
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and intramolecular ring transformation of N,N′-dialkylated 2,6,9-triazabicyclo[3.3.1]nonadienes†
Yumi
Nakaike
 a,
Yusuke
Yoshida
b,
Soichi
Yokoyama
bcd,
Akitaka
Ito
bc and
Nagatoshi
Nishiwaki
*bc
a,
Yusuke
Yoshida
b,
Soichi
Yokoyama
bcd,
Akitaka
Ito
bc and
Nagatoshi
Nishiwaki
*bc
aDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Yoshidahonmachi, Sakyo-ku, Kyoto 606-8501, Japan
bSchool of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: nishiwaki.nagatoshi@kochi-tech.ac.jp
cResearch Center for Molecular Design, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
dThe Institute of Scientific and Industrial Research, Osaka University, Mihogaoka, Ibaraki, Osaka 567-0047, Japan
First published on 28th October 2020
Abstract
The first and facile synthesis of N,N′-dialkylated 2,6,9-triazabicyclo[3.3.1]nonadienes was achieved by the [4 + 4] self-condensation of β-formyl-β-nitroenamine in the presence of ammonium acetate. The 2,6- and 2,9-dialkylated products were found to be interconvertible when dissolved in a solvent. This isomerization proceeds through intramolecular ring transformation via a common intermediate under equilibrium.
Introduction
β-Formyl-β-nitroenamine 1 possesses multiple functionalities such as a formyl, nitro, and amino group, and a carbon–carbon double bond with biased electron density, which bring about diverse reactivities.1 Two electrophilic sites of 1 react with dinucleophiles such as hydrazines, 1,2-diamines, and ketones to afford nitrated pyrazoles,2 diazepines2 and phenols (Scheme 1, a),3 respectively, in which nitroenamine 1 serves as a synthetic equivalent of nitromalonaldehyde (NMA-H). Its sodium salt (NMA-Na) component has been used for this purpose for a long time. However, NMA-Na should be handled carefully because of the explosive impurity, and aqueous or alcoholic media should be used.4 In contrast, nitroenamine 1 is safely handleable, and its high solubility in common organic solvents allows its reaction with versatile reagents in organic media.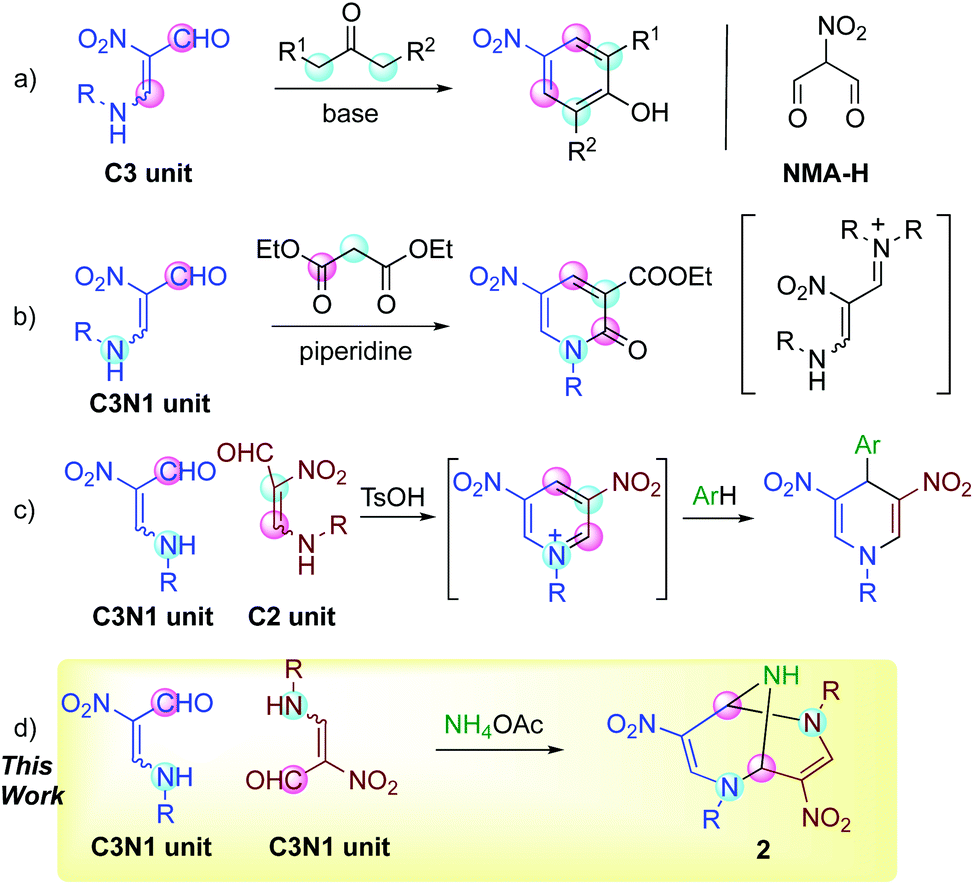 | ||
| Scheme 1 Reactions using nitroenamine 1, in which the pink circles and blue circles indicate electrophilic and nucleophilic sites, respectively. | ||
The dipolar properties of nitroenamine 1 facilitate its reactions with 1,3-dicarbonyl compounds possessing both nucleophilic and electrophilic sites, and polyfunctionalized pyridones are synthesized through this process.5 In these reactions, a secondary amine such as piperidine is found to be a suitable base for condensation, while a tertiary amine such as triethylamine causes no reaction, indicating that the intermediate formation of iminium species is crucial (Scheme 1, b). Furthermore, the [4 + 2] self-condensation of 1 proceeds in the presence of sulfonic acid to form a dinitropyridinium salt in situ, which can be trapped by electron-rich benzenes leading to the formation of 4-arylated 1,4-dihydropyridines (Scheme 1, c).6
These results prompted us to design a new synthetic method for the triazabicyclic framework, including the [4 + 4] self-condensation of nitroenamine 1. Tröger's base7 (3,7-dibenzo-2,6-diazabicyclo[3.3.1]nonane) with a V-shaped structure has attracted much attention because of its chirality,8 biological activity,9 molecular recognition ability,10 and optoelectronic properties.11 However, the bridgehead nitrogen atoms cannot be modified anymore. Hence, researchers’ attention has recently turned towards its aza-analogue, 2,6,9-triazabicyclo[3.3.1]nonane (TABN), because of the availability of further interactions and modifiability of nitrogen atoms in the scaffold.12 However, it is still difficult to introduce a functional group into the bicyclic framework,13 except for two examples.14,15 We focused on one of the examples, Ostercamp's work. The functionalized TABN derivative was first synthesized by the treatment of NMA-Na with methylamine (Scheme 2, a).15 The use of safely handleable nitroenamine 1 was considered to represent a general synthetic method for diverse TABNs because the N-substituent of 1 can be easily modified. In Ostercamp's work, methylamine also serves as a nitrogen source to form a nitrogen bridge, which indicates that the addition of a nitrogen source to our reaction is also necessary. In our study of three-component ring transformation using dinitropyridone, ammonium acetate was found to serve as an excellent nitrogen source and an activator of the substrate (Scheme 2, b),16 which prompted us to study the synthesis of TABNs using nitroenamine 1 and ammonium acetate (Scheme 1, d).
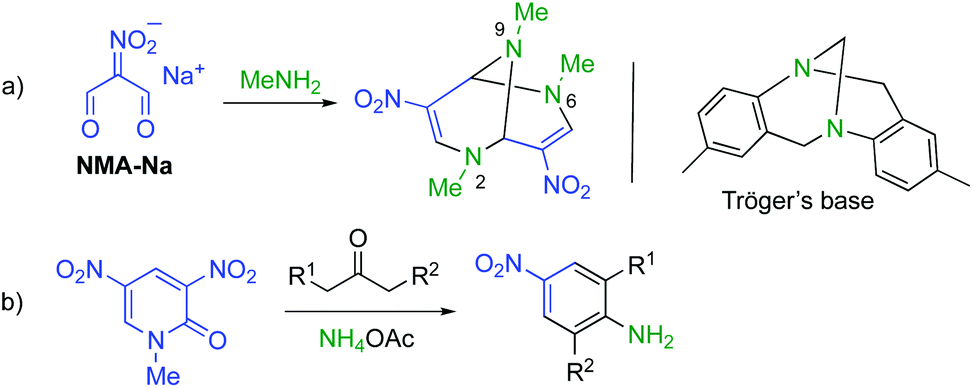 | ||
| Scheme 2 Synthetic equivalents of NMA-H: (a) sodium salt NMA-Na and (b) 1-methyl-3,5-dinitro-2-pyridone. | ||
Results and discussion
When a solution of nitroenamine 1a in ethanol was heated at 80 °C for 1 d in the presence of ammonium acetate, four kinds of TABNs 2a–5a were formed, among which 2a and 3a could not be separated by column chromatography (Table 1, entry 1). In the 1H NMR spectrum of one major product 2a, a singlet signal assigned to two equivalent olefinic protons and an NH signal showing coupling with the adjacent bridgehead protons were observed. Hence, this isomer was determined to be a 2,6-dipropyl derivative. The other major product 3a possesses an unsymmetrical structure; one of the olefinic protons was observed as a singlet, and the other was observed as a doublet coupled with the adjacent NH proton, which was also supported by the 1H–1H COSY 2D NMR spectrum. The recrystallization of the 2a and 3a mixture afforded a single crystal of 2a, which was subjected to X-ray crystallography to confirm the structure (Fig. 1, left). Fortunately, a single crystal of dibenzyl derivative 3d was obtained, and the structure was confirmed to be a 2,9-disubstituted isomer (Fig. 1, right). The use of lower reaction temperature increased the total yield of products 2a–5a, presumably because the competitive decomposition of ammonium acetate was suppressed (entry 2). Although this reaction proceeded even at room temperature, the total yield decreased to an extent (entry 3). The yields of dealkylated products 4a and 5a increased when larger amounts of ammonium acetate were used (entries 4 and 5). | ||
| Fig. 1 ORTEP views of 2a (left, 2,6-di-Pr) and 3d (right, 2,9-di-Bn). Thermal ellipsoids are represented at probably the 50% level. CCDC: 2027731 (2a) and 2027735 (3d).† | ||
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | NH4OAc (equiv.) | Temp. (°C) | Yielda (%) | Recovery of 1a (%) | |||||
| 2 | 3 | 4 | 5 | Total | ||||||
| a Determined by 1H NMR. b Non-alkylated bicyclic compound 6 was obtained in 26% yield. | ||||||||||
| 1 | Pr | a | 1 | 80 | 25 | 12 | 7 | 2 | 46 | 20 |
| 2 | Pr | a | 1 | 50 | 51 | 20 | 19 | 1 | 90 | 10 |
| 3 | Pr | a | 1 | rt | 45 | 12 | 18 | 0 | 75 | 18 |
| 4 | Pr | a | 2 | 50 | 47 | 21 | 23 | 2 | 93 | 4 |
| 5 | Pr | a | 4 | 50 | 39 | 18 | 39 | 1 | 97 | 1 |
| 6 | i-Pr | b | 1 | 50 | 27 | 4 | 20 | 0 | 51 | 29 |
| 7b | t-Bu | c | 1 | 50 | 0 | 0 | 0 | 0 | 0 | 30 |
| 8 | –CH2Ph | d | 1 | 50 | 26 | 53 | 11 | 3 | 93 | 4 |
| 9 | –CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
e | 1 | 50 | 21 | 41 | 15 | 3 | 80 | 6 |
| 10 | –CH2CH2OH | f | 1 | 50 | 37 | 24 | 8 | 12 | 81 | 4 |
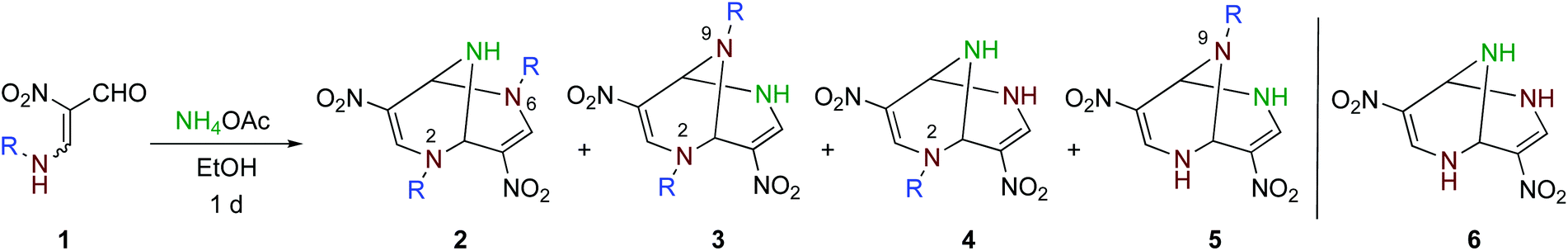
The optimized conditions were applied to other nitroenamines 1b–f. Bicyclic products 2b–5b possessing bulkier isopropyl groups could be prepared by altering nitroenamine to 1b (entry 6). When tert-butyl-substituted nitroenamine 1c was employed, the corresponding bicyclic products 2c–5c were not formed, and non-alkylated bicyclic product 6 was obtained with 26% yield, which was due to the easy elimination of the stable tert-butyl cation under acidic conditions (entry 7). It was also possible to introduce functional groups to the ring nitrogen atoms, such as benzyl, allyl, and 2-hydroxyethyl groups, efficiently (entries 8–10).
All attempts to control the selectivity between 2 and 3 failed. During this study, we noticed that the 2/3 ratio changed each time we performed 1H NMR. This observation suggests that there is a relationship of equilibrium between 2 and 3. To confirm this, the single crystals of 2a and 3d used for X-ray crystallography were dissolved in deuterated solvents, and the change in the mole fraction ([2]/[2] + [3]) was monitored by 1H NMR at intervals of several minutes/hours (Fig. 2). Conversion from 2a to 3a was observed by only dissolving them in the solvents at room temperature, irrespective of the solvent, without the formation of any detectable byproduct. A different behavior was observed when 2a was dissolved in chloroform-d, which might be due to a trace amount of hydrochloric acid in the solvent. Conversely, the formation of 2d was also observed when 3d was dissolved in deuterated solvents. The equilibrium constants (K = [3]/[2]) were temperature-dependent, as shown in Fig. 3. The K value for 3a/2a increased upon increasing the temperature, whereas that for 3d/2d decreased. The Gibbs energy differences between 2 and 3 determined by the Arrhenius-type fittings (ln K = −ΔG/RT) reveal that 3d is more stable than 2d (ΔG < 0) in contrast to the 2a/3a system (Table 2). The difference originated from the existence of π-stacking between the benzyl groups in 3d. In addition, the K values in CD3CN were smaller than the relevant values in CDCl3, presumably due to the polarity and/or Lewis acidity/basicity of the solvent. These tendencies were qualitatively reproduced by theoretical calculations. Furthermore, when a mixture of 2a and 3a was heated in the presence of ammonium acetate, nitroenamine 1a was formed in 10% yield in the reaction mixture, indicating the existence of equilibrium between 1a and TABNs 2a and 3a.
 | ||
| Fig. 2 Time courses of the mole fraction of 2a (closed squares) and 3d (open squares) in C6D6 (black), CDCl3 (green) and CD3CN (red). | ||
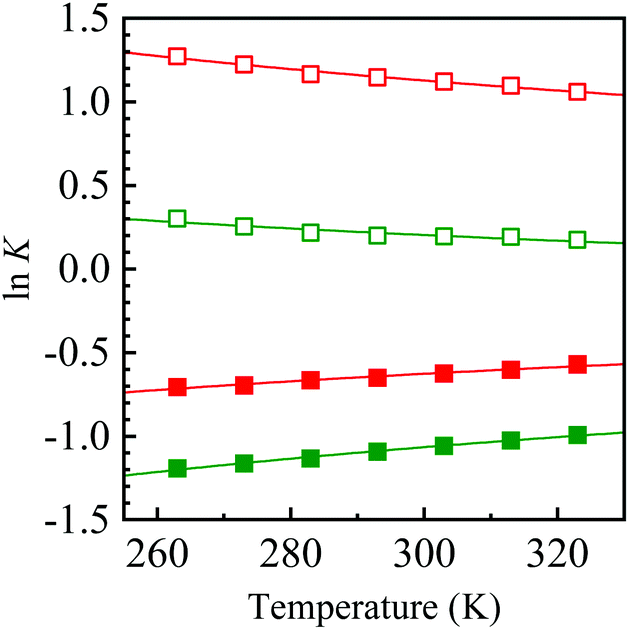 | ||
| Fig. 3 Temperature dependences of equilibrium constants K for 3a/2a (closed squares) and 3d/2d (open squares) in CDCl3 (green) and CD3CN (red). | ||
Based on these results, bicyclic products 2 and 3 are considered to be formed as shown in Scheme 3. The reaction is initiated by the conversion of a formyl group of 1 to iminium ion 7, which facilitates the attack of the less nucleophilic amino group of another molecule of 1 forming 8. After the elimination of ammonia, the formed iminium ion is attacked by the imino group intramolecularly to form a six-membered ring. Dihydropyrimidine 10 is a common intermediate of 2 and 3. When the amino group in the side chain attacks the 4-position (Path A), 2,6-dialkyl product 2 is formed. On the other hand, 2,9-dialkyl product 3 is formed when the amino group attacks the 6-position of 10 (Path B). A direct rearrangement between 2 and 3 is also possible if a stable cation such as a benzyl and allyl cation is released. However, ring transformation via intermediate 10 is considered to be more plausible based on our previous findings; 4,6-dinitro-2,8-diazabicyclo[3.3.1]nona-2-ene and 3-nitropyridine are interconvertible under equilibrium in the presence of ammonium acetate.17 The use of larger amounts of ammonium acetate is subject to the conversion of nitroenamine 1 or iminium ion 7 to nitroenamine 11 (Scheme 4), which affords dealkylated products 4 and 5 in a manner similar to that shown in Scheme 3. Products 4 and 5 are also inseparable because there is a relationship between these compounds.
 | ||
| Scheme 3 A plausible mechanism for the formation of bicyclic compounds 2 and 3. | ||
 | ||
| Scheme 4 Conversion of an alkylamino group to an amino group in the nitroenamine moiety. | ||
Conclusions
TABN frameworks were efficiently constructed by the [4 + 4] self-condensation of nitroenamine 1 in the presence of ammonium acetate. Since the N-alkyl group of 1 is easily modified, versatile TABNs were available by this protocol. The major products, 2,6- and 2,9-dialkylated TABNs 2 and 3, were found to be interconvertible when dissolved in a solvent and this isomerization proceeds through intramolecular ring transformation under equilibrium via the common intermediate 10. Further studies on the physical properties of TABNs and chemical conversion using the functionalities are in progress, and the results will be presented in due course.Experimental section
General
The melting points were determined using an SRS-Optimelt automated melting point system. All the reagents and solvents were commercially available and used as received. The 1H NMR spectra were measured on a Bruker Ascend-400 at 400 MHz with tetramethylsilane as an internal standard. The 13C NMR spectra were measured on a Bruker Ascend-400 at 100 MHz, and the assignments of the 13C NMR spectra were performed by DEPT experiments. The high-resolution mass spectra were measured on an AB SCIEX Triple TOF 4600. The IR spectra were recorded on a JASCO FT/IR-4200 spectrometer.Preparation of nitroenamines 1a–f
Nitroenamines 1a–f were prepared according to our established method.18 Nitropyrimidinone was prepared in 70% overall yield by the condensation of commercially available 1,1,3,3-tetramethoxypropane and N-methylurea in 12 M hydrochloric acid followed by nitration with nitric acid in sulfuric acid. To a solution of pyrimidinone (310 mg, 2 mmol) in methanol (40 mL), propylamine (410 μL, 5 mmol) was added, and the mixture was heated under reflux for 3 h. After evaporation, the residue was extracted with hexane (3 × 30 mL), and the removal of hexane afforded NMR pure azadienamine (390 mg, 1.96 mmol, 98%) as a pale yellow oil. A solution of azadienamine (200 mg, 1 mmol) in chloroform (5 mL) was charged on silica gel (20 g) in a column and allowed to stand at room temperature for 1 day, and then it was eluted with chloroform. The solvent was removed under reduced pressure to give nitroenamine 1a (150 mg, 93%). Other nitroenamines 1b–f were prepared in the same way by using the corresponding amines instead of propylamine (Scheme 5).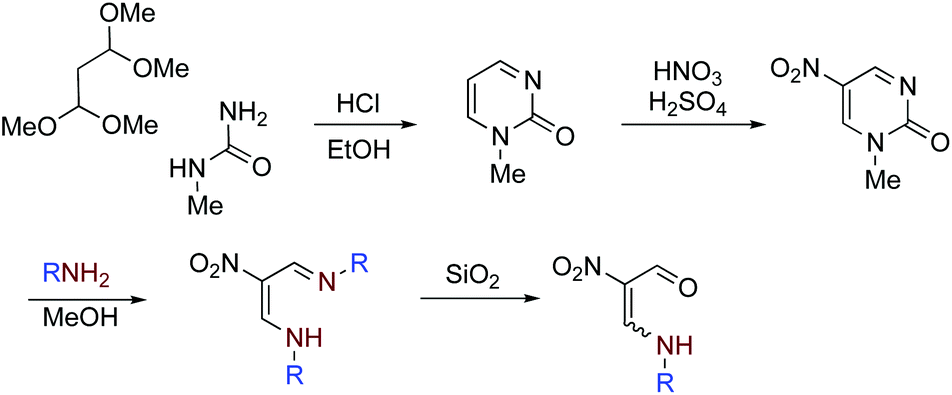 | ||
| Scheme 5 Outline of the preparative method for nitroenamines 1a–f. | ||
4-Aza-2-nitro-2-heptenal (1a)18
Brown solid (E/Z = 76/24). (E)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 3H), 1.74 (tq, J = 7.3, 7.4 Hz, 2H), 3.45 (dt, J = 6.8, 7.3 Hz, 2H), 8.48 (dd, J = 3.6, 14.5 Hz, 1H), 10.14 (d, J = 3.6 Hz, 1H), 10.3–10.8 (br, 1H). (Z)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 3H), 1.76 (tq, J = 7.3, 7.4 Hz, 2H), 3.53 (dt, J = 6.8, 7.3 Hz, 2H), 8.48 (d, J = 15.3 Hz, 1H), 9.5–9.9 (br, 1H), 10.05 (s, 1H).4-Aza-5-methyl-2-nitro-2-hexenal (1b)19
Brown solid (E/Z = 76/24). (E)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.40 (d, J = 6.6 Hz, 6H), 3.75 (sep, J = 6.6 Hz, 1H), 8.52 (ddd, J = 0.6, 3.6, 14.6 Hz, 1H), 10.15 (d, J = 3.6 Hz, 1H), 10.3–10.8 (br, 1H). (Z)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.42 (d, J = 6.6 Hz, 6H), 3.84 (sep, J = 6.6 Hz, 2H), 7.95 (d, J = 15.4 Hz, 1H), 9.4–9.9 (br, 1H), 10.56 (s, 1H).4-Aza-5,5-dimethyl-2-nitro-2-hexenal (1c)18
White solid (E/Z = 79/21). (E)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.45 (s, 9H), 8.57 (dd, J = 3.6, 14.9 Hz, 1H), 10.15 (d, J = 3.6 Hz, 1H), 10.3–10.8 (br, 1H). (Z)-Isomer; 1H NMR (400 MHz, CDCl3) δ 1.47 (s, 9H), 7.99 (d, J = 15.7 Hz, 1H), 9.7–10.1 (br, 1H), 9.94 (s, 1H).4-Aza-2-nitro-5-phenyl-2-pentenal (1d)
White solid; mp 119.2–120.0 °C (E/Z = 77/23). (E)-Isomer; 1H NMR (400 MHz, CDCl3) δ 4.64 (d, J = 5.9 Hz, 2H), 7.2–7.5 (m, 5H), 8.54 (dd, J = 3.6, 14.3 Hz, 1H), 10.16 (d, J = 3.6 Hz, 1H), 10.6–11.0 (br, 1H); 13C NMR (100 MHz, CDCl3) δ 54.5 (CH2), 126.3 (C), 127.8 (CH), 129.1 (CH), 129.5 (CH), 133.9 (CH), 155.0 (C), 186.4 (CH). (Z)-Isomer; 1H NMR (400 MHz, CDCl3) δ 4.71 (d, J = 6.0 Hz, 2H), 7.2–7.5 (m, 5H), 7.98 (d, J = 15.1 Hz, 1H), 9.7–10.0 (br, 1H), 10.07 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 54.7 (CH2), 125.1 (C), 127.9 (CH), 129.2 (CH), 129.5 (CH), 133.9 (CH), 150.3 (C), 183.4 (CH). HRMS (ESI/TOF) calcd for C10H10N2O3(M − H+): 205.0619, found: 205.0624; IR (KBr, cm−1) 3212, 1656, 1606, 1505, 1317.4-Aza-2-nitro-2,6-pentadienal (1e)5
Yellow oil (E/Z = 78/22). (E)-Isomer; 1H NMR (400 MHz, CDCl3) δ 4.09 (br dd, J = 5.7, 5.7 Hz, 2H), 5.3–5.5 (m, 2H), 5.90 (ddt, J = 5.7, 11.2, 17.0 Hz, 1H), 8.48 (dd, J = 3.6, 14.4 Hz, 1H), 10.15 (d, J = 3.6 Hz, 1H), 10.3–10.8 (br, 1H). (Z)-Isomer; 1H NMR (400 MHz, CDCl3) δ 4.16 (br dd, J = 5.8, 5.8 Hz, 2H), 5.3–5.5 (m, 2H), 5.8–6.0 (m, 1H, overlapped with signals of E-isomer), 7.91 (d, J = 15.2 Hz, 1H), 9.5–9.9 (br, 1H), 10.06 (s, 1H).4-Aza-6-hydroxy-2-nitro-2-hexenal (1f)
Brown solid; mp 125.4–126.5 °C (E/Z = 71/29). (E)-Isomer; 1H NMR (400 MHz, CD3CN) δ 3.1–3.4 (br, 1H), 3.5–3.6 (m, 2H), 3.70 (t, J = 5.2 Hz, 2H), 8.58 (br d, J = 8.8 Hz, 1H), 10.10 (d, J = 3.6 Hz, 1H), 10.3–10.7 (br, 1H); 13C NMR (100 MHz, CD3CN) δ 53.3 (CH2), 60.8 (CH2), 126.8 (C), 157.4 (CH), 186.3 (CH). (Z)-Isomer; 1H NMR (400 MHz, CD3CN) δ 3.2–3.4 (br, 1H), 3.62 (t, J = 4.7 Hz, 2H), 3.6–3.8 (m, 2H), 7.96 (d, J = 15.2 Hz, 1H), 9.5–9.9 (br, 1H), 9.98 (s, 1H); 13C NMR (100 MHz, CD3CN) δ 53.4 (CH2), 60.8 (CH2), 125.6 (C), 152.7 (CH), 183.8 (CH). HRMS (ESI/TOF) calcd for C5H8N2O4(M − H+): 159.0411, found: 159.0416; IR (KBr, cm−1) 3494, 3216, 1674, 1589, 1491, 1314.Synthesis of triazabicyclic compounds
To a solution of formylnitroenamine 1a (158.2 mg, 1.0 mmol) in ethanol (4 mL), ammonium acetate (78.5 mg, 1.0 mmol) was added, and the resultant mixture was heated at 50 °C for 1 day. After the removal of the solvent under reduced pressure, the residual brown solid was dissolved in dichloromethane (30 mL) and washed with water (30 mL × 1). The aqueous layer was extracted with dichloromethane (30 mL × 3). The combined organic layer was dried over magnesium sulfate and evaporated to afford a brown solid as a residue. The solid was subjected to column chromatography on silica gel using ethyl acetate/dichloromethane (1/9) as an eluent to afford a mixture of 2a and 3a as a yellow solid (81.0 mg, 0.27 mmol, 55%) and a mixture of 3a and 4a as a brown solid (17.9 mg, 0.07 mmol, 14%). However, further separation could not be achieved. When other nitroenamines 1b–f were used, the experiments were performed in the same way. In the cases of 1b and 1f, ethyl acetate was used as an extraction solvent instead of dichloromethane. In the case of 1f, ethyl acetate was used as an eluent for column chromatography.The NMR spectra were analyzed using a mixture of 2 and 3 because these products are interconvertible under equilibrium when dissolved in solvents. Hence, the content ratio varied in each measurement. With regard to bicyclic products 3 and 4, the NMR spectra were measured using a mixture because they were inseparable despite several attempts. The yields of 3 and 4 were determined on the basis of the integral ratio.
2,6,9-Triaza-4,8-dinitro-2,6-dipropylbicyclo[3.3.1]nona-3,7-diene (2a) and 2,6,9-triaza-4,8-dinitro-2,9-dipropylbicyclo[3.3.1]nona-3,7-diene (3a)
Yellow solid, mp 152.5–153.3 °C, Rf = 0.43 (SiO2, dichloromethane/methanol = 20/1). 2a; 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J = 7.4 Hz, 6H), 1.6–1.9 (m, 4H), 2.2–2.3 (br, 1H), 3.43 (ddd, J = 7.7, 7.7, 13.9 Hz, 2H), 3.98 (ddd, J = 5.8, 7.8, 13.9 Hz, 2H), 5.68 (d, J = 2.8 Hz, 2H), 8.20 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 11.1 (CH3), 23.3 (CH2), 56.9 (CH2), 61.9 (CH), 122.4 (C), 144.6 (CH). 3a; 1H NMR (400 MHz, CDCl3) δ 0.93 (t, J = 7.4 Hz, 3H), 1.00 (t, J = 7.4 Hz, 3H), 1.56 (ddq, J = 7.2, 7.2, 7.4 Hz, 2H), 1.6–1.9 (m, 2H), 2.37 (dt, J = 7.2, 12.6 Hz, 1H), 2.41 (dt, J = 7.4, 12.6 Hz, 1H), 3.39 (ddd, J = 7.8, 7.8, 14.0 Hz, 1H), 3.95 (ddd, J = 5.4, 8.1, 14.0 Hz, 1H), 5.34 (d, J = 1.4 Hz, 1H), 5.32 (s, 1H), 7.0–7.2 (br, 1H), 8.16 (s, 1H), 8.29 (d, J = 4.7, 1H); 13C NMR (100 MHz, CDCl3) δ 11.1 (CH3), 11.6 (CH3), 20.8 (CH2), 23.2 (CH2), 51.4 (CH2), 56.8 (CH2), 61.3 (CH), 66.9 (CH), 122.2 (C), 122.7 (C), 142.2 (CH), 144.4 (CH). HRMS (ESI/TOF) calcd for C12H19N5O4 (M + H+): 298.1510, found: 298.1515; IR (KBr, cm−1) 3291, 2965, 1613, 1304, 1226. A single crystal of 2a was obtained as yellow needles by recrystallization from dichloromethane/hexane.2,6,9-Triaza-4,8-dinitro-2-propylbicyclo[3.3.1]nona-3,7-diene (4a) and 2,6,9-triaza-4,8-dinitro-9-propylbicyclo[3.3.1]nona-3,7-diene (5a)
Yellow solid, mp 91.5–93.6 °C (4a/5a = 89/11). 4a; 1H NMR (400 MHz, CD3CN) δ 0.92 (t, J = 7.4 Hz, 3H), 1.6–1.9 (m, 2H), 2.7–2.9 (br, 1H), 3.45 (ddd, J = 7.7, 7.7, 13.8 Hz, 1H), 3.87 (ddd, J = 5.5, 7.8, 13.8 Hz, 1H), 5.57 (d, J = 1.6 Hz, 1H), 5.68 (d, J = 2.0 Hz, 1H), 7.6–8.1 (br, 1H), 8.20 (s, 1H), 8.22 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 11.1 (CH3), 23.6 (CH2), 55.6 (CH2), 55.7 (CH), 61.5 (CH), 124.0 (C), 124.3 (C), 143.5 (CH), 145.35 (CH). 5a; 1H NMR (400 MHz, CD3CN) δ 0.87 (t, J = 7.4 Hz, 3H), 1.51 (ddq, J = 6.7, 6.8, 7.4 Hz, 2H), 2.34 (dt, J = 6.7, 14.7 Hz, 1H), 2.36 (dt, J = 6.8, 14.7 Hz, 1H), 5.33 (s, 2H), 7.6–8.1 (br, 2H), 8.22 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 11.7 (CH3), 21.2 (CH2), 51.3 (CH2), 62.0 (CH), 124.0 (C), 142.8 (CH). HRMS (ESI/TOF) calcd for C9H13N5O4 (M − H+): 254.0895, found: 254.0890; IR (KBr, cm−1) 3283, 2965, 1611, 1340, 1225, 1139, 926.2,6,9-Triaza-2,6-di(2-methylethyl)-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (2b) and 2,6,9-triaza-2,9-di(2-methylethyl)-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (3b)
Yellow solid, mp 160.6–162.1 °C. Signals of 3b were too small to be assigned. 2b; 1H NMR (400 MHz, CDCl3) δ 1.36 (d, J = 6.6 Hz, 12H), 2.1–2.3 (br, 1H), 4.36 (sep, J = 6.6 Hz, 2H), 5.69 (d, J = 2.7 Hz, 2H), 8.27 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 21.3 (CH3), 24.2 (CH3), 55.5 (CH), 63.4 (CH), 122.7 (C), 140.9 (CH); HRMS (ESI/TOF) calcd for C12H18N5O4 (M − H+): 296.1364, found: 296.1375; IR (KBr, cm−1) 3291, 2973, 1609, 1338, 1259, 1230, 1181.2,6,9-Triaza-2-(2-methylethyl)-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (4b)
Brown solid, mp 126.2–127.9 °C. Signals of 5b were too small to be assigned. 1H NMR (400 MHz, CD3CN) δ 1.30 (d, J = 6.6 Hz, 3H), 1.33 (d, J = 6.6 Hz, 3H), 2.7–2.9 (br, 1H), 4.24 (qq, J = 6.6, 6.6 Hz, 1H), 5.57 (d, J = 1.1 Hz, 1H), 5.70 (d, J = 1.2 Hz, 1H), 7.7–8.0 (br, 1H), 8.21 (s, 1H), 8.26 (s, 1H); 13C NMR (100 MHz, CD3CN) δ 21.2 (CH3), 23.9 (CH3), 56.2 (CH), 57.4 (CH), 64.4 (CH), 124.1 (C), 124.9 (C), 141.4 (CH), 143.4 (CH); HRMS (ESI/TOF) calcd for C9H12N5O4 (M − H+): 254.0895, found: 254.0898; IR (KBr, cm−1) 3283, 1609, 1342, 1232.2,6,9-Triaza-4,8-dinitro[3.3.1]nona-3,7-diene (6)
Yellow solid, mp 207.0–208.1 °C (dec.). 1H NMR (400 MHz, DMSO-d6) δ 3.72 (t, J = 2.7 Hz, 1H), 5.51 (d, J = 2.7 Hz, 2H), 8.24 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 56.2 (CH), 123.1 (C), 142.8 (CH); HRMS (ESI/TOF) calcd for C6H7N5O4 (M − H+): 212.0425, found: 212.0427; IR (KBr, cm−1) 3166, 3012, 1607, 1353, 1223.2,6,9-Triaza-2,6-dibenzyl-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (2d) and 2,6,9-triaza-2,9-dibenzyl-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (3d)
Brown solid, mp 155.8–157.5 °C, Rf = 0.29 (SiO2, hexane/ethyl acetate/triethylamine = 6/4/0.5). 2d; 1H NMR (400 MHz, CDCl3) δ 1.9–2.1 (br, 1H), 4.68 (d, J = 14.8 Hz, 2H), 5.08 (d, J = 14.8 Hz, 2H), 5.65 (d, J = 2.6 Hz, 2H), 6.9–7.5 (m, 10H), 8.26 (s, 2H). 3d; 1H NMR (400 MHz, CDCl3) δ 3.29 (d, J = 13.4 Hz, 1H), 3.35 (d, J = 13.4 Hz, 1H), 4.63 (d, J = 14.7 Hz, 1H), 4.99 (d, J = 14.7 Hz, 1H), 5.24 (d, J = 1.3 Hz, 1H), 5.35 (d, J = 1.3 Hz, 1H), 6.9–7.5 (m, 10H), 8.30 (s, 1H), 8.38 (s, 1H). A signal of N–H was absent, which was presumably due to overlapping. The 13C NMR (100 MHz, CDCl3) measurement was performed using a mixture of two isomers 2d and 3d. δ 53.7 (CH2), 59.1 (CH2), 59.1 (CH2), 60.7 (CH), 61.1 (CH), 65.8 (CH), 121.8 (C), 122.8 (C), 123.0 (C), 128.2 (CH), 128.4 (CH), 128.6 (CH), 128.7 (CH), 128.9 (CH), 129.0 (CH), 129.0 (CH), 129.4 (CH), 129.4 (CH), 134.5 (C), 134.7 (C), 135.0 (C), 142.8 (CH), 144.6 (CH), 144.8 (CH); HRMS (ESI/TOF) calcd for C20H19N5O4(M + H+): 394.1510, found: 394.1525; IR (KBr, cm−1) 3279, 3057, 1614, 1318, 1209. A single crystal of 3d was obtained as yellow blocks by recrystallization from ethanol/diethyl ether.2,6,9-Triaza-2-benzyl-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (4d) and 2,6,9-triaza-9-benzyl-4,8-dinitrobicyclo[3.3.1]nona-3,7-diene (5d)
Yellow solid, mp 155.0–155.8 °C. 4d; 1H NMR (400 MHz, CD3CN) δ 2.6–2.8 (br, 1H), 4.78 (d, J = 15.0 Hz, 1H), 4.99 (d, J = 15.0 Hz, 1H), 5.56 (dd, J = 1.0, 2.7 Hz, 1H), 5.63 (d, J = 2.7 Hz, 1H), 7.1–7.5 (m, 5H), 8.24 (s, 1H), 8.26 (s, 1H). 5d; 1H NMR (400 MHz, CD3CN) δ 3.54 (d, J = 13.3 Hz, 1H), 3.59 (d, J = 13.3 Hz, 1H), 5.25 (s, 2H), 7.1–7.5 (m, 5H), 8.27 (s, 2H). A signal of N–H was absent, which was presumably due to overlapping. The 13C NMR (100 MHz, CD3CN) measurement was performed using a mixture of two isomers 4d and 5d. δ 53.7 (CH2), 57.1 (CH), 58.9 (CH2), 61.5 (CH), 62.5 (CH), 123.8 (C), 124.1 (C), 124.9 (C), 128.8 (CH), 129.2 (CH), 129.3 (CH), 129.6 (CH), 129.9 (CH), 130.0 (CH), 136.9 (C), 137.4 (C), 142.9 (CH), 143.5 (CH), 145.3 (CH); HRMS (ESI/TOF) calcd for C13H13N5O4 (M − H+): 302.0895, found: 302.0895; IR (KBr, cm−1) 3276, 1705, 1610, 1320, 1204, 928.2,6,9-Triaza-4,8-dinitro-2,6-di(3-propen-1-yl)bicyclo[3.3.1]nona-3,7-diene (2e) and 2,6,9-triaza-4,8-dinitro-2,9-di(3-propen-1-yl)bicyclo[3.3.1]nona-3,7-diene (3e)
Brown solid, mp 60.5–62.0 °C. 2e; 1H NMR (400 MHz, CDCl3) δ 2.20 (br, 1H), 4.17 (dddd, J = 1.5, 1.5, 5.0, 15.4 Hz, 2H), 3.60 (br dd, J = 7.1, 15.4 Hz, 2H), 5.82 (dddd, J = 1.2, 1.2, 1.2, 17.2 Hz, 2H), 5.3–5.5 (m, 2H), 5.69 (d, J = 2.8 Hz, 2H), 5.87 (dddd, J = 5.0, 7.1, 10.2, 17.2 Hz, 2H), 8.18 (s, 2H). 3e; 1H NMR (400 MHz, CDCl3) δ 3.01 (dddd, J = 1.2, 1.2, 6.4, 13.5 Hz, 1H), 3.08 (dddd, J = 1.2, 1.2, 6.3, 13.5 Hz, 1H), 4.13 (dddd, J = 1.3, 1.3, 5.1, 15.3 Hz, 1H), 4.44 (br dd, J = 7.2, 15.3 Hz, 1H), 5.26 (dddd, J = 1.2, 1.2, 1.2, 17.1 Hz, 1H), 5.29 (dddd, J = 1.3, 1.3, 1.3, 10.2 Hz, 1H), 5.3–5.5 (m, 2H), 5.47 (dddd, J = 1.3, 1.3, 1.3, 17.0 Hz, 1H), 5.34 (d, J = 1.5 Hz, 1H), 5.38 (d, J = 1.5 Hz, 1H), 5.82 (dddd, J = 6.3, 6.4, 10.2, 17.1 Hz, 1H), 5.86 (dddd, J = 5.1, 7.2, 10.2, 17.0 Hz, 1H), 6.2–6.8 (br, 1H), 8.18 (s, 1H), 8.30 (s, 1H). The 13C NMR (100 MHz, CDCl3) measurement was performed using a mixture of two isomers 2e and 3e. δ 52.6 (CH2), 57.5 (CH2), 57.5 (CH2), 60.7 (CH), 61.7 (CH), 66.2 (CH), 120.6 (CH2), 121.1 (CH2), 121.4 (CH2), 121.8 (C), 122.8 (C), 122.9 (C), 131.5 (CH), 131.7 (CH), 132.4 (CH), 142.8 (CH), 144.3 (CH), 144.6 (CH); HRMS (ESI/TOF) calcd for C12H15N5O4 (M − H+): 292.1051, found: 292.1054; IR (KBr, cm−1) 3283, 1613, 1482, 1416, 1309, 920, 760.2,6,9-Triaza-4,8-dinitro-2-(3-propen-1-yl)bicyclo[3.3.1]nona-3,7-diene (4e) and 2,6,9-triaza-4,8-dinitro-9-(3-propen-1-yl)bicyclo[3.3.1]nona-3,7-diene (5e)
Yellow solid, mp 135.4–137.5 °C. 4e; 1H NMR (400 MHz, CD3CN) δ 2.7–2.9 (br, 1H), 4.19 (dddd, J = 1.4, 1.4, 5.0, 15.6 Hz, 1H), 4.43 (dddd, J = 1.4, 1.4, 6.8, 15.6 Hz, 1H), 5.30 (dddd, J = 1.4, 1.4, 1.4, 10.2 Hz, 1H), 5.39 (dddd, J = 1.4, 1.4, 1.4, 17.1 Hz, 1H), 5.57 (dd, J = 1.1, 2.7 Hz, 1H), 5.65 (br d, J = 2.8 Hz, 1H), 5.94 (dddd, J = 5.0, 6.9, 10.2, 17.1 Hz, 1H), 7.5–8.1 (br, 1H), 8.18 (s, 1H), 8.22 (s, 1H). 5e; 1H NMR (400 MHz, CD3CN) δ 3.00 (dddd, J = 1.3, 1.3, 6.4, 13.7 Hz, 1H), 3.05 (dddd, J = 1.3, 1.3, 6.4, 13.7 Hz, 1H), 5.30 (dddd, J = 1.3, 1.3, 1.3, 10.2 Hz, 1H), 5.39 (dddd, J = 1.3, 1.3, 1.3, 17.1 Hz, 1H), 5.33 (s, 2H), 5.85 (dddd, J = 6.4, 6.4, 10.2, 17.1 Hz, 1H), 7.5–8.1 (br, 2H), 8.23 (s, 2H). The 13C NMR (100 MHz, CD3CN) measurement was performed using a mixture of two isomers 4e and 5e. δ 52.6 (CH2), 57.1 (CH), 57.7 (CH2), 61.5 (CH), 62.8 (CH), 119.9 (CH2), 119.9 (CH2), 123.8 (C), 124.1 (C), 124.9 (C), 134.0 (CH), 134.4 (CH), 142.8 (CH), 143.4 (CH), 145.1 (CH); HRMS (ESI/TOF) calcd for C9H11N5O4 (M − H+): 252.0738, found: 252.0740; IR (KBr, cm−1) 3278, 1610, 1423, 1334, 1215, 927.2,6,9-Triaza-2,6-di(2-hydroxyethyl)-4,8-dinitro[3.3.1]nona-3,7-diene (2f) and 2,6,9-triaza-2,9-di(2-hydroxyethyl)-4,8-dinitro[3.3.1]nona-3,7-diene (3f)
Brown solid, mp 68.1–70.7 °C. 2f; 1H NMR (400 MHz, CD3CN) δ 2.8–3.0 (br, 1H), 3.0–3.3 (br, 2H), 3.44 (ddd, J = 3.8, 7.0, 14.2 Hz, 2H), 3.69 (ddd, J = 3.8, 6.0, 11.8 Hz, 2H), 3.77 (ddd, J = 3.4, 7.0, 11.8 Hz, 2H), 4.01 (ddd, J = 3.4, 6.0, 14.2 Hz, 2H), 5.71 (d, J = 1.0 Hz, 2H), 8.24 (s, 2H); 13C NMR (100 MHz, CD3CN) δ57.4 (CH2), 61.5 (CH2), 63.3 (CH), 123.5 (C), 144.6 (CH). 3f; 1H NMR (400 MHz, CD3CN) δ 2.49 (ddd, J = 5.2, 5.2, 13.7 Hz, 1H), 2.64 (ddd, J = 5.0, 5.0, 13.7 Hz, 1H), 2.9–3.1 (br, 1H), 3.0–3.3 (br, 1H), 3.5–3.8 (m, 5H, overlapped with signals of 3f), 3.97 (ddd, J = 3.3, 6.0, 14.7 Hz, 1H), 5.42 (d, J = 1.5 Hz, 1H), 5.61 (d, J = 1.5 Hz, 1H), 7.5–8.2 (br, 1H), 8.26 (s, 1H), 8.27 (s, 1H); 13C NMR (100 MHz, CD3CN) δ 51.9 (CH2), 57.3 (CH2), 60.7 (CH2), 61.4 (CH2), 62.1 (CH), 68.4 (CH), 122.9 (C), 123.1 (C), 143.4 (CH), 145.9 (CH). HRMS (ESI/TOF) calcd for C10H15N5O6 (M − H+): 300.0950, found: 300.0954; IR (KBr, cm−1)1209, 1307, 1612, 2359, 3307.2,6,9-Triaza-2-(2-hydroxyethyl)-4,8-dinitro[3.3.1]nona-3,7-diene (4f) and 2,6,9-triaza-9-(2-hydroxyethyl)-4,8-dinitro[3.3.1]nona-3,7-diene (5f)
Brown solid, mp 95.6–97.2 °C. 4f; 1H NMR (400 MHz, CD3CN) δ 2.8–3.0 (br, 1H), 2.9–3.4 (br, 1H), 3.57 (ddd, J = 3.8, 6.9, 14.3 Hz, 1H), 3.67 (ddd, J = 3.8, 6.2, 11.6 Hz, 1H), 3.77 (ddd, J = 3.5, 6.9, 11.6 Hz, 1H), 3.99 (ddd, J = 3.5, 6.2, 14.3 Hz, 1H), 5.57 (s, 1H), 5.73 (s, 1H), 7.4–8.2 (br, 1H), 8.21 (s, 1H), 8.23 (s, 1H); 13C NMR (100 MHz, CD3CN) δ 57.0 (CH2), 57.2 (CH), 61.5 (CH2), 63.8 (CH), 124.0 (C), 124.7 (C), 143.5 (CH), 145.6 (CH). 5f; 1H NMR (400 MHz, CD3CN) δ 2.49 (ddd, J = 5.5, 5.5, 13.4 Hz, 1H), 2.55 (ddd, J = 5.1, 5.1, 13.4 Hz, 1H), 2.9–3.4 (br, 1H), 3.5–3.7 (m, 2H, overlapped with signals of 4f), 5.44 (s, 2H), 7.4–8.2 (br, 2H), 8.23 (s, 2H); 13C NMR (100 MHz, CD3CN) δ 51.9 (CH2), 60.7 (CH2), 62.5 (CH), 124.0 (C), 142.7 (CH). HRMS (ESI/TOF) calcd for C8H11N5O5 (M − H+): 256.0687, found: 256.0685; IR (KBr, cm−1) 1208, 1226, 1317, 1609, 2359, 3251.X-Ray crystallographic analysis
Diffraction data were collected at 93 or 123 K under a cold N2-gas stream using a Rigaku XtaLAB Synergy-S/Mo system (λ = 0.71073 Å (Mo-Kα)). The integrated data were analyzed using the Olex2 crystallographic software package.20 The structures were solved with the ShelXT structure solution program21 using Intrinsic Phasing and refined with the ShelXL refinement package22 using least-squares minimization. Anisotropic refinement was performed for all non-hydrogen atoms, and all the hydrogen atoms were placed at the calculated positions.Temperature dependent NMR spectra
1H NMR spectra were obtained by using the variable-temperature (VT) NMR technique. The sample solutions were stored at −10–50 °C for over 12 h prior to the measurements.Theoretical calculations
Density functional theory (DFT) calculations for 2a, 2d, 3a and 3d were conducted using Gaussian 16 W software (revision A.03)23 using the restricted Becke three-parameter hybrid functionals with the Lee–Yang–Parr correlation (RB3LYP)24,25 and 6-311+G(d,p) basis set.26,27 Ground-state geometries were optimized and, then, the Gibbs energies of the molecules were calculated. Solvation was considered using the conductor-like polarizable continuum model.28,29Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Nakaike, H. Asahara and N. Nishiwaki, Russ. Chem. Bull. Int. Ed., 2016, 65, 2129 CrossRef CAS.
- N. Nishiwaki, T. Ogihara, T. Takami, M. Tamura and M. Ariga, J. Org. Chem., 2004, 69, 8382 CrossRef CAS.
- Y. Nakaike, Y. Kamijo, S. Mori, M. Tamura, N. Nishiwaki and M. Ariga, J. Org. Chem., 2005, 70, 10169 CrossRef CAS.
- P. E. Fanta and R. A. Stein, Chem. Rev., 1960, 60, 261 CrossRef CAS.
- Y. Nakaike, D. Hayashi, N. Nishiwaki, Y. Tobe and M. Ariga, Org. Biomol. Chem., 2009, 7, 325 RSC.
- H. Asahara, M. Hamada, Y. Nakaike and N. Nishiwaki, RSC Adv., 2015, 5, 90778 RSC; Y. Nakaike, N. Nishiwaki, M. Ariga and Y. Tobe, J. Org. Chem., 2014, 79, 2163 CrossRef CAS.
- J. Tröger, J. Prakt. Chem., 1887, 36, 225 CrossRef.
- V. Prelog and P. Wieland, Helv. Chim. Acta, 1944, 27, 1127 CrossRef CAS.
- N. Classens, F. Perard, C. Bresson, C. Moucheron and A. K. D. Mesmaeker, J. Inorg. Biochem., 2007, 101, 987 CrossRef; M. Valík, J. Malina, L. Palivec, J. Foltýnová, M. Tkadlecová, M. Urbanová, V. Brabec and V. Král, Tetrahedron, 2006, 62, 8591 CrossRef.
- S. Satishkuma and M. Periasamy, Tetrahedron: Asymmetry, 2009, 20, 2257 CrossRef CAS; Y. Kubo, T. Ohno, J. Yamanaka, S. Takita, T. Iida and Y. Ishimaru, J. Am. Chem. Soc., 2001, 123, 12700 CrossRef; M. J. Crossley, L. G. Mackay and A. C. Try, J. Chem. Soc., Chem. Commun., 1995, 1925 RSC.
- R. Yuan, M. Li, J. Xu, S. Huang, S. Zhou, P. Zhang, J. Liu and H. Wu, Tetrahedron, 2016, 72, 4081 CrossRef CAS.
- A. Leganza, C. Bezze, C. Zonta, F. Fabris, O. De Lucci and A. Linden, Eur. J. Org. Chem., 2006, 2987 CrossRef CAS; L. Y. Ukhin and L. G. Kuzmina, Russ. Chem. Bull., 2004, 53, 2262 CrossRef; P. Molina, A. Arques, A. Tarraga, M. Obon, N. Jagerovic and J. Elguero, Tetrahedron, 1998, 54, 997 CrossRef.
- I. Muthukrishnan, M. Karuppasamy, S. Nagarajan, C. U. Maheswari, V. Pace, J. C. Menéndez and V. Sridharan, J. Org. Chem., 2016, 81, 9687 CrossRef CAS; C. Su, R. Tandiana, J. Balapanuru, W. Tang, K. Pareek, C. T. Nai, T. Hayashi and K. P. Loh, J. Am. Chem. Soc., 2015, 137, 685 CrossRef; A. Tsutsui, R. Imamaki, S. Kitazume, S. Hanashima, Y. Yamaguchi, M. Kaneda, S. Oishi, N. Fujii, A. Kurbangalieva, N. Taniguchi and K. Tanaka, Org. Biomol. Chem., 2014, 12, 5151 RSC; B. Pettersson, J. Bergman and P. H. Svensson, Tetrahedron, 2013, 69, 2647 CrossRef; D. Mao, J. Tang, W. Wang, S. Wu, X. Liu, J. Yu and L. Wang, J. Org. Chem., 2013, 78, 12848 CrossRef.
- R. Paredes, R. Abonia, J. Cadavid, R. Moreno-Fuquen, A. Jaramilo, A. Hormaza, A. Ramirez and A. Kennedy, Tetrahedron, 2002, 58, 55 CrossRef CAS.
- D. L. Ostercamp and S. Wiles, J. Heterocycl. Chem., 2000, 37, 1357 CrossRef CAS.
- S. T. Le, H. Asahara and N. Nishiwaki, Eur. J. Org. Chem., 2015, 1203 CrossRef CAS; S. T. Le, H. Asahara and N. Nishiwaki, J. Org. Chem., 2015, 80, 8856 CrossRef.
- S. T. Le, H. Asahara, K. Kobiro, R. Sugimoto, K. Saigo and N. Nishiwaki, Asian J. Org. Chem., 2014, 3, 297 CrossRef CAS.
- N. Nishiwaki, Y. Tohda and M. Ariga, Bull. Chem. Soc. Jpn., 1996, 69, 1997 CrossRef CAS.
- S. M. Kvito, Y. V. Maksimov, T. Y. Paperno and V. V. Perekalin, Zh. Org. Khim., 1973, 9, 471 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, GAUSSIAN 16 W (Revision A.03), Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data and copies of NMR spectra of compounds 2–6, crystallographic data of 2a and 3d, and temperature-dependent NMR spectra. CCDC 2027731 and 2027735. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01950j |
| This journal is © The Royal Society of Chemistry 2020 |