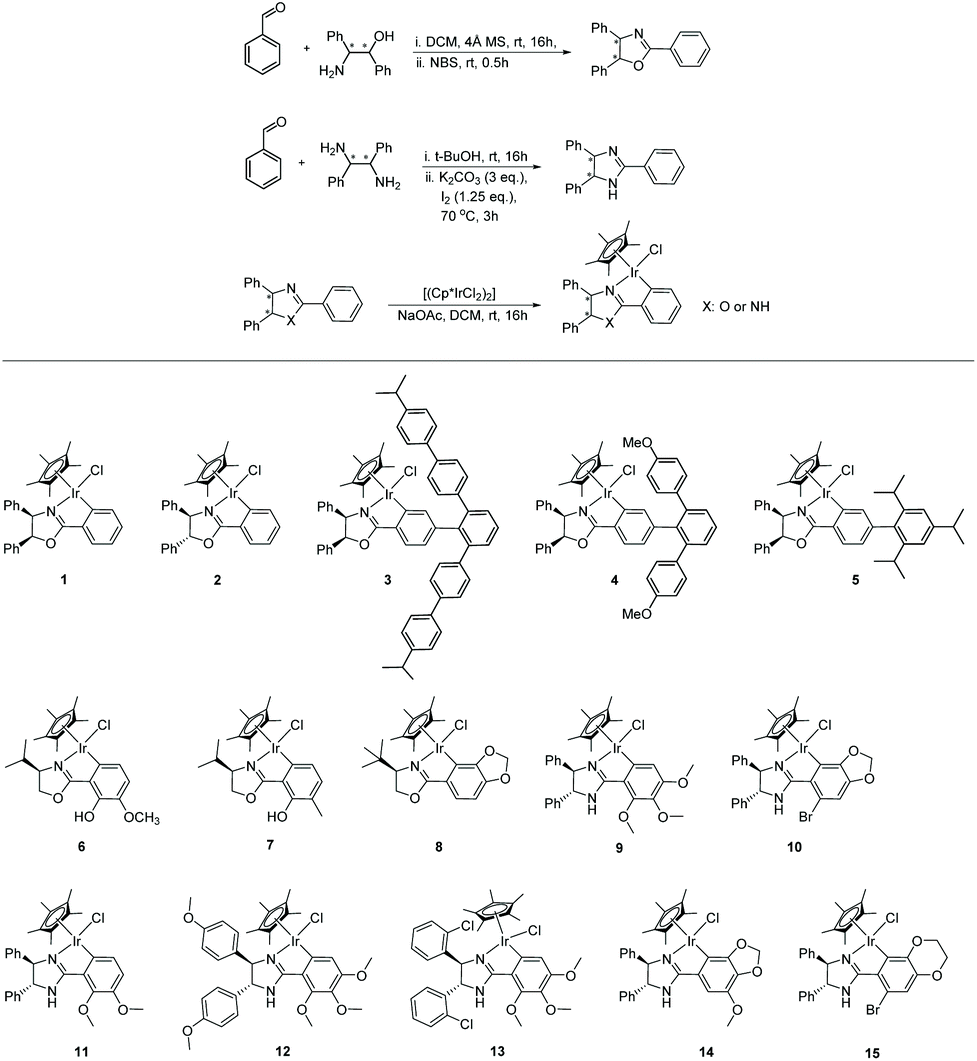 | ||
| Scheme 2 Synthesis of oxazoline and imidazoline ligands and related chiral iridacycles. | ||
The formation of the iridacycles was confirmed by NMR and HRMS analysis and by X-ray diffraction in the case of complexes 1 and 2. Chelation of the ligands onto iridium introduces chirality at the metal centre. However, the iridacycles are formed generally as a mixture of two diastereoisomers, presumably due to the small difference between their thermodynamic stability, as has been observed in analogous cases.13,20c For example, 1 and 2 exist as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 mixture, respectively, in chloroform. The X-Ray diffraction structures of 1 and 2 (Fig. 1) were obtained from single crystals by diffusing n-hexane into a dichloromethane solution of the complexes. As expected, piano-stool style geometries are observed for both complexes. The structure of 1, which bears the cis- or (S,R)-oxazoline ligand, features a S-configured iridium centre in the solid state. In contrast, the iridium centre of 2, which bears the trans- or (R,R)-oxazoline ligand, is R-configured. The bond distances involving the iridium show no notable difference between the two complexes except for a slightly longer Ir–N distance in 2 and are in the expected range. A CH/π interaction appears present between the methyl of the Cp* ring and the phenyl ring from the oxazoline. For example, in 1, the distance between a Cp* hydrogen and the closest hydrogen on the phenyl ring is only 2.640 Å.
:1 and 1:1 mixture, respectively, in chloroform. The X-Ray diffraction structures of 1 and 2 (Fig. 1) were obtained from single crystals by diffusing n-hexane into a dichloromethane solution of the complexes. As expected, piano-stool style geometries are observed for both complexes. The structure of 1, which bears the cis- or (S,R)-oxazoline ligand, features a S-configured iridium centre in the solid state. In contrast, the iridium centre of 2, which bears the trans- or (R,R)-oxazoline ligand, is R-configured. The bond distances involving the iridium show no notable difference between the two complexes except for a slightly longer Ir–N distance in 2 and are in the expected range. A CH/π interaction appears present between the methyl of the Cp* ring and the phenyl ring from the oxazoline. For example, in 1, the distance between a Cp* hydrogen and the closest hydrogen on the phenyl ring is only 2.640 Å.
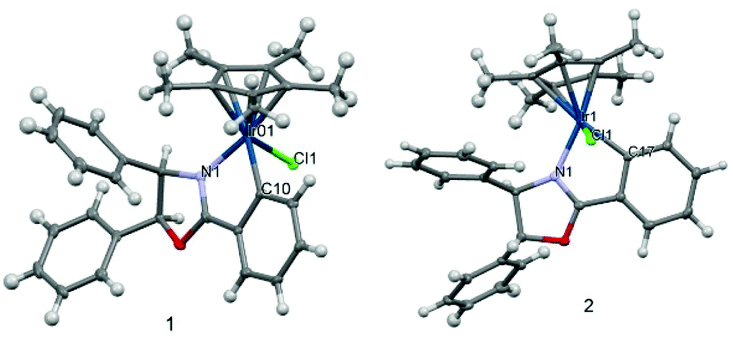 | ||
| Fig. 1 Molecular structures of 1 and 2 determined by single crystal X-ray diffraction. For 1: selected bond distances (Å): Ir1–Cl1 2.402(1), Ir1–N1 2.084(2), Ir1–C10 2.052(3). Selected bond angles (°): Cl1–Ir1–N1 85.1(1), Cl1–Ir1–C10 83.2(1), N1–Ir1–C10 77.4(2). For 2: selected bond distances (Å): Ir1–Cl1 2.4056(6), Ir1–N1 2.101(2), Ir1–C17 2.062. Selected bond angles (°): Cl1–Ir1–N1 86.57(5), Cl1–Ir1–C17 84.18, N1–Ir1–C17 77.48(8). | ||
2.2. Asymmetric reductive amination of ketones
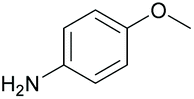
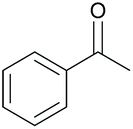
Chiral amines have received considerable attention in fine chemical and pharmaceutical applications.22 DARA represents a convenient pathway for their synthesis and can be accomplished through metal-catalysed, organocatalytic or biocatalytic approaches, using a range of hydrogen sources.22c,23 Among them, DARA reactions utilising hydrogen gas have been the most widely reported,24 whilst ATH systems,25 exploiting other hydrogen sources, have remained underdeveloped. DARA via transfer hydrogenation is, however, highly appealing, as it is likely to be easier and safer in operation than using hydrogen gas.Iridacycles 1–15 were initially screened for the DARA of acetophenone with p-methoxyaniline in isopropanol (IPA), using formic acid as the hydrogen source in the form of formic acid-triethylamine azeotrope (FT) (Table 1). All the complexes showed good to high catalytic activities; however, the enantioselectivities varied considerably, ranging from 0 to 56% ee. There appears to be little correlation between the ee's and the ligand structure in general, partially due to the diversity of the ligand structures. Of those screened, complex 9, a 3,4,5-trimethoxy imidazoline iridicycle, yielded the highest enantioselectivity (56% ee) for the DARA (Table 1, entry 9). Therefore, 9 was subjected to a range of different solvents, additives, hydride sources and temperatures to determine the optimal conditions (ESI, Table S1†). Finally, it was determined that using the FT in IPA at 20–25 °C would provide the best conversion and enantioselectivity (Table 1, entry 10).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | İridacycle | Conv. (%) | ee (%) | Entry | İridacycle | Conv. (%) | ee (%) |
| Reaction conditions: 0.5 mmol acetophenone, 0.6 mmol p-methoxy aniline, 1 mol% iridacycle, 2.5 mL anhydrous IPA, 0.5 mL FT, sealed in air, ambient temperature; PMP: p-methoxyphenyl. The product is of R configuration.a Optimization at 23 °C. | |||||||
| 1 | 1 | 68 | 23 | 9 | 9 | 78 | 56 |
| 2 | 2 | 83 | 14 | 10 | 9 | 92 | 56 |
| 3 | 3 | 62 | 10 | 11 | 10 | 72 | 50 |
| 4 | 4 | 60 | 0 | 12 | 11 | 75 | 42 |
| 5 | 5 | 95 | 25 | 13 | 12 | 78 | 21 |
| 6 | 6 | 50 | 30 | 14 | 13 | 75 | 39 |
| 7 | 7 | 50 | 17 | 15 | 14 | 65 | 43 |
| 8 | 8 | 80 | 30 | 16 | 15 | 75 | 43 |
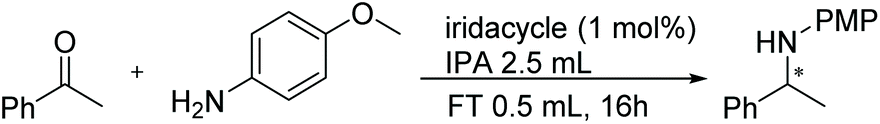
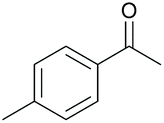
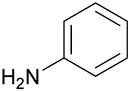
Subsequently, a range of ketones and amines were investigated to determine whether the iridacycle 9 could be exploited for DARA. As can be seen from Table 2, whilst 9 catalysed efficient reductive amination, affording the amine products generally in high yields, the enantioselectivities were only moderate in most cases. More specifically, utilising p-methoxyaniline as the amine source, the para- and meta-substituted acetophenones provided very high yields and moderate enantioselectivities (Table 2, entries 2–6). However, an ortho-substituted ketone displayed a lower enantioselectivity (Table 2, entry 7), indicating that the ortho substituent interferes with the enantioselectivity-determining step. Changing R′ to an ethyl or a phenyl group led to enhanced selectivity (Table 2, entries 8 and 10); however, lengthening the alkyl group resulted in a lower selectivity (Table 2, entry 9), demonstrating a limit to the functionalisation that can be tolerated in this position. The reaction with an alkyl ketone afforded a very high yield (97%), but racemic amine (Table 2, entry 11). In the case of other aniline derivatives coupling with acetophenone, the yields decreased significantly (Table 2, entries 12–16), indicating that the electron donating methoxy group on the amine is crucial, probably assisting in the imine formation. With the more nucleophilic benzylamines (Table 2, entries 17–20), the yields were high; but the enantioselectivity was disappointingly low, presumably as a result of reduced steric rigidity in the imine intermediate.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Ketone | Amine | Yieldd (%) | eee (%) | Entry | Ketone | Amine | Yieldd (%) | eee (%) |
| a Reaction conditions (unless otherwise stated) (i): 0.5 mmol ketone, 0.6 mmol amine, 1 mol% 9, 2.5 ml anhydrous IPA, 0.5 mL FT, sealed in air, 16 h. We assume the products to be of the same configuration, i.e. R. b Reaction conditions (ii): 0.5 mmol ketone, 0.6 mmol amine, 1 mol% 9, 3 mL HCO2Na/HCO2H pH 4.5, 0.3 mL 2-MeTHF, sealed in air, 8 h. c The same reaction condition with [b] but 16 h. d Yield of isolated product. e Determined by HPLC. | |||||||||
| 1 |
|
|
90 | 56 | 11 |
|
|
97 97b | 0 5b |
| 2 |
|
|
95 | 50 | 12 |
|
|
37 82c | 53 48c |
| 3 |
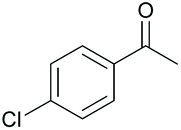
|
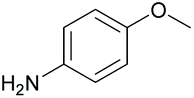
|
94 80b | 54 48b | 13 |
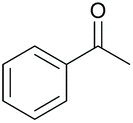
|
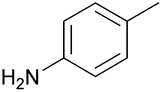
|
37 84c | 55 53c |
| 4 |
|
|
80 | 54 | 14 |
|
|
8 42c | 52 54c |
| 5 |
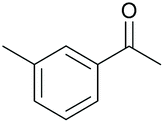
|
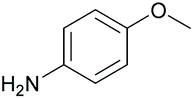
|
96 | 56 | 15 |
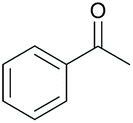
|
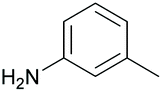
|
47 | 53 |
| 6 |
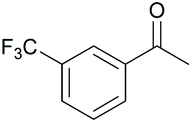
|
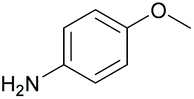
|
95 | 56 | 16 |
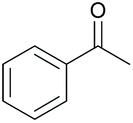
|
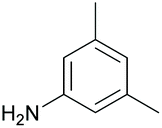
|
50 65c | 62 60c |
| 7 |
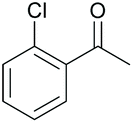
|
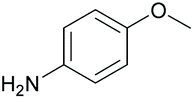
|
88 | 34 | 17 |
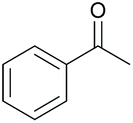
|
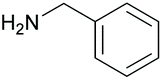
|
80 97c | 16 10c |
| 8 |
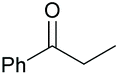
|
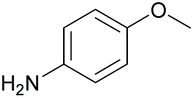
|
88 | 63 | 18 |
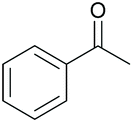
|
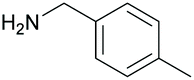
|
66 94c | 14 16c |
| 9 |
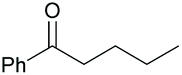
|
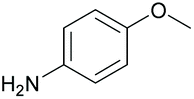
|
75 84c | 38 36c | 19 |
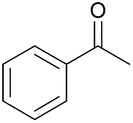
|
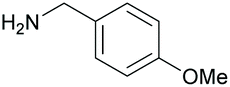
|
77 93c | 17 22c |
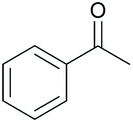
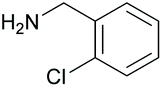
| 10 |
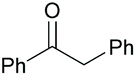
|
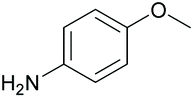
|
38 76c | 63 59c | 20 |
|
|
90 93c | 10 2c |
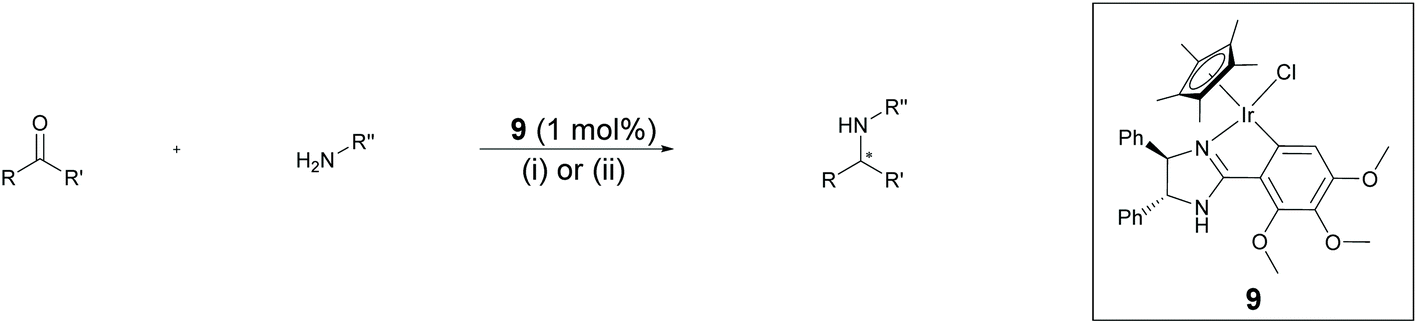
Aiming to improve the yield and enantioselectivity, the impact of aqueous conditions was also investigated, utilising NaCO2H/HCO2H(aq.) at pH 4.5 as a hydrogen source for DARA, with 2-methyltetrahydrofuran (2-MeTHF) as a co-solvent. Under such conditions, some of the yields were improved; however, the enantioselectivities showed little change (Table 2, entries 3, 9–14, 16). In particular, benzylamines afforded higher yields but still low enantioselectivities (Table 2, entries 17–20).
2.3. ATH of pyridinium salts
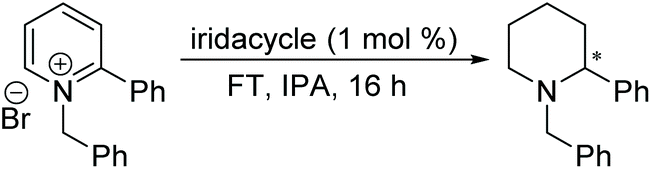
Chiral piperidines are valuable building blocks for natural products and synthetic bioactive molecules. Asymmetric hydrogenation of pyridines can be used to obtain chiral piperidines.26 However, these reactions are generally difficult to carry out, due to the coordination ability and the resonance stability of the pyridines. To overcome these challenges, pyridines can be activated, in the form of pyridinium salts or auxiliary-substituted pyridines.26b,27 Whilst transfer hydrogenation of pyridines has been reported,4a,28 very little has been reported for ATH of pyridines. In 2007 Rueping et al. developed the first organocatalytic ATH system, utilising a Brønsted acid to activate the substrate and induce chirality.29 With the chiral iridacycles in hand, we also examined their potential for ATH of pyridinium salts.The iridacycles were first examined for ATH of N-benzyl-2-phenylpyridinium bromide salt, using FT in IPA, as shown Table 3. It appears that the oxazoline-containing iridacycles (Table 3, entries 1–6,) generally exhibited a low enantioselectivity (up to 34% ee). Among the imidazoline ligands, the previously used 9 provided 44% ee, and the best enantioselectivity was achieved with the dioxoleiridacycle 10, which afforded 51% ee (Table 3, entry 8). Using 10 as catalyst at a lower temperature of −10 °C, the selectivity was increased and the piperidine was isolated in 97% yield with an ee of 77% (Table 3, entry 9)
|
|
|||||
|---|---|---|---|---|---|
| Entry | Iridacycle | ee (%) | Entry | Iridacycle | ee % |
| Reaction conditions: 0.5 mmol pyridinium salt, 1 mol% Ir cat, 2.5 mL anhydrous IPA, 0.5 mL FT, sealed, ambient temperature. Enantiomeric excess determined by chiral HPLC.a Reaction conducted at −10 °C.b Isolated yield in parentheses. | |||||
| 1 | 3 | 20 | 8 | 10 | 51 |
| 2 | 4 | 10 | 9a | 10 | 77(97)b |
| 3 | 5 | 15 | 10 | 11 | 33 |
| 4 | 6 | 6 | 11 | 12 | 24 |
| 5 | 7 | 34 | 12 | 13 | 20 |
| 6 | 8 | 25 | 13 | 14 | 46 |
| 7 | 9 | 44 | 14 | 15 | 38 |
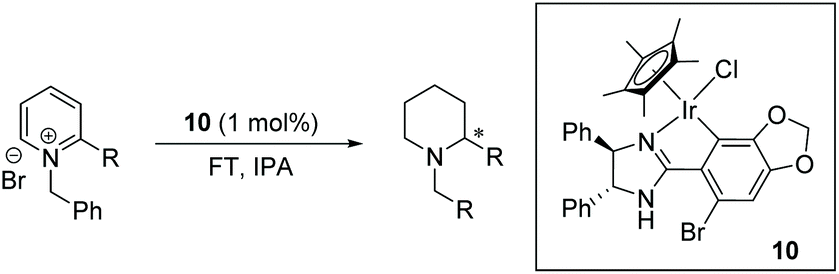
Under the conditions identified, we examined the ATH of a range of pyridinium salts. Selected examples are shown in Table 4.‡ As can be seen, the iridacycle 10 is active for the substituted pyridinium salts, affording the corresponding piperidines in high yields. However, the enantioselectivity varied considerably, from 77% ee for the 2-aryl substituted substrates to 10% ee for the less sterically hindered 2-benzyl substituted one (Table 4, entries 1–3).
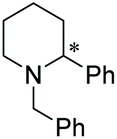
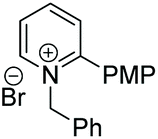
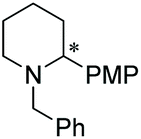
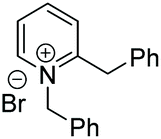
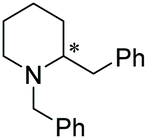
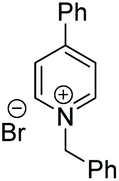
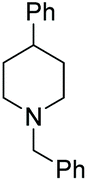
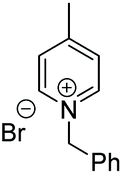
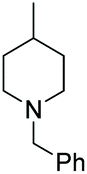
3. Conclusions
Chiral iridacycles can be easily accessed via cyclometalation reaction of [Cp*IrCl2]2 with a ligand that undergoes C–H activation and such complexes could serve as catalysts for asymmetric reactions. In this study, we have synthesised a wide range of chiral iridacycles and examined their potential application in DARA via transfer hydrogenation. Among the iridacycles, the 3,4,5-trimethoxy imidazoline-bearing 9 proved to be the most effective, affording moderate enantioselectivities in the DARA of acetophenones with aniline derivatives. The iridacycles are also active in catalysing the ATH of pyridiniums, albeit with only good ee's in the case of 2-aryl substituted substrates. Research within our group continues to develop chiral iridacycles, aiming for better enantioselective catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank AstraZeneca (JS) and the Scientific and Technological Research Council of Turkey (TUBİTAK) for support and Dr Ramachandran Gunasekar for technical assistance.Notes and references
- C. Wang and J. Xiao, Chem. Commun., 2017, 53, 3399 RSC.
- C. Michon, K. MacIntyre, Y. Corre and F. Agbossou-Niedercorn, ChemCatChem, 2016, 8, 1755 CrossRef CAS.
- (a) J. Wu, J. H. Barnard, Y. Zhang, D. Talwar, C. M. Robertson and J. Xiao, Chem. Commun., 2013, 49, 7052 RSC; (b) B. Villa-Marcos, W. Tang, X. Wu and J. Xiao, Org. Biomol. Chem., 2013, 11, 6934 RSC; (c) W. Tang, C. Lau, X. Wu and J. Xiao, Synlett, 2014, 25, 81 CAS; (d) E. Salomo, P. Rojo, P. Hernandez-Llado, A. Riera and X. Verdaguer, J. Org. Chem., 2018, 83, 4618 CrossRef CAS; (e) Y. Schramm, F. Barrios-Landeros and A. Pfaltz, Chem. Sci., 2013, 4, 2760 RSC.
- (a) D. Talwar, H. Y. Li, E. Durham and J. Xiao, Chem. – Eur. J., 2015, 21, 5370 CrossRef CAS; (b) D. Talwar, X. Wu, O. Saidi, N. P. Salguero and J. Xiao, Chem. – Eur. J., 2014, 20, 12835 CrossRef CAS; (c) Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629 RSC.
- (a) J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234 RSC; (b) J. Wu, D. Talwar, S. Johnston, M. Yan and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 6983 CrossRef CAS; (c) X. Jiang, W. Tang, D. Xue, J. Xiao and C. Wang, ACS Catal., 2017, 7, 1831 CrossRef CAS; (d) K. Fujita, T. Yoshida, Y. Imori and R. Yamaguchi, Org. Lett., 2011, 13, 2278 CrossRef CAS.
- (a) D. Gulcemal, S. Gulcemal, C. M. Robertson and J. Xiao, Organometallics, 2015, 34, 4394 CrossRef CAS; (b) Q. Lei, Y. Wei, D. Talwar, C. Wang, D. Xue and J. Xiao, Chem. – Eur. J., 2013, 19, 4021 CrossRef CAS; (c) D. Talwar, N. P. Salguero, C. M. Robertson and J. Xiao, Chem. – Eur. J., 2014, 20, 245 CrossRef CAS; (d) Y. Wei, C. Wang, X. Jiang, D. Xue, J. Li and J. Xiao, Chem. Commun., 2013, 49, 5408 RSC; (e) C. Wang, A. Pettman, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548 CrossRef CAS.
- (a) Q. Zou, C. Wang, J. Smith, D. Xue and J. Xiao, Chem. – Eur. J., 2015, 21, 9656 CrossRef CAS; (b) D. Wang, K. Zhao, C. Xu, H. Miao and Y. Ding, ACS Catal., 2014, 4, 3910 CrossRef CAS.
- (a) Y. Corre, V. Rysak, X. Trivelli, F. Agbossou-Niedercorn and C. Michon, Eur. J. Org. Chem., 2017, 4820 CrossRef CAS; (b) Y. Corre, W. Iali, M. Hamdaoui, X. Trivelli, J. P. Djukic, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2015, 5, 1452 RSC; (c) M. Hamdaoui, C. Desrousseaux, H. Habbita and J. P. Djukic, Organometallics, 2017, 36, 4864 CrossRef CAS.
- (a) T. Jerphagnon, R. Haak, F. Berthiol, A. J. A. Gayet, V. Ritleng, A. Holuigue, N. Pannetier, M. Pfeffer, A. Voelklin, L. Lefort, G. Verzijl, C. Tarabiono, D. B. Janssen, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Top. Catal., 2010, 53, 1002 CrossRef CAS; (b) T. Jerphagnon, A. J. A. Gayet, F. Berthiol, V. Ritleng, N. Mrsic, A. Meetsma, M. Pfeffer, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Chem. – Eur. J., 2009, 15, 12780 CrossRef CAS.
- W. Iali, F. La Paglia, X. F. Le Goff, D. Sredojevic, M. Pfeffer and J. P. Djukic, Chem. Commun., 2012, 48, 10310 RSC.
- S. Arita, T. Koike, Y. Kayaki and T. Ikariya, Angew. Chem., Int. Ed., 2008, 47, 2447 CrossRef CAS.
- S. Arita, T. Koike, Y. Kayaki and T. Ikariya, Organometallics, 2008, 27, 2795 CrossRef CAS.
- (a) E. Feghali, L. Barloy, J. T. Issenhuth, L. Karmazin-Brelot, C. Bailly and M. Pfeffer, Organometallics, 2013, 32, 6186 CrossRef CAS; (b) N. Pannetier, J. B. Sortais, J. T. Issenhuth, L. Barloy, C. Sirlin, A. Holuigue, L. Lefort, L. Panella, J. G. de Vries and M. Pfeffer, Adv. Synth. Catal., 2011, 353, 2844 CrossRef CAS.
- S. Sabater, M. Baya and J. A. Mata, Organometallics, 2014, 33, 6830 CrossRef CAS.
- Y. Boutadla, D. L. Davies, R. C. Jones and K. Singh, Chem. – Eur. J., 2011, 17, 3438 CrossRef CAS.
- H. J. Chen, R. H. X. Teo, Y. Li, S. A. Pullarkat and P. H. Leung, Organometallics, 2018, 37, 99 CrossRef CAS.
- (a) R. A. Arthurs, D. L. Hughes, P. N. Horton, S. J. Coles and C. J. Richards, Organometallics, 2019, 38, 1099 CrossRef CAS; (b) R. A. Arthurs, C. C. Prior, D. L. Hughes, V. S. Oganesyan and C. J. Richards, Organometallics, 2018, 37, 4204 CrossRef CAS; (c) R. A. Arthurs, P. N. Horton, S. J. Coles and C. J. Richards, Eur. J. Inorg. Chem., 2017, 229 CrossRef; (d) R. A. Arthurs, M. Ismail, C. C. Prior, V. S. Oganesyan, P. N. Horton, S. J. Coles and C. J. Richards, Chem. – Eur. J., 2016, 22, 3065 CrossRef CAS.
- (a) M. G. Avello, M. C. de la Torre, M. A. Sierra, H. Gornitzka and C. Hemmert, Chem. – Eur. J., 2019, 25, 13344 CrossRef CAS; (b) J. Mas-Roselló, T. Smejkal and N. Cramer, Science, 2020, 368, 1098 CrossRef.
- G. Zhou, A. H. Ahmed, C. M. Robertson, R. Liu, Z. Li, K. Luzyanin, N. G. Berry, W. Chen and J. Xiao, ACS Catal., 2018, 8, 8020 CrossRef CAS.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS; (b) W. Bauer, M. Prem, K. Polborn, K. Suenkel, W. Steglich and W. Beck, J. Inorg. Chem., 1998, 485 CAS; (c) D. L. Davies, O. Al-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132 RSC; (d) L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492 CrossRef CAS.
- (a) K. Schwekendiek and F. Glorius, Synthesis, 2006, 2996 CAS; (b) M. Ishihara and H. Togo, Tetrahedron, 2007, 63, 1474 CrossRef CAS.
- (a) S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985 CrossRef CAS; (b) M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS; (c) C. Wang and J. Xiao, Top. Curr. Chem., 2014, 343, 261 CrossRef CAS.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753 CrossRef CAS.
- (a) H. U. Blaser, H. P. Buser, H. P. Jalett, B. Pugin and F. Spindler, Synlett, 1999, 867 CrossRef CAS; (b) V. I. Tararov, R. Kadyrov, T. H. Riermeier and A. Börner, Chem. Commun., 2000, 1867 RSC; (c) R. Kadyrov, T. H. Riermeier, U. Dingerdissen, V. Tararov and A. Borner, J. Org. Chem., 2003, 68, 4067 CrossRef CAS; (d) Y. Chi, Y. G. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120 CrossRef CAS; (e) V. I. Tararov, R. Kadyrov, T. H. Riermeier, C. Fischer and A. Borner, Adv. Synth. Catal., 2004, 346, 561 CrossRef CAS; (f) L. Rubio-Perez;, F. J. Perez-Flores;, P. Sharma;, L. Velasco and A. Cabrera, Org. Lett., 2009, 11, 265 CrossRef; (g) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967 CrossRef CAS; (h) D. Steinhuebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316 CrossRef CAS; (i) B. Villa-Marcos, C. Li, K. R. Mulholland, P. J. Hogan and J. Xiao, Molecules, 2010, 15, 2453 CrossRef CAS; (j) S. Zhou, S. Fleischer, H. Jiao, K. Junge and M. Beller, Adv. Synth. Catal., 2014, 356, 3451 CrossRef CAS; (k) H. Huang, X. Liu, L. Zhou, M. Chang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5309 CrossRef CAS; (l) G. Gao, S. Du, Y. Yang, X. Lei, H. Huang and M. Chang, Molecules, 2018, 23, 2207 CrossRef; (m) L. Hu, Y. Zhang, Q. W. Zhang, Q. Yin and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5321 CrossRef CAS.
- (a) R. Kadyrov and T. H. Riermeier, Angew. Chem., Int. Ed., 2003, 42, 5472 CrossRef CAS; (b) N. A. Strotman, C. A. Baxter, K. M. J. Brands, E. Cleator, S. W. Krska, R. A. Reamer, D. J. Wallace and T. J. Wright, J. Am. Chem. Soc., 2011, 133, 8362 CrossRef CAS; (c) G. D. Williams, R. A. Pike, C. E. Wade and M. Wills, Org. Lett., 2003, 5, 4227 CrossRef CAS.
- (a) M. W. Chen, Y. Ji, J. Wang, Q. A. Chen, L. Shi and Y. G. Zhou, Org. Lett., 2017, 19, 4988 CrossRef CAS; (b) Z. S. Ye, M. W. Chen, Q. A. Chen, L. Shi, Y. Duan and Y. G. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10181 CrossRef CAS.
- (a) M. Chang, Y. Huang, S. Liu, Y. Chen, S. W. Krska, I. W. Davies and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 12761 CrossRef CAS; (b) B. Qu, H. P. R. Mangunuru, X. Wei, K. R. Fandrick, J. N. Desrosiers, J. D. Sieber, D. Kurouski, N. Haddad, L. P. Samankumara, H. Lee, J. Savoie, S. Ma, N. Grinberg, M. Sarvestani, N. K. Yee, J. J. Song and C. H. Senanayake, Org. Lett., 2016, 18, 4920 CrossRef CAS; (c) M. Renom-Carrasco, P. Gajewski, L. Pignataro, J. G. de Vries, U. Piarulli, C. Gennari and L. Lefort, Chem. – Eur. J., 2016, 22, 9528 CrossRef CAS; (d) F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 2850 CrossRef CAS; (e) F. Glorius, Org. Biomol. Chem., 2005, 3, 4171 RSC; (f) C. Y. Legault and A. B. Charette, J. Am. Chem. Soc., 2005, 127, 8966 CrossRef CAS.
- (a) Q. Zhou, L. Zhang, W. Meng, X. Feng, J. Yang and H. Du, Org. Lett., 2016, 18(20), 5189 CrossRef CAS; (b) P. Frediani, L. Rosi, L. Cetarini and M. Frediani, Inorg. Chim. Acta, 2006, 359, 2650 CrossRef CAS; (c) J. Wu, W. Tang, A. Pettman and J. Xiao, Adv. Synth. Catal., 2013, 355, 35 CrossRef CAS.
- M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2007, 46, 4562 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterization data. CCDC 2031296 and 2031297. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob02049d |
| ‡ The enantioselectivity of the other products could not be determined due to the lack of chiral HPLC columns at the time. |
| This journal is © The Royal Society of Chemistry 2021 |