
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transient imine directing groups for the C–H functionalisation of aldehydes, ketones and amines: an update 2018–2020
Joe I.
Higham
 and
James A.
Bull
*
and
James A.
Bull
*
Department of Chemistry, Imperial College London, South Kensington, London SW7 2AZ, UK. E-mail: j.bull@imperial.ac.uk
First published on 8th September 2020
Abstract
The use of pre-installed directing groups has become a popular and powerful strategy to control site selectivity in transition metal catalysed C–H functionalisation reactions. However, the necessity for directing group installation and removal reduces the efficiency of a directed C–H functionalisation method. To overcome this limitation, taking inspiration from organocatalytic methodologies, the use of transient directing groups has arisen. These methods allow for a transient ligand to be used, potentially in catalytic quantities, without the need for discrete installation or removal steps, enabling the discovery of more efficient, and mechanistically intriguing, dual catalytic methods. This review summarises recent developments in this fast moving field covering >70 new methodologies, highlighting new directing group designs and advances in mechanistic understanding. It covers progress since 2018, providing an update to our previous review of the field.
1. Introduction
C–H functionalisation has caused a paradigm shift in thinking about synthetic disconnections, enabling streamlined syntheses of complex organic molecules in fewer steps and improved efficiency.1 A robust and reliable approach to achieving regiocontrol in C–H activation reactions is the use of a pre-installed directing group (DG) to bind and position a metal centre. Important examples include bidentate 8-aminoquinoline introduced by Daugulis,2 and Yu's more weakly coordinating acidic amides.3 However, an inherent limitation of this strategy is the need to install and remove the directing group in discrete steps, adding at least two additional steps to a synthetic sequence (Scheme 1a). Directing group removal can also be challenging, or impossible, limiting the tolerance/utility of this approach for intricate substrates and functionalisation at a late stage. Recently, innovative approaches have been developed to correctly position catalysts and avoid the requirement for these additional steps, such as the use of native directing groups4 and non-covalent interactions.5 This review is concerned with the rapidly expanding area of C–H functionalisation using transient directing groups, exploiting existing common aldehyde and amine functionality in the substrate to form a directing imine.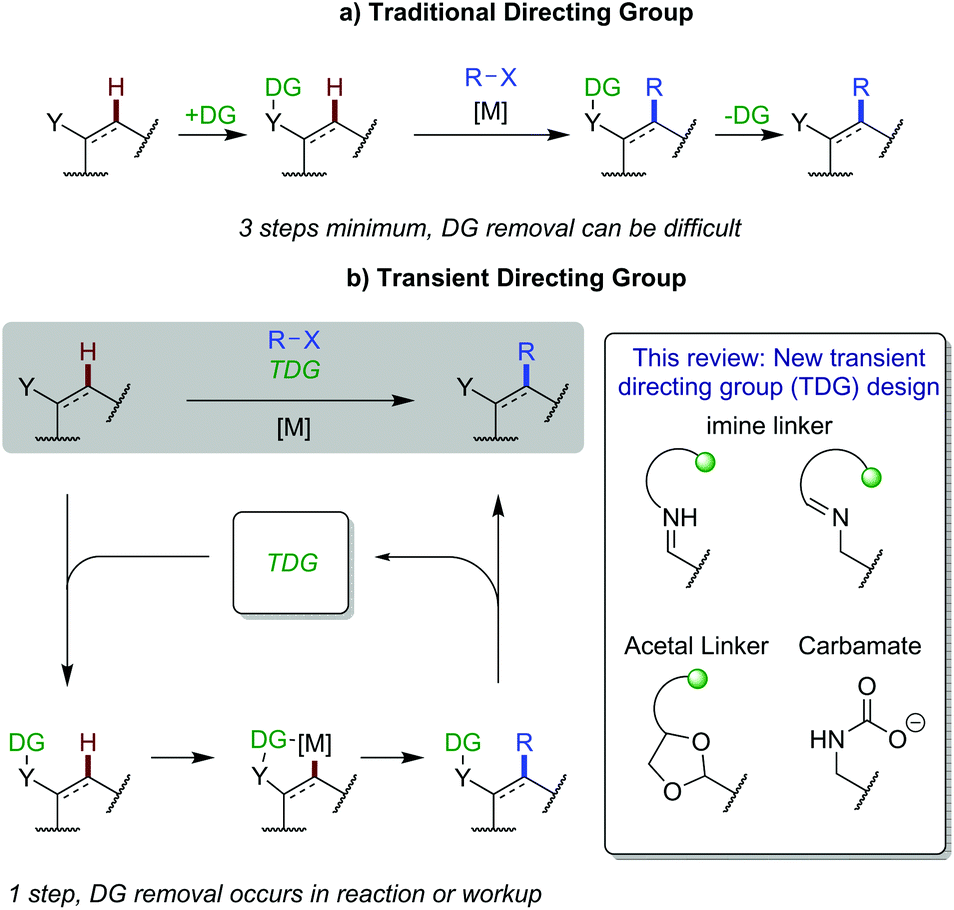 | ||
| Scheme 1 Schematic approaches to C–H activation by traditional directing group installation and using a transient directing group. | ||
Transient C–H functionalisation involves the formation of a labile linkage to attach a directing group, often with the linkage being part of an in situ generated directing group. This can coordinate to a metal catalyst to promote C–H activation and new bond formation, followed by cleavage, all in the same reaction pot (Scheme 1b). This idea was first demonstrated by Jun in 1997,6 to achieve alkylation of aldehydic C–H bonds. Later, in 2016, Yu showed the first example of this strategy applied to C(sp3)–H functionalisation of aldehydes and ketones involving a transient imine directing group.7
The interest in this field has prompted several reviews,8 including one published by us (Bull).8e However, there have been a large number of very recent reports of transient C–H activation including extensive advances in mechanistic understanding, indeed the field has doubled since our previous review. This review is an update for the period from April 2018–July 2020 covering over 70 new examples. References discussed in our previous review will not be duplicated. There have been key advancements in the development of new C–X bond forming methodologies, and enantioselective reactions. This has been achieved by the continuing development of new transient directing groups, bearing new secondary binding sites.
This review is structured to cover reactions which achieve C–H functionalisation of aldehydes, ketones and then amines through a transient directing group approach. The majority involve imine DGs, using the complementary amine or aldehyde. Additionally, other acid labile directing groups (i.e. acetals or carbamates), which can be installed and removed in a stoichiometric telescoped one pot manner, will be discussed. All directing groups used to date in transient C–H functionalisation are summarised in Fig. 1 and 2, with previously unreported directing groups highlighted.
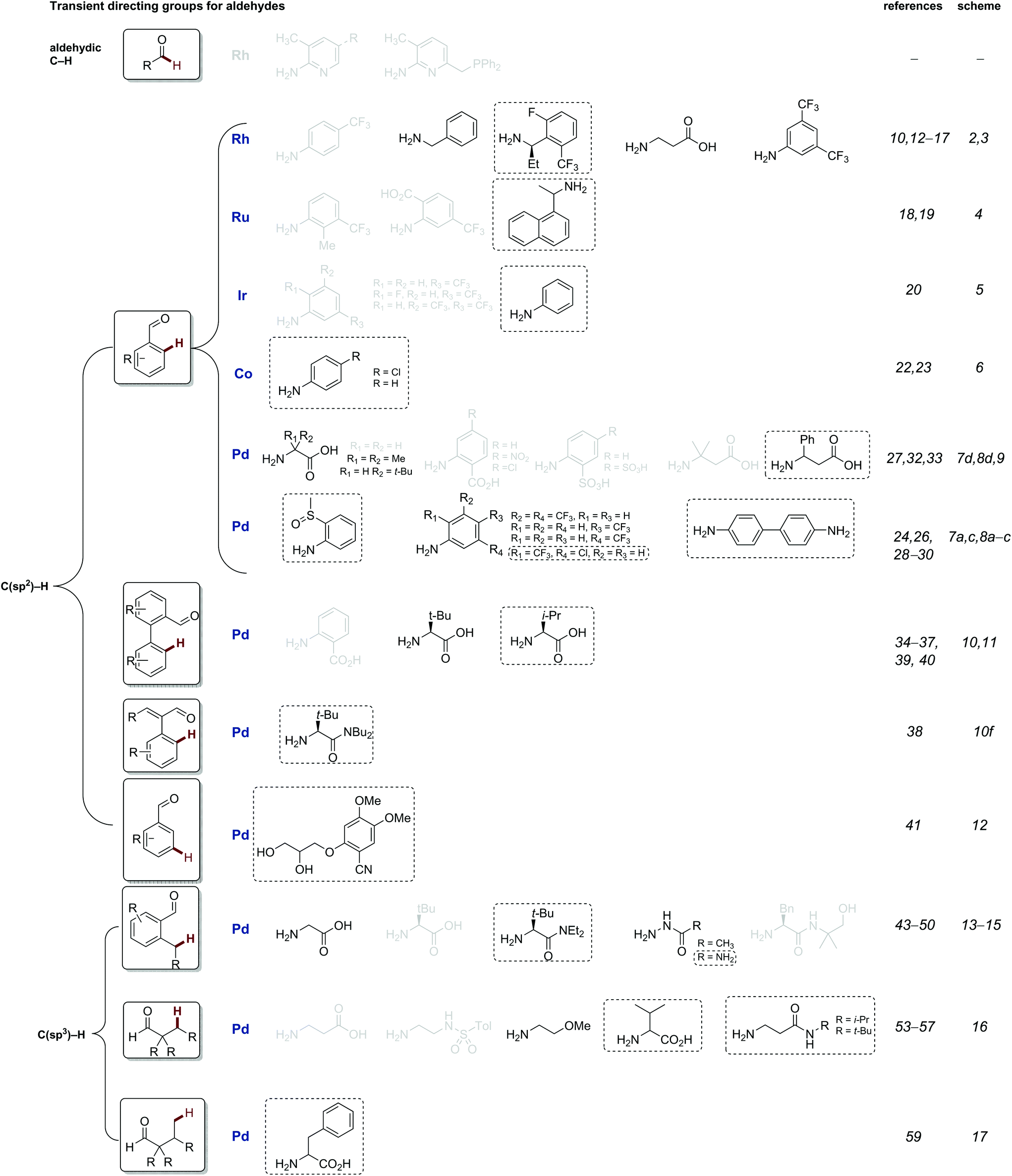 | ||
| Fig. 1 Summary of all transient imine directing groups for the functionalisation of aldehyde C–H bonds. Greyed TDGs are not discussed in this review, see previous review.8c Directing groups in black will be discussed in this review, and novel directing groups introduced in the last 2 years are boxed. | ||
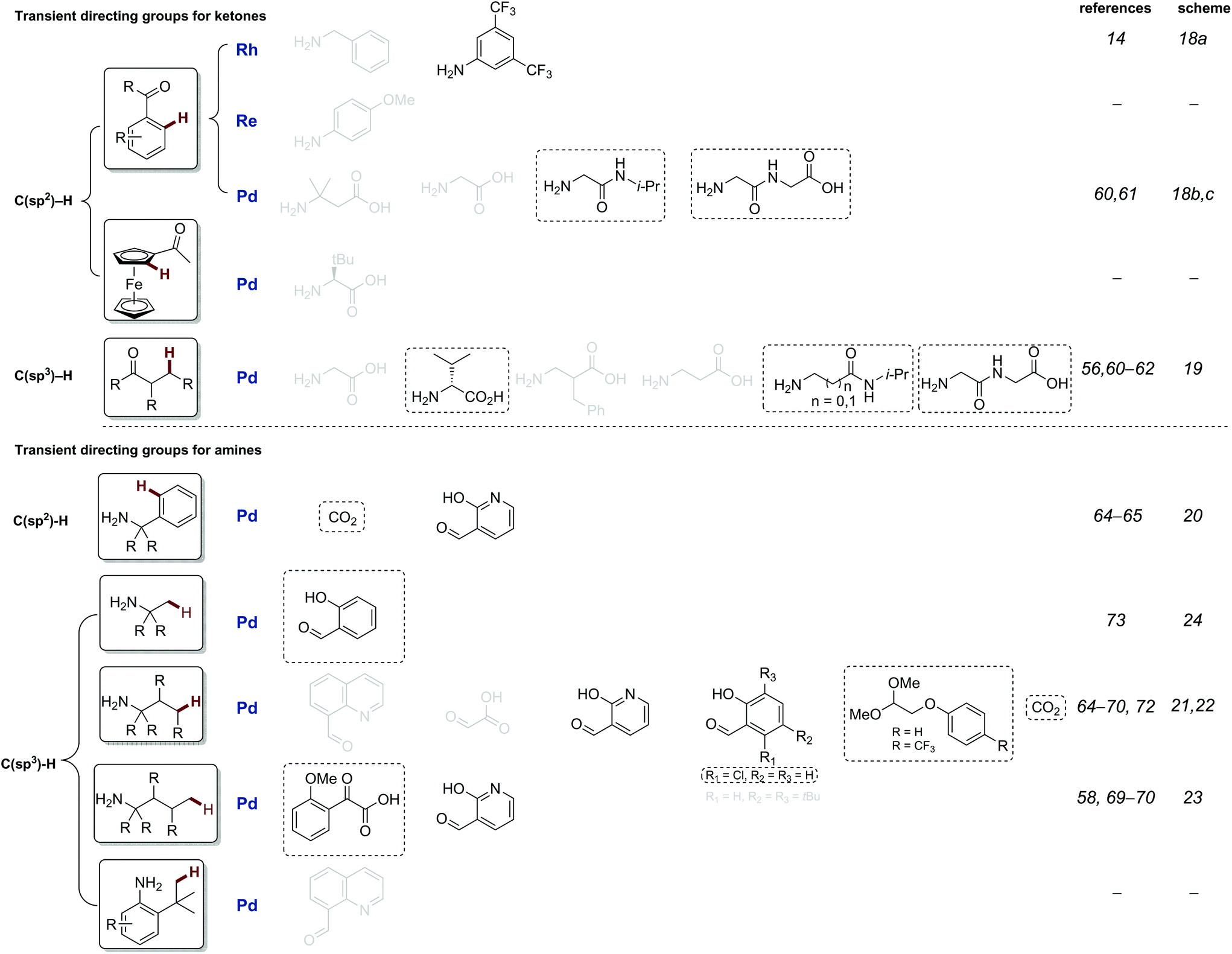 | ||
| Fig. 2 Summary of all transient imine directing groups for the functionalisation of ketone and amine C–H bonds. Greyed TDGs are not discussed in this review, see previous review.8c Directing groups in black will be discussed in this review, and novel directing introduced in the last 2 years groups are boxed. | ||
Additionally, advances in directing group free approaches achieving amine functionalisation will also be discussed, as well as the broader use of a transient directing group for alkene functionalisation. The inclusion of these aspects hints at the potential for a broader expansion of this approach. This work has been ordered chronologically within each section, separated by hybridisation of C–H bond, metal catalyst and regioselectivity.
2. C–H functionalisation of aldehydes
Aldehydes are exceptionally useful synthetic building blocks owing to the range of functional groups to which they can be converted. Therefore, there has been a concerted effort for the development of new methodologies which can functionalise aldehydic systems. In addition, the facile and reversible formation of aldimines greatly facilitates a transient directing group approach with aldehyde substrates. Notable recent advances include novel directing group development, new enantioselective methods and the development of milder more sustainable methods.This section deals with the C–H functionalisation of aldehyde substrates and is divided into the following sections: C(sp2)–H functionalisation of benzaldehydes, benzylic C(sp3)–H functionalisation of benzaldehydes, and C(sp3)–H functionalisation of aldehydes. In addition to ortho functionalisation, distal meta-functionalisation will also be discussed.
C(sp2)–H functionalisation of benzaldehydes
C(sp2)–H functionalisation with a transient directing group has developed greatly in the past couple of years. This class of substrates has seen the greatest exploration into the use of new alternative metal catalysts, experimentation with the construction of C–heteroatom bonds and development of milder and more environmentally benign conditions. Additionally, ortho-arylation has been successfully used as a key step in the total synthesis of Boletopsin 11.9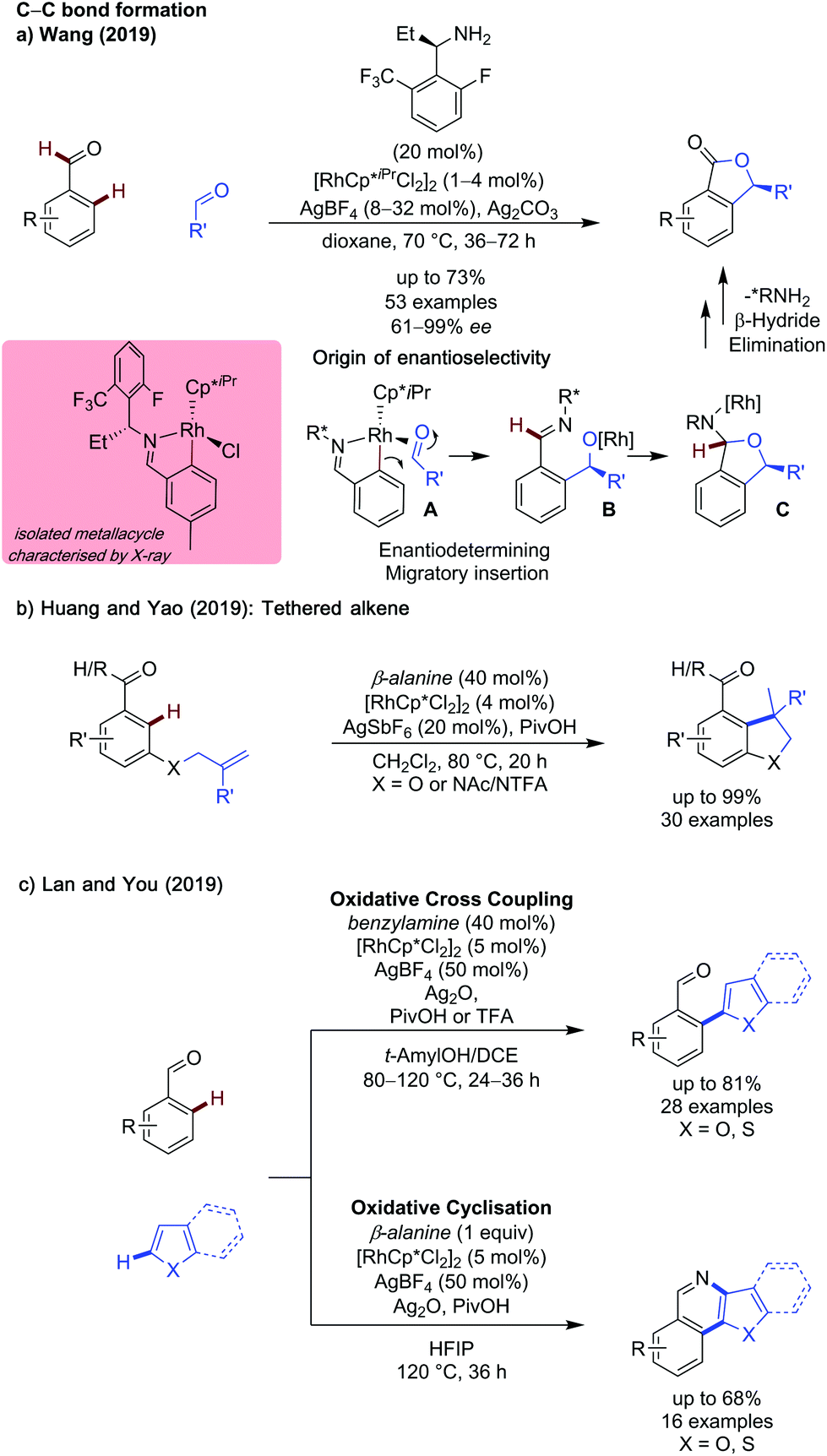 | ||
| Scheme 2 Rhodium catalysed C–C bond forming reactions. | ||
While monodentate TDGs are most common with rhodium catalysis, Huang and Yao showed β-alanine as an effective directing group for the rhodium catalysed intramolecular hydroarylation of benzaldehydes (Scheme 2b).12 A range of electronically disparate aldehydes (and ketones) reacted smoothly, including a heteroaromatic pyridine example. Substituents on the tethered alkene were well tolerated, with electron-poor and sterically hindered examples functioning well. The heteroatom tether could be changed from O to NAc or NTFA to form indolines, albeit in lower yield.
A rhodium metallacycle was proposed to form after imine formation and a C–H activation event. The substrate-tethered alkene binds to the rhodium centre to undergo carbo-metalation then protodemetalation to give the product. In this system, a KIE of 1.1 was observed indicating the C–H activation step was not rate determining, and deuterium incorporation at the ortho position of the starting material implied this step was also reversible. Further studies at varying temperatures showed deuteration of the ortho C–H positions of the starting material could occur at temperatures as low as 40 °C, though hydroarylation was only observed at elevated temperatures.
Lan and You reported rhodium catalysed methods for oxidative cross dehydrogenative coupling (CDC) of benzaldehydes and (benzo)thiophenes and benzofurans, in which subtle changes in reaction conditions could allow access to the functionalised aldehyde or a cyclised pyridine product (Scheme 2c).13 Monodentate benzylamine was shown to promote formation of the ortho-functionalised product. The use of t-amylOH with DCE as a co-solvent was also key to ensure the free aldehyde product was formed selectively. This CDC coupling was shown to be compatible with a variety of ortho- and meta-substituted aldehydes in up to 81% when m-tolualdehyde was employed. However, para-substituted and systems unbiased to mono-heteroarylation were not explored.
Additionally, a limited scope of thiophenes and benzofurans were demonstrated.
This report also showed that by changing the directing group to β-alanine and the solvent to HFIP, it was possible to achieve a one pot heteroarylation, cyclisation reaction to give pyridine product. A modest scope of pyridines were reported, in yields up to 68% when using p-phenyl benzaldehyde and benzothiophene as coupling partners. Control experiments indicated that the mechanism of cyclisation was independent to the presence of rhodium, however the presence of Ag(I) was required for cyclisation to occur, and TEMPO shuts down reactivity, thus a silver mediated radical cyclisation mechanism was proposed.
In addition to the advances in C–C bond formation, rhodium catalysis has been shown effective for C–N bond formation. Prabhu developed an improved method for C–H amidation of aldehydes using dioxazolones as coupling partners under ambient conditions (Scheme 3a).14 The reaction functions in the same manner to Jiao's earlier work,15 however the exceptionally mild conditions allowed for a broader substrate scope, including the use of dioxazolones bearing alkyl substituents.
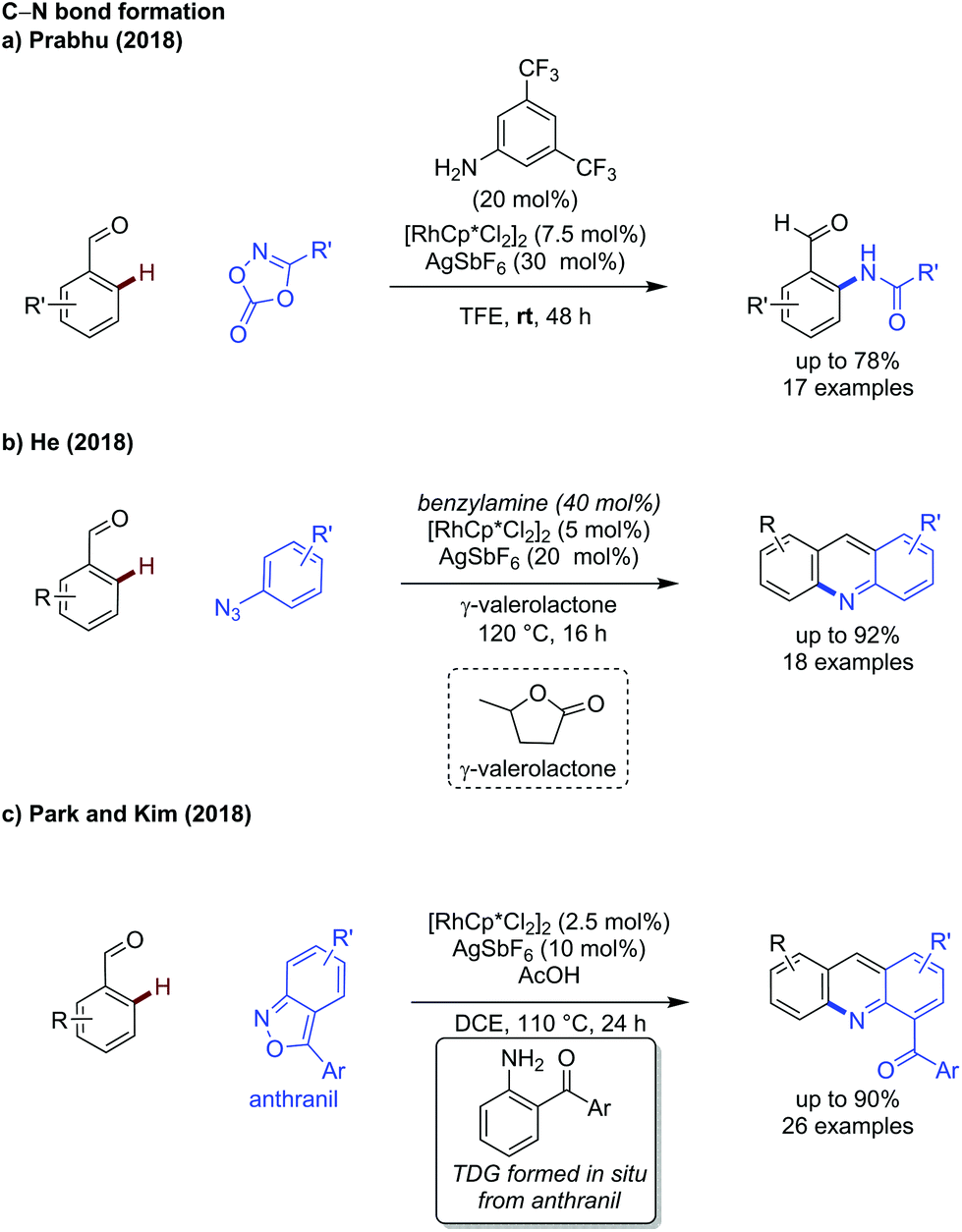 | ||
| Scheme 3 Rhodium catalysed transient C–N bond forming reactions. | ||
Two further reports of C–N bond formation detail the synthesis of acridines using azides or anthranils as coupling partners, by He and Kim respectively. In the former case, benzylamine was used to effectively promote the reaction (Scheme 3b),16 while in the latter case the TDG was formed in situ from the acid mediated decomposition of anthranil coupling partner (Scheme 3c).17 In He's work, γ-valerolactone (a biomass derived solvent) was used due to its favourable environmental profile, showing a transient C–H activation methodology can be compatible with greener solvent choices.
In Kim's work, the in situ generated amino ketone acts as a bidentate directing group to promote formation of the rhodium metallacycle, after which the anthranil can act as a nitrene equivalent to insert into the C–Rh bond and form an ortho aminated intermediate. In both He and Kim's studies, the aminated intermediates underwent an intramolecular cyclisation, then elimination, to form the acridine core.
 | ||
| Scheme 4 Ruthenium catalysed enantioselective transient C–H functionalisation of aldehydes. | ||
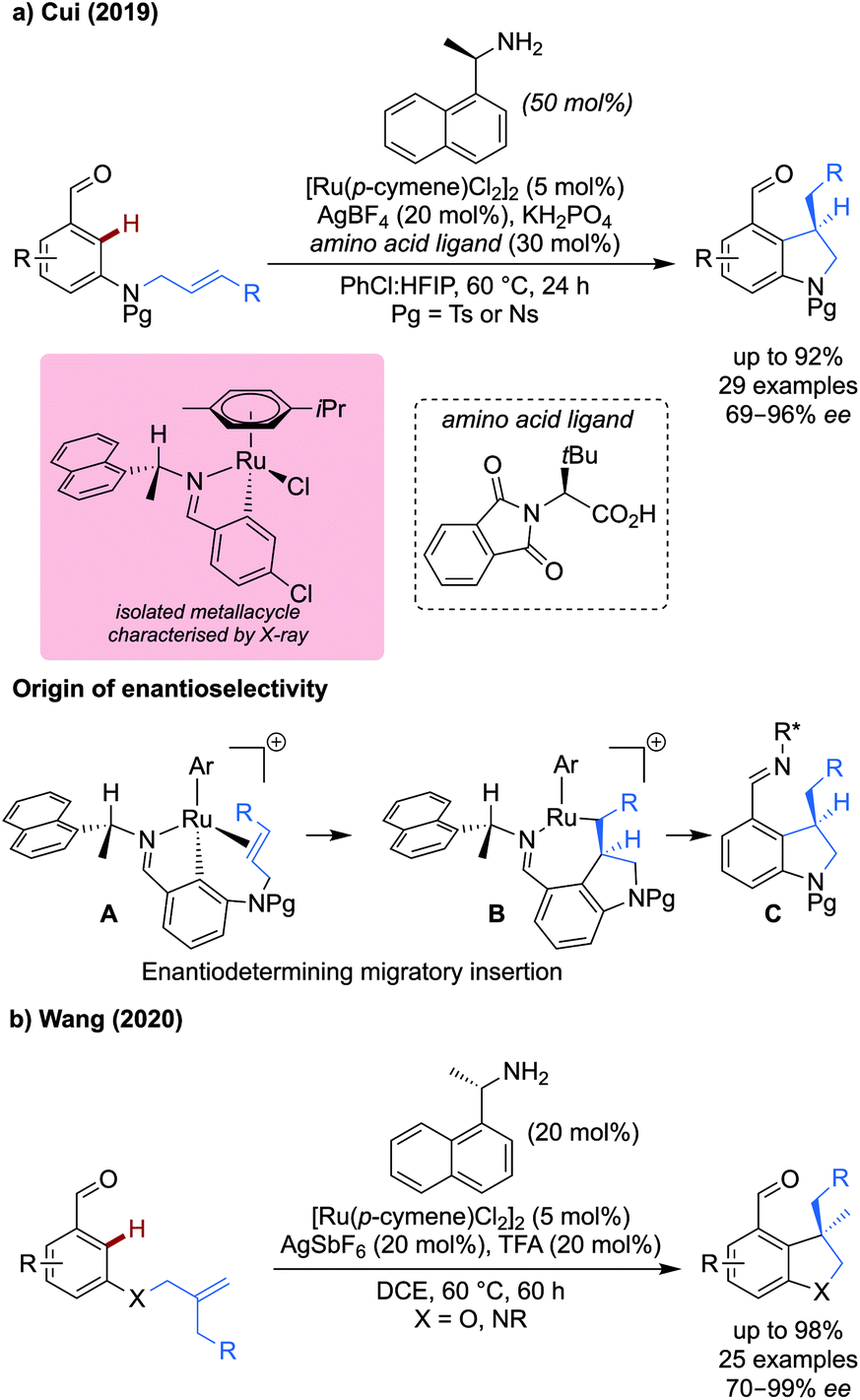
Using 1-(naphthalenyl)ethan-1-amine as a transient directing group, both procedures exhibited facial selectivity during the migratory insertion step. The p-cymene ligand on ruthenium restricts rotation of the directing group C–N single bond, leading to the orientation of the naphthyl and methyl groups to be fixed, exemplified by intermediate A. The smaller alkene then preferentially coordinates to the same side as the methyl group, rather than the naphthyl group, to avoid a steric clash, which lead to the stereochemical outcome observed.
Cui's work used an additional chiral amino acid derived ligand to further improve the enantioselectivity. A close ion pair formed between the chiral carboxylate and the cationic ruthenium metallacycle was proposed. An isolated metallacycle with a related aldehyde was obtained and characterised by X-ray analysis. Aldehydes bearing electron-donating and withdrawing ortho-, meta- and para-substituents (OMe, F and CF3) reacted effectively to give the corresponding indolines in good to excellent ee, up to 89% yield and 94% ee for the meta-methoxy example. More functionalised substituents on the tethered alkene were well tolerated, with silyl protecting groups and esters compatible with the reaction.
Wang demonstrated under related conditions that dihydrobenzofurans could be synthesised from the corresponding oxygen tethered alkenyl aldehyde (Scheme 4b).19 The amine transient DG was the sole source of enantioselectivity, achieving ring closure in yields and ees up to 98% for a range of substituted aldehydes bearing methoxy, nitro, aryl and halide substituents. The yields were unaffected by electron-rich, neutral, and poor substituents on the aldehyde moiety. The reaction was more sensitive to changes to the tethered alkene, with much reduced yield when R = Cl. Neither method was successful when using trisubstituted alkenes, or when X = CH2, restricting the scope to the synthesis of heterocycles.
 | ||
| Scheme 5 Iridium catalysed alkylation of (hetero)aromatics. | ||
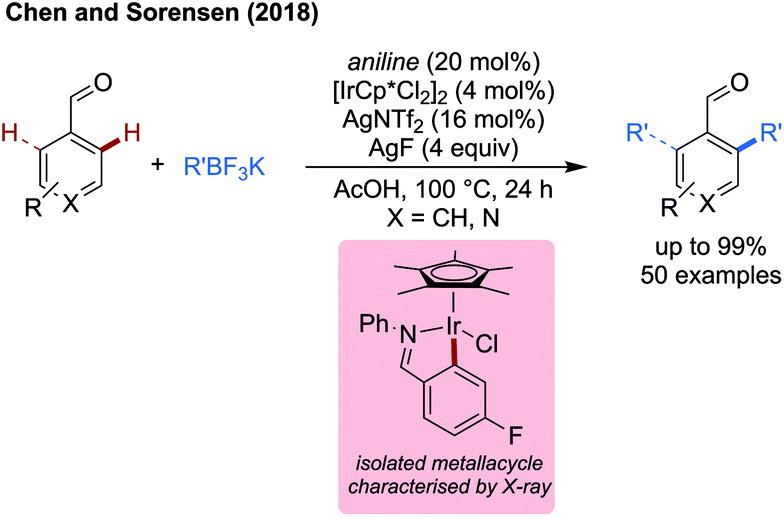
This work complements an earlier study on palladium catalysed methylation,21 allowing the installation of longer and more elaborate alkyl groups and achieving up to 99% yield. Monoalkylation was observed in strongly sterically biased and ortho-substituted systems in good to excellent yields in most cases. Without a strong steric bias, dialkylation predominates. Most impressive, was the ability to functionalise pyridines and other heteroaromatics (e.g. 99% with 4-pyridinecarboxaldehyde) which would otherwise be challenging with palladium catalysis.
An iridacycle was isolated which was characterised by X-ray crystallography. This could be effectively converted to cationic complex and then could be alkylated by treatment with the alkyl boron reagent and silver fluoride in AcOH, indicating the iridacycle to be a competent intermediate in the reaction.
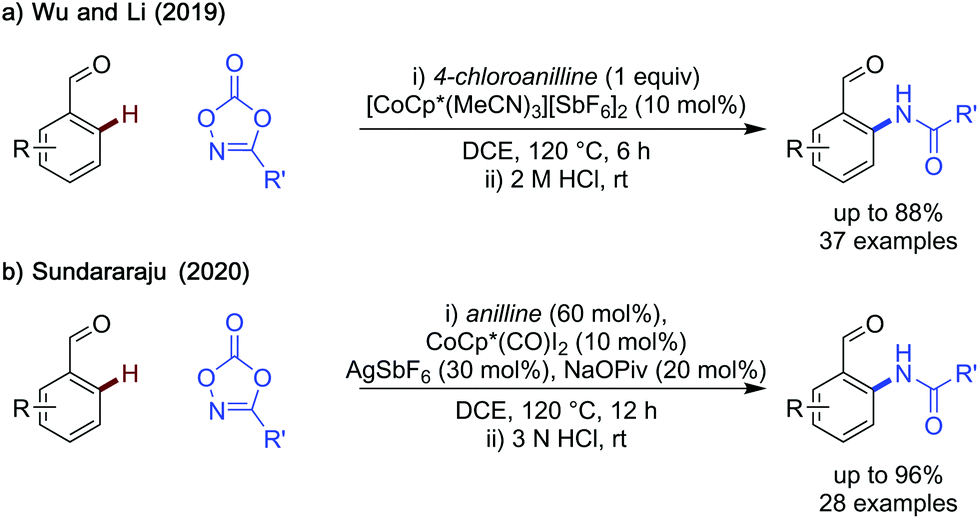 | ||
| Scheme 6 Cobalt catalysed C–H amination. | ||
Both methods displayed a wide substrate scope, with electron-neutral and rich benzaldehydes being particularly effective substrates. Li's method struggles to amidate aldehydes bearing inductively electron-withdrawing substituents (i.e. CF3) however, Sundararaju's method can amidate these substrates more effectively in several cases. A limitation shared by both methods, is that the catalysts are not commercially available and can require up to two steps to synthesise. Additionally, silver salts are required either in the synthesis of the catalyst, used by Li, or as a reaction additive, in Sundararaju's method, which may detract from the benefits of using a base metal catalyst. Fluctuations in cobalt supply can be a limitation for any method relying on this catalyst.
ortho-C(sp2)–H Functionalisation. Palladium catalysis is by far the most commonly used in transient C–H functionalisation procedures. This can be attributed to the high degree of mechanistic understanding that has aided the development of new more versatile bond forming methodologies in recent years.
2-Methylsulfinyl aniline was shown to be an effective directing group for arylation of benzaldehydes (Scheme 7a).24 This directing group promotes ortho-arylation selectively, with benzylic C–H arylation not observed. The isolated palladacycle showed the directing group to bind as a L-type ligand through the lone pair on the sulfur atom. This palladacycle reacts with ArI to form the product aldehyde. Biphenyls were prepared in moderate to good yields, with electronically distinct aryl iodides being reactive coupling partners. Substituted benzaldehydes could be arylated effectively, with higher yields observed for more electron-rich systems.
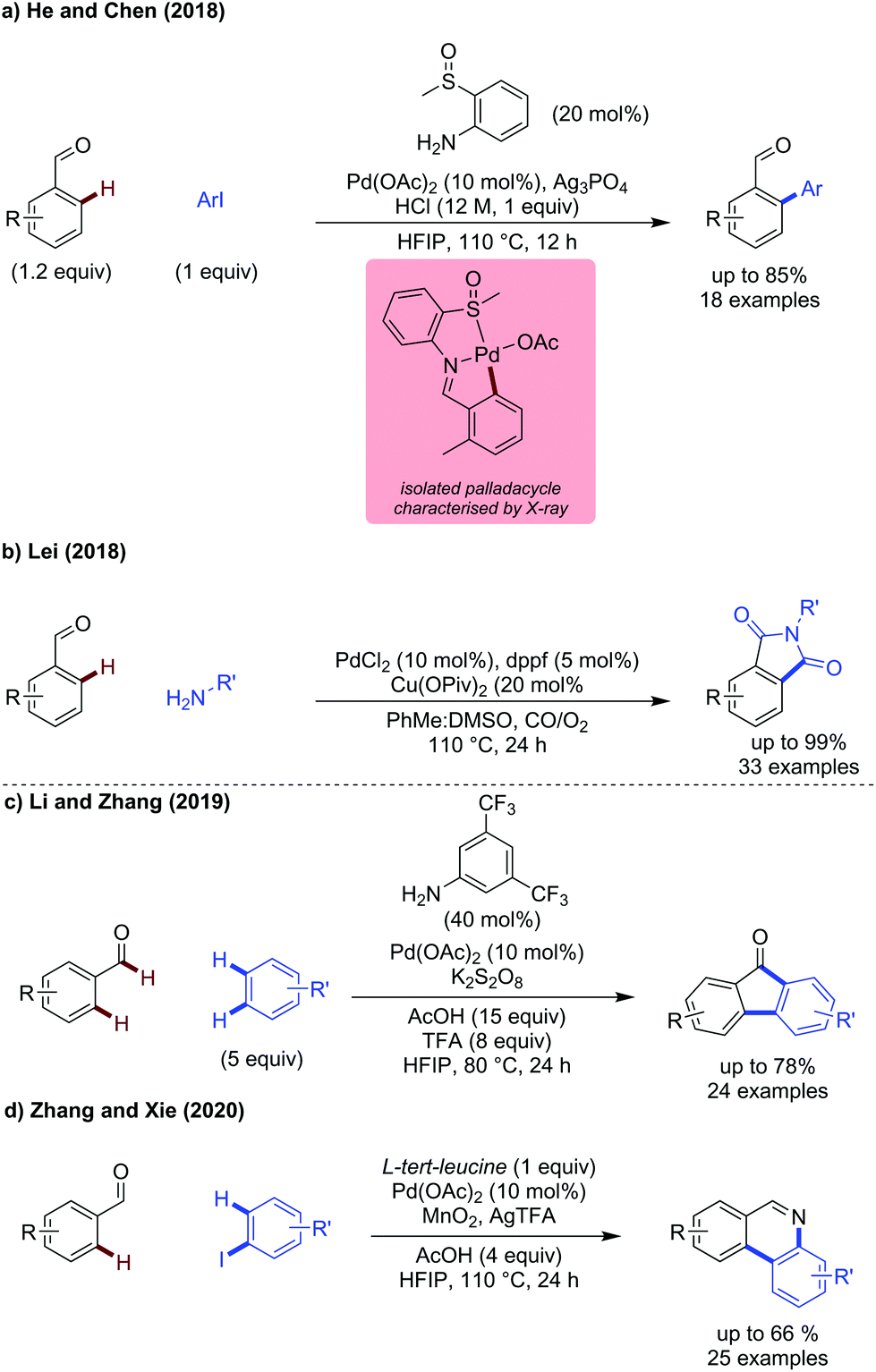 | ||
| Scheme 7 Palladium catalysed transient C–C bond forming reactions. | ||
Lei demonstrated the synthesis of substituted phthalimides from aldehydes via a transient dual palladium/copper catalysed carbonylative strategy (Scheme 7b).25 This method proceeds by a PdII/Pd0 catalytic cycle, with copper pivalate as a catalytic reoxidant and oxygen as the terminal oxidant. Several substituted benzaldehydes were reactive but lower yields were observed with systems bearing electron-withdrawing groups. The amine portion could be changed to electron-rich and electron-poor anilines without significant erosion in yield. Alkyl amines could also be used allowing diverse substitution at the nitrogen, also indicating the compatibility of this reaction to changes in the directing group.
Zhang showed direct cross-dehydrogenative coupling reaction of aldehyde and aromatics to access fluorenones using 3,5-bistrifluoromethylaniline as the transient directing group and potassium persulfate as the terminal oxidant (Scheme 7c).26 Electron-poor aldehydes and aromatic coupling partners were most effective, with bromoanisole particularly effective.
Zhang and Xie used manganese(IV) oxide as an oxidant and aryl iodides as coupling partners to make phenanthridines. The amino acid acts as both transient directing group and nitrogen source (Scheme 7d).27 The reaction is proposed to proceed by initial ortho arylation, followed by MnO2 mediated oxidation/decarboxylation. Moreover, an imine radical cation intermediate was proposed, which undergoes a radical cyclisation then oxidation to form the phenanthridine product.
In addition to developments in the formation of novel C–C bond forming reactions, there have been a few advances in the development of transient C–X bond forming methodologies using palladium catalysis (Scheme 8).28,29
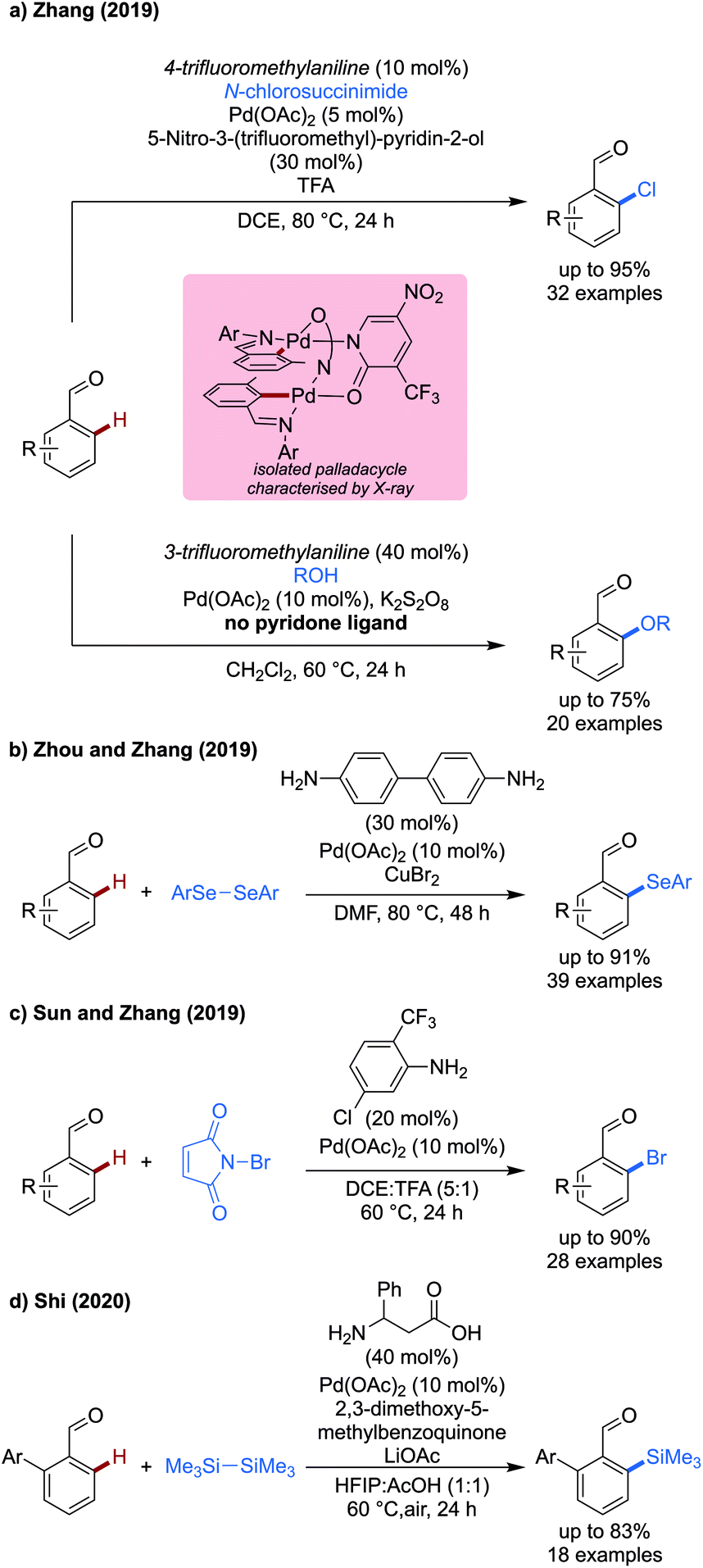 | ||
| Scheme 8 Palladium catalysed transient C–X bond forming reactions. | ||
Zhang contributed both chlorination and methoxylation methods using NCS and methanol/potassium persulfate as coupling partners to achieve effective functionalisation of a range of substituted benzaldehydes in high yields (Scheme 8a).28 4-Trifluoromethylaniline used in combination with a pyridone ligand enabled chlorination of benzaldehydes in excellent yields. The reaction exhibited a broad functional group tolerance allowing the inclusion of nitrile, ester and sulfonamide functionalities on the aldehyde component. Additionally, a celecoxib derivative could be chlorinated in a late stage manner, the imine directing group overriding the effect of any other coordinating functionalities on the substrate. Pyrroles, indoles and benzothiophene carboxaldehydes were also chlorinated, though a lower yield was observed.
By using an alcohol coupling partner, and potassium persulfate as a stoichiometric oxidant, methoxylation could also be achieved on a range of different benzaldehydes, though the yields of the methoxylation were lower than that of the chlorination reaction.
A dimeric palladacyclic intermediate was isolated which could be both chlorinated and methoxylated under their respective standard conditions.
In this same year, Zhou and Zhang showed that a transient C–H activation methodology was compatible with the use of diselenides as coupling partners to achieve the first example of ortho selenation of benzaldehydes (Scheme 8b).29 Benzidine was used as the TDG in this case, which presumably acts as a monodentate directing group to direct palladium. The extra amine site on the TDG effectively doubles its loading. Copper bromide was used as an additive in the reaction, potentially to reoxidise Pd0 in a presumed PdII/Pd0 mechanism. Alternatively, CuII could react with selenols formed as byproducts in the reaction, preventing catalyst poisoning. High selectivity for mono selenation was observed in all cases, and the reaction functioned effectively with electron-rich and electron-poor substrates. Thiophenes and indoles could be selenated, however furans and pyrroles were unreactive. Many different aryl diselenides could be used which all reacted in excellent yields, however alkyl diselenides were unreactive.
Sun and Zhang demonstrated a monodentate transient directing group strategy for the ortho-bromination of benzaldehydes (Scheme 8c).30 This work is related to an earlier work by Yu using a bidentate anthranilic acid directing group,31 with a slightly broader scope investigated in this later work. The monodentate 2-trifluoromethyl-5-chloroaniline directing group was shown to be effective for the transformation, with a range of substrates biased to mono-bromination reacting in 20–98% yield. Ortho- and meta-halides reacted effectively (54–88%), however a severely reduced yield of 20% was observed when a meta-methoxy substrate was used. When using substrates unbiased to mono-bromination, and higher equivalents of NBS (2.5 equiv. vs. 1.2 equiv.) dibromination was observed in good to excellent yields (67–82%).
In 2020, Shi demonstrated the first example of C–H silylation using a transient directing group strategy (Scheme 8d).32 This method used phenyl substituted β-alanine as a directing group and hexamethyldisilane as a silylating reagent. A benzoquinone-type oxidant was used to promote the coupling. While the scope consisted exclusively of 2-aryl substituted benzaldehydes, the reaction proceeded in up to 78%. Only hexamethyldisilane could be used as a coupling partner, with other similar reagents (hexaphenyldisilane) being unreactive.
γ-Arylation of heteroaromatic carboxaldehydes. In 2019, Maiti and Volla published a method to arylate indole carboxaldehydes selectively at the γ-position, extending the scope of the method to include aza-indoles (Scheme 9).33 While the benzyl protecting group was most effective, unprotected indole could also be arylated, albeit in lower yield. Indoles bearing aryl groups and halides at C6 all reacted in good to excellent yields. Importantly, 7-azaindoles reacted reasonably well, with the added coordination site on the substrate not poisoning the catalyst. Additionally, the reaction exhibited a broad scope of aryl iodides, with electron-rich and electron-poor aromatics functioning effectively in yields up to 92%. The reaction was not catalytic in transient directing group, with 2 equivalents of glycine required for reactivity.
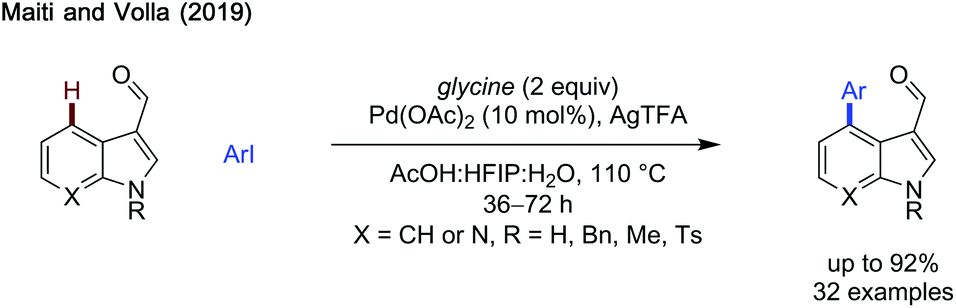 | ||
| Scheme 9 Arylation of indole carboxaldehydes. | ||
Atroposelective C(sp2)–H functionalisation. Several new reports of atroposelective allylation,34 naphthylation,35 alkynylation36 allylation and alkenylation37–39 for the synthesis of axially chiral products have been disclosed (Scheme 10). All methods used either L-tert-leucine, L-valine or derivatives as a transient directing group, in which chiral information is effectively transferred to give high enantioselectivities under relatively mild reaction conditions.
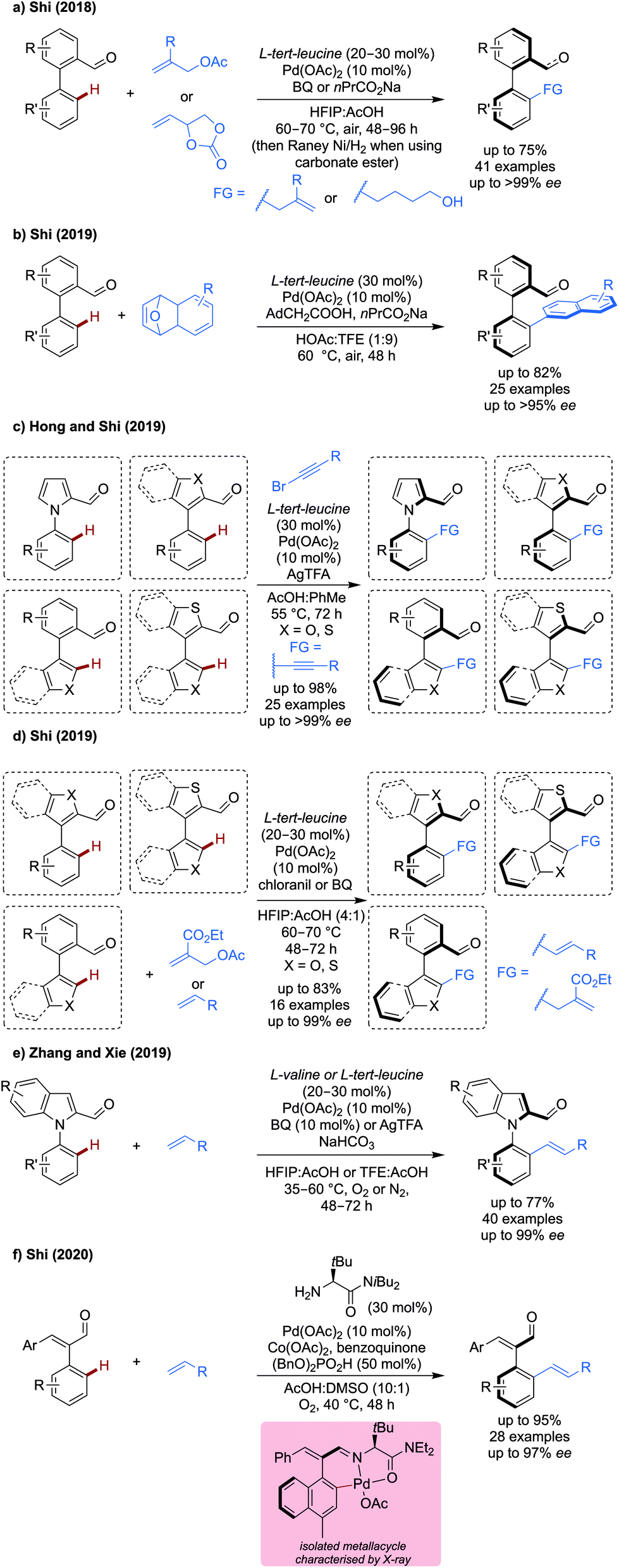 | ||
| Scheme 10 Asymmetric C–H functionalisation reactions of biaryls and styrenes. | ||
Allyl acetates and carbonate esters were demonstrated as viable coupling partners for the enantioselective synthesis of biaryls (Scheme 10a).34 Allyl acetate could be effectively coupling using benzoquinone as an additive in up to 73% yield and >99% ee with a variety of substituted biaryl systems. Only a small range of allyl acetates were demonstrated, almost all with R = CO2Et. Allyl carbonate esters could also be used as coupling partners. Slightly modified conditions were employed, whereby the BQ is omitted and a sodium carboxylate additive is used. After the reaction, a RANEY® nickel reduction of the allylic alcohol intermediate (and simultaneous reduction of aldehyde functional group) gave a diol product in good to excellent yields and ees.
Naphthylation was achieved using unusual 7-oxabenzonorbornadienes as coupling partners (Scheme 10b),35 which can react with the palladacycle in a redox neutral manner by migratory insertion into the C–Pd bond followed by β-oxygen elimination, protodepalladation and dehydration to give the napthylated product. While slight reductions in yield were observed when changing substituents on the aldehyde portion, the ee remained extremely high in all cases (>95%). The aldehyde products could be further derivatised to generate effective organocatalysts, showing this utility of this strategy for the synthesis potentially challenging synthetic targets.
Later, Hong and Shi demonstrated an enantioselective alkynylation of more challenging, less rotationally constrained, heteroaromatic pentaatomic biaryl systems (Scheme 10c).36 High yields and ees were observed under their standard conditions when using N-1-naphthyl pyrrole carboxaldehydes, despite the lower energy conformation barrier. When using an aryl-benzothiophene system, good to high yields and excellent ees were observed, however changing the heteroatom from S to O, resulted in a dramatic drop in ee, to the point where no enantioselectivity is seen for a couple of cases. A limited scope of alkynes was demonstrated.
Shi also demonstrated that on similar pentaatomic biaryls, allylation and alkenylation could be achieved using allyl acetates and alkenes as coupling partners respectively (Scheme 10d).37 With benzoquinone (BQ) based oxidants (chloranil for allylation and BQ for alkenylation) a silver free protocol of atroposelective C–H functionalisation was achieved. However, only the acrylate bearing an acetoxymethyl group was demonstrated to be an effective coupling partner for allylation on a modest range of aldehydes. Other acrylates and styrenes could be used as coupling partners in moderate (50%) to good yield (70%).
Xie reported the alkenylation of indole and pyrrole carboxaldehydes using either L-valine or L-tert-leucine as a transient directing group (Scheme 10e).38 Using a set of mild, silver-free conditions (L-valine, BQ 10 mol%, HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH, 35 °C, under O2) the desymmetrisation and dynamic kinetic resolution of pyrrole and indole carboxaldehydes proceeded in good yields and excellent ee (90–99% ee). A set of slightly modified conditions enabled the kinetic resolution of indole carboxaldehydes (L-tert-leucine, AgTFA 2equiv., TFE:AcOH, 60 °C and under N2) in up to 99% ee and s factors of up to >400. A reasonably broad scope of aldehydes and alkenes was shown, with the unsubstituted N-phenylindole carboxaldehyde being particularly reactive with nbutyl acrylate (77%, 94% ee).
:AcOH, 35 °C, under O2) the desymmetrisation and dynamic kinetic resolution of pyrrole and indole carboxaldehydes proceeded in good yields and excellent ee (90–99% ee). A set of slightly modified conditions enabled the kinetic resolution of indole carboxaldehydes (L-tert-leucine, AgTFA 2equiv., TFE:AcOH, 60 °C and under N2) in up to 99% ee and s factors of up to >400. A reasonably broad scope of aldehydes and alkenes was shown, with the unsubstituted N-phenylindole carboxaldehyde being particularly reactive with nbutyl acrylate (77%, 94% ee).
The use of styrenes, less conformationally stable substrates, was achieved using an amide derivative of L-tert-leucine (Scheme 10f).39 A silver-free system was developed, using cobalt acetate, benzoquinone under an oxygen atmosphere, presumably to promote reoxidation of palladium, though mechanistic discussion of the specific roles of these reagents was not elaborated. When using substituted styrene aldehydes with a high enough barrier to interconversion, high yields and ees could be obtained. When using phenyl substituted styrenes, a bulky group was necessary to retain ee, as the barrier to interconversion of the two enantiomers becomes too low. A broad range of Michael acceptors functioned well in the reaction, with esters, sulfonate ester, phosphonates and even unactivated alkenes being reactive. Alkenes derived from biologically relevant molecules, including esterone and menthol, could also be tethered via this strategy in high diasteromeric ratios.
Ackermann demonstrated the combination of an electrochemical approach with a transient C–H activation methodology to achieve an atroposelective alkenylation of biaryl systems (Scheme 11).40 Good yields and high ees were obtained in most cases, with several biaryl and N-naphthyl pyrroles functioning well. Vinyl sulfones, phosphonates, amides and ketones all proceeded in good yields and excellent enantioselectivity. Mechanistic studies indicate the concerted-metalation-deprotonation (CMD) is rate determining. This method overcomes one of the main issues of these oxidative alkenylations, the requirement for stoichiometric oxidants, representing an important step forward for more sustainable transient C–H activation. The chiral biaryl aldehydes were also effectively transformed into highly enantioenriched helicene derivatives.
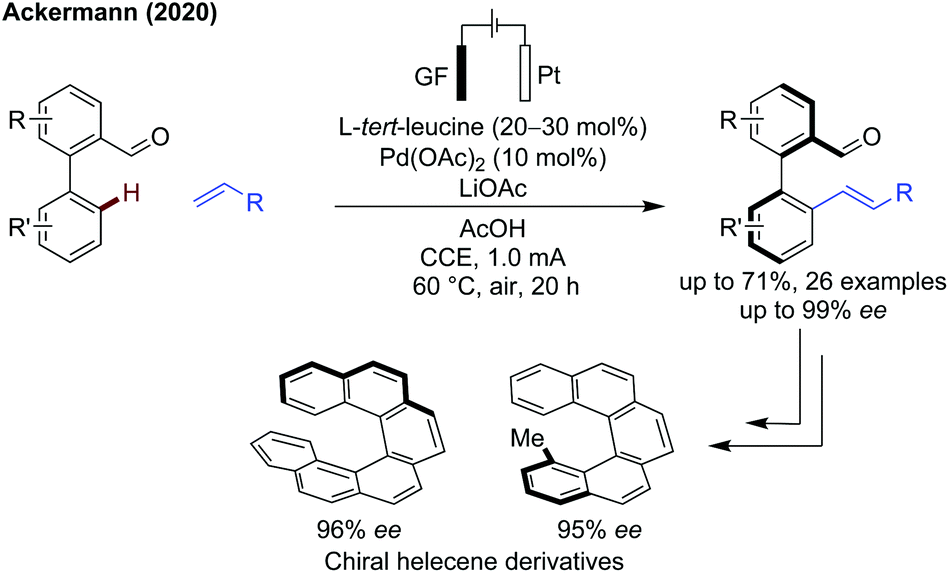 | ||
| Scheme 11 Electrocatalytic atroposelective C–H alkenylation. GF = graphite felt electrode. CCE = constant current electrolysis. | ||
meta-C(sp2)–H functionalisation. meta-Functionalisation is more challenging, especially when using an imine as the linking functionality as this sets up an ideal binding site for ortho functionalisation. While a method catalytic in directing group has yet to be reported, Xu and Jin used a telescoped DG installation and cleavage protocol with an acetal directing group to achieve meta-alkenylation of aldehydes (Scheme 12).41 The higher binding affinity of the nitrile compared to the acetal oxygen leads to selective coordination of palladium to this site, and meta alkenylation of the masked aldehyde substrate. This telescoped method involves first an acid catalysed acetal protection with the diol directing group, followed by palladium catalysed alkenylation using silver acetate as the terminal oxidant. Subsequent hydrolysis of the alkenylated product acetal furnished the aldehyde functionality. This method effectively alkenylated benzaldehydes (and acetophenones) bearing a broad range of different functional groups in a reasonably high yield and selectivity for the meta position. Other alkenes could be used as effective coupling partners, provided they were electronically activated by either an ester or sulfone substituent. Finally, the alkenylated aldehydes were demonstrated to be versatile synthetic building blocks, and extensive derivatisation was performed which proceeded in 52–81% yield, accessing functional groups including but not limited to hydroxyketones, oxazoles, allenes and alkenes.
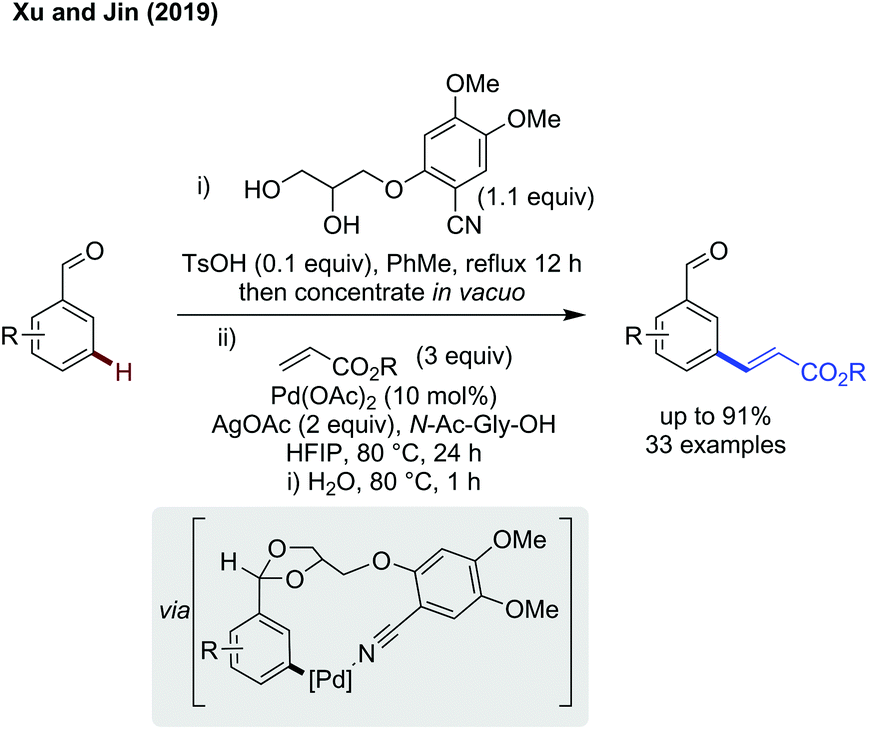 | ||
| Scheme 12 Acetal template directing meta C(sp2)–H alkenylation. | ||
In a preprint, Maiti has disclosed an alternative means of achieving meta-selective functionalisation of aldehydes/amines, using a stoichiometric imine with a pyrimidine directing group to position the catalyst at a remote position for alkenylation.42
Benzylic C(sp3)–H functionalisation of benzaldehydes
The activation of benzylic C(sp3)–H has seen some key advances in the past two years, with both new directing groups and novel coupling partners being reported.43–50 The benzylic C–H bonds are electronically activated by the presence of the aryl group, and are relatively constrained geometrically.Yi and Zhang reported a method to construct polyaromatic hydrocarbons from o-tolualdehydes and di/tri-iodoarenes (Scheme 13a).43 While similar to earlier work by Kim,44 this method used a simple and commercial TDG (glycine) to access a broader range of polycycle aromatic hydrocarbons in higher yields. Diiodoarenes with 1,2-, 1,3-, and 1,4-substitution along with 1,3,5-triiodoarenes could be used successfully to construct di/trialdehyde products which were then cyclised by treatment with triflic acid to give polyaromatic products. o-Tolualdehyde itself gave the dialdehyde in 65% yield and the subsequent cyclisation gave a polyaromatic product in 83% yield. Yu and Zhang also reported a modified set of conditions to achieve the synthesis of substituted polyaromatic hydrocarbons in a one pot method from o-tolualdehydes and iodoarenes (Scheme 13b).45
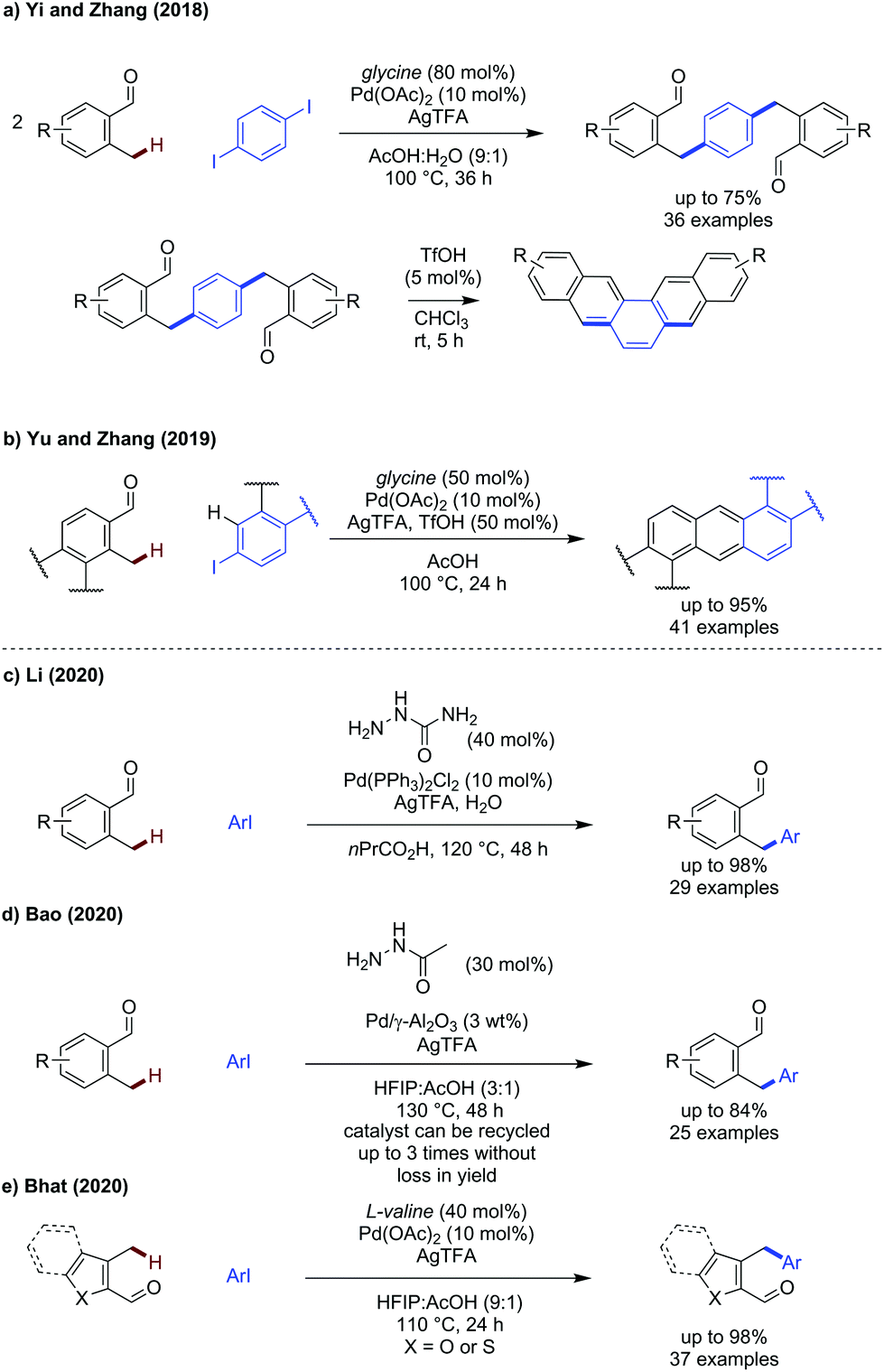 | ||
| Scheme 13 Palladium catalysed benzylic C(sp3)–H activation forming C–C bonds. | ||
Li described the use of semicarbazide as a transient directing group for the arylation of benzylic C(sp3)–H bonds.46 Similar to acetohydrazide, semicarbazide possesses a hydrazine moiety whereby the primary amine is more nucleophilic due to the α-effect, promoting hydrazone formation. However, semicarbazide has NH2 in place of CH3 which could change the strength of the secondary binding site and allow for either an L,L or L,X binding mode to be operational. No C(sp2)–H arylation was observed.
To improve the sustainability of the transient C–H arylation strategy, Bao demonstrated that palladium nanoparticles impregnated on Al2O3 could be used to promote the reaction as a heterogeneous catalyst (Scheme 13d).47 The inherent advantages of easy catalyst removal and recyclability make this an attractive strategy from a sustainability point of view, and the catalyst could be reused up to 3 times before the yield declined (74% 3rd cycle, 63% 4th cycle, 47% 5th cycle). Yields obtained were comparable to a homogenous process (30–63%) using acetohydrazide as the transient directing group.
The arylation of 3-methyl(benzo)thiophene carboxaldehydes and3-methylbenzofuran carboxaldehydes were demonstrated by Bhat, using valine as an effective transient directing group for this type of benzylic substrate bearing a pentaatomic aromatic backbone (Scheme 13e).48 A broad range of aromatics could be installed in high yields, up to 98% using 4-CO2Me iodobenzene as a coupling partner. Several substituted iodopyridines could also be successfully used to install heteroaromatics. An indole carboxaldehyde example was included in the scope, however the yield was very low (<10%).
Yu described the first example of benzylic C(sp3)–H fluorination (and acetoxylation) using a transient directing group in 2018 (Scheme 14).49 This seminal work used an N-fluoropyridinium salt as an oxidant and fluorine source (or as a bystanding oxidant for the acetoxylation). The fluorination reaction pathway was found to be promoted by the use of a hindered amino amide directing group possessing an L,L type binding (confirmed by isolated palladacycle that amide binds via oxygen). This binding mode was proposed to facilitate the formation of a cationic five coordinate PdIV intermediate which favours inner sphere reductive elimination (Scheme 14b). The inclusion of pentafluorobenzoic acid as a less nucleophilic carboxylate was also important for the fluorination to proceed in good yields and high ees. Only aldehydes bearing electron-withdrawing ortho- or meta-substituents (nitro, trifluoromethyl) were compatible with the reaction conditions, affording fluorinated products in 36–61% yield and up to 99% ee. When using unsubstituted 2-ethylbenzaldehyde, ortho-C(sp2)–H acyloxylation occurred in addition to benzylic fluorination to give a doubly functionalised product in a low 16% yield. On substrates bearing either electron-rich OMe group or on the 2-methylbenzaldehyde, acyloxylation was observed exclusively.
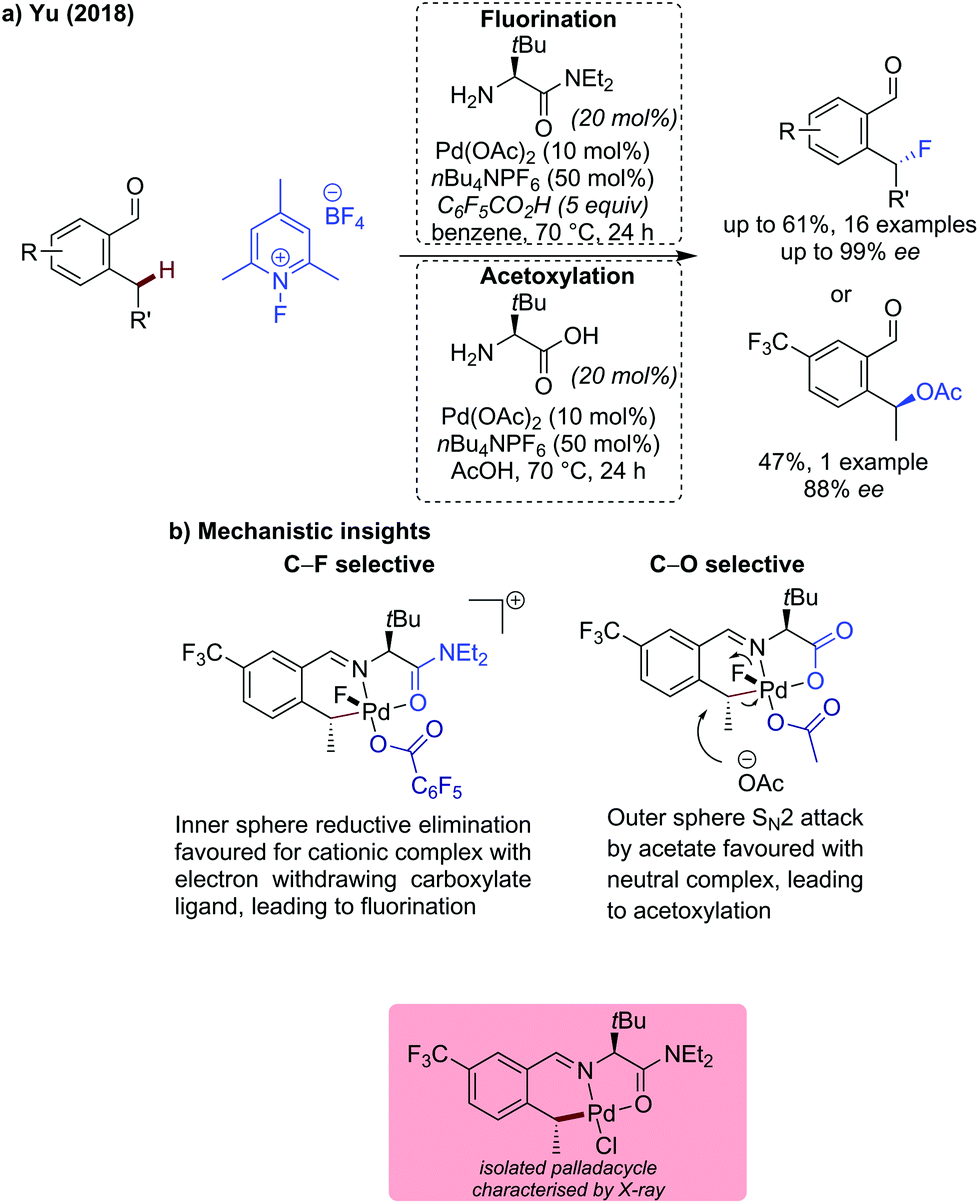 | ||
| Scheme 14 Palladium catalysed benzylic C(sp3)–H activation forming C–F and C–O bonds via selective reductive elimination pathways. | ||
The directing group was proposed to induce chirality by favouring the CMD transition state in which the palladacycle has the directing group substituent on the opposite side to the benzylic substituent, thus avoiding a potential steric clash. This configuration was observed in the isolated palladacycle. The bulky t-Bu group on the directing group was key to achieving enantioselectivity. In the C–F bond formation pathway, there is an inner sphere reductive elimination and retention of configuration, hence the observed product stereochemical outcome.
Acetoxylation on the other hand, was proposed to go via outer sphere SN2 attack by acetate, favoured by the use of L,X-type bidentate transient directing group (i.e. glycine), to form a neutral PdIV intermediate. In combination with acetic acid as the solvent, selective C–O bond formation was achieved. This change in mechanism was confirmed by subjecting the isolated Pd(II)-palladacycle to the F+ oxidant in acetic acid. The acetoxylated product of the opposite stereochemical outcome to the fluorinated product was observed, indicating an inversion of configuration due to an SN2 attack. With the focus on the fluorination protocol, the reaction scope for the acetoxylation was not expanded.
Following Yu's work, Bao recently published an acetoxylation protocol using acetohydrazide as a transient directing group and potassium persulfate as the terminal oxidant (Scheme 15).50 MCM-48, a type of silica based molecular sieve, is proposed to improve solubility of the potassium persulfate enabling acetoxylation. While good yields were obtained for relatively electron-neutral aldehydes, reduced yields were observed for both more electron-rich and electron-poor substrates.
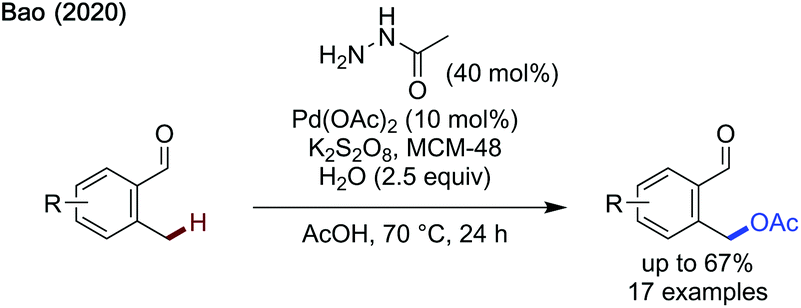 | ||
| Scheme 15 Benzylic acetoxylation of aldehydes using acetohydrazide as a transient directing group. | ||
C(sp3)–H functionalisation of aldehydes
More challenging is the functionalisation of C(sp3)–H bonds, as the increased flexibility and lack of electronic activation both make a CMD mechanism more difficult. With few previous reports, examples remain relatively sparse on aldehyde substrates.8,51There were few early reports achieving C(sp3)–H functionalisation,52 2019 saw several publications achieving β-arylation of aliphatic aldehydes. We (Bull) demonstrated the use of 2-methoxyethylamine as a directing group for the intramolecular arylation of tertiary aldehydes, forming fragrant indane derivatives (Scheme 16a).53 By tethering the coupling partner, it was possible to use unactivated aryl bromides as opposed to aryl iodides commonly employed in transient C–H arylation protocols. While yields were modest, a range of aldehydes could be cyclised to their indane analogues with electron-poor aromatics performing better than the more electron-rich examples.
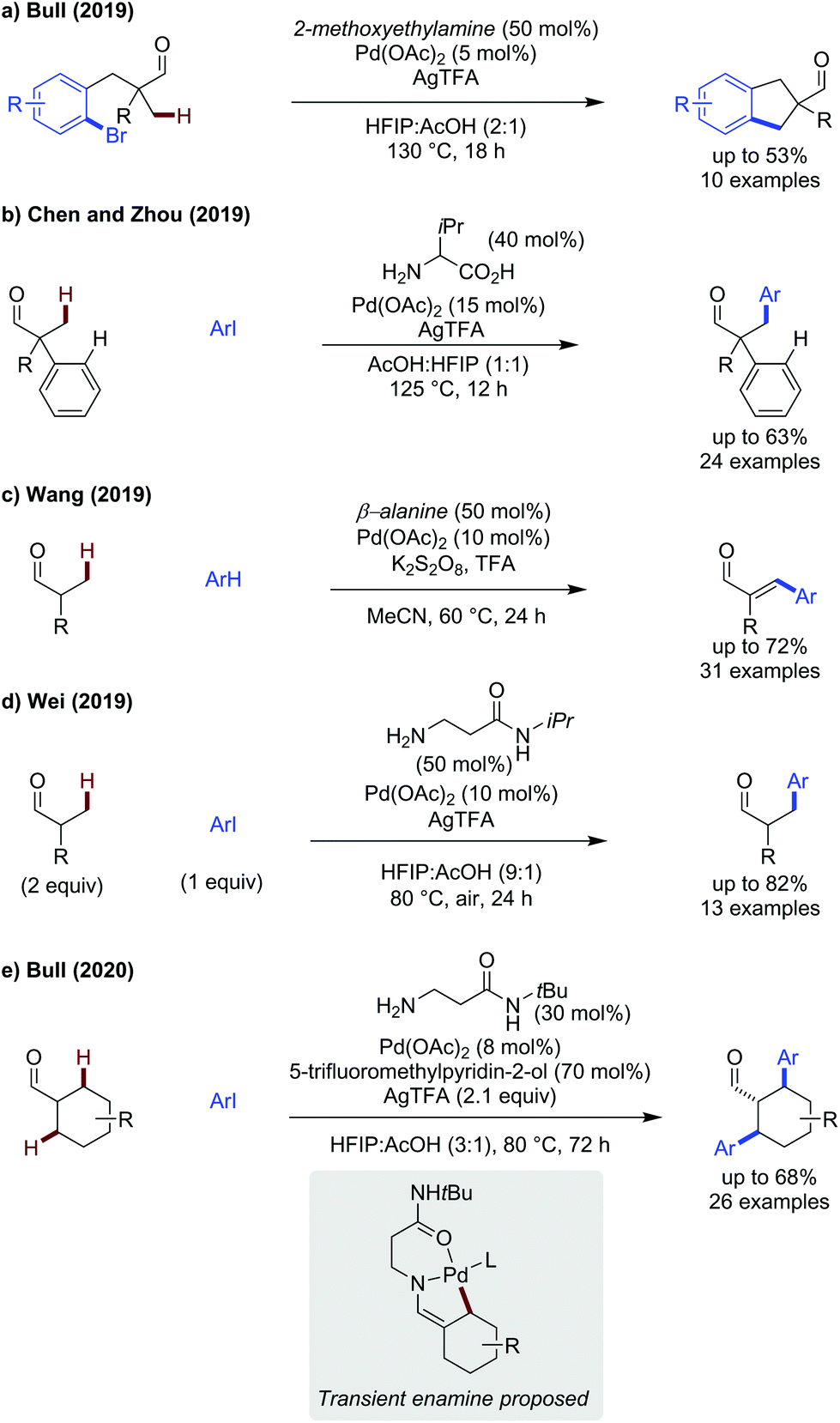 | ||
| Scheme 16 Palladium catalysed β-C(sp3)–H activation of aldehydes. | ||
Valine was shown to be an effective directing group for the selective C(sp3)–H arylation of tertiary phenylacetaldehydes (Scheme 16b).54 Overall the reaction proceeded in modest to good yield (35–63%) with a preference for the mono arylated product. While the reaction was tolerant to aryl iodides of varying electronic properties (OMe, halide, nitro), use of more sterically hindered ortho substituted aryl iodides led to lower yields (o-F, 38% yield). The aromatic pre-installed on the aldehyde could be changed readily to electron-withdrawing, donating and sterically bulky substitution patterns. A decrease in yield was observed when inductively electron-withdrawing substituents or bulky ortho-substituents were introduced. When using an enantioenriched L-tert-leucine directing group, a modest 40% ee was obtained.
Wang realised a cross dehydrogenative coupling using secondary aldehydes and aromatics as coupling partners to form cinnamaldehyde products (Scheme 16c).55 Potassium persulfate was used as an effective oxidant to promote this transformation, using β-alanine as the transient directing group. Ethoxybenzene was the most effective aromatic coupling partner, with isobutyraldehyde, giving 72% of the cinnamaldehyde product with only the para-substituted product observed. While the aromatic scope was predominantly consisting of electron-rich examples, which react effectively, electron-neutral benzene and electron-poor chlorobenzene both could be used as coupling partners, albeit in moderate yield. Additionally, a mixture of ortho-, meta-, and para-products were seen in the latter case. Propanal could be arylated in 59% yield when using 2-methylanisole as a coupling partner.
The reaction was proposed to proceed by first forming methacrolein via a Saegusa oxidation (R = Me), which was observed forming in the first 3 hours of the reaction. Given the products obtained are exclusively of E-stereochemistry, a carbopalladation pathway is more likely. Presumably, an aryl palladium species could form independently, and undergo carbopalladation with the acrolein intermediate, followed by β-H elimination.
Bulky β-alanine derived amino-amides have been shown to be effective directing groups for the arylation of several aliphatic aldehydes. Wei showed, using an iPr substituted amino amide, and the aldehyde in excess, the arylation of aldehydes and ketones (discussed later) could be achieved (Scheme 16d).56 Electron-poor aryl iodides tended to react more effectively (4-CO2Et gave 82% while 4-iodoanisole gave 60%). Like Yu's work detailing benzylic fluorination,49 the amide was proposed to bind as a neutral L-type ligand through the oxygen lone pair.
We (Bull) detailed the diarylation of cyclohexane carboxaldehyde (Scheme 16e),57 using a combination of a bulky amino-amide and a pyridone ligand additive, previously described by Yu to enhance amine functionalisation.58 A thorough screen of β-alanine derivatives indicated bulky amino amides were most effective for promoting reactivity. The inclusion of the 5-trifluoromethylpyridone ligand improved reactivity, potentially by acting as a carboxylate surrogate to accelerate CMD. The reaction proceeded in modest to good yields, giving exclusively trans-diarylated products (confirmed by X-ray crystal structure), with electron-neutral and electron-rich aryl iodides being most reactive. Relatively complex cyclohexane scaffolds could be accessed by arylation of substituted cyclohexane carboxaldehydes which would otherwise be challenging to synthesise.
The origin of the trans stereochemistry was an intriguing result and led to the proposal of a transient enamine acting as the directing group in this reaction which was hydrolysed after diarylation to give the thermodynamically favourable trans product. Using tertiary aldehydes completely shuts off reactivity, presumably as enamine formation is no longer possible.
Finally, Li and Ge demonstrated that a transient C–H activation methodology could be used to access more distal C–H bonds when using substrates biased to γ-arylation (Scheme 17).59 A pyridone ligand was key to promoting reactivity, along with phenylalanine as a transient directing group, proposed to go via a 6-membered palladacycle intermediate. The reaction functions well for tertiary alkyl aldehyde examples, giving access to the γ-arylated products in up to 72% yield (e.g. R,R′ = n-Bu) and more functionalised aldehydes with fluoro (50%) or methoxy substituents (63%), as well as aromatic ethers in 61%. When using a substrate where there is the choice of reacting at either β- or γ-positions, diarylation is observed predominantly (i.e. both β and γ) with the β-position preferred to the γ-position in a 51:23:5 ratio (di:β:γ). The scope of aryl iodide proved to be broad, with the yield being insensitive to the substituents on the aromatic. Additionally, even hindered ortho substituted systems were effective.
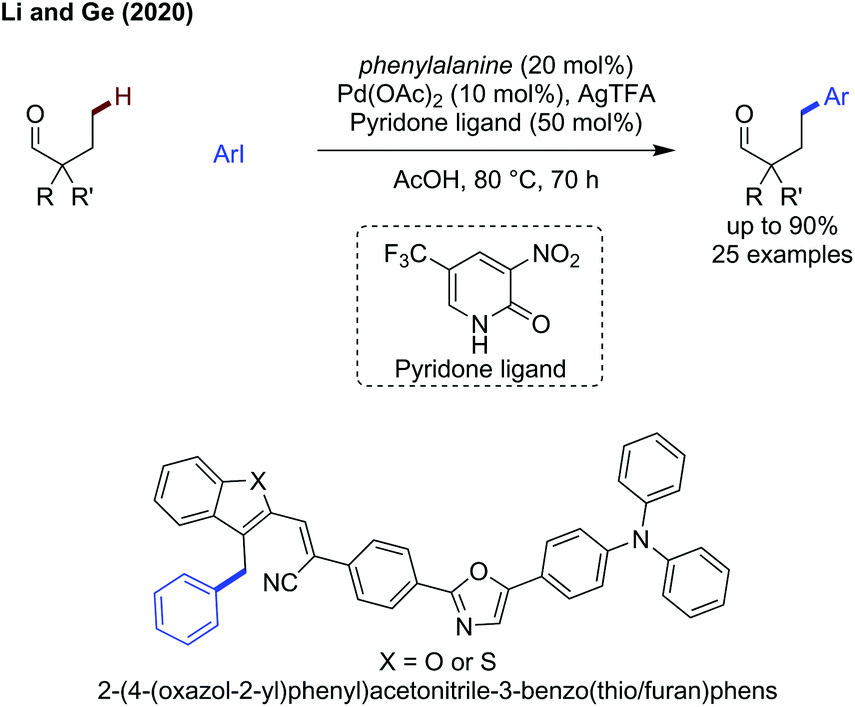 | ||
| Scheme 17 γ-C(sp3)–H arylation of aldehydes. | ||
This method was applied to the arylation of benzylic heteroaromatic substrates (benzothiophenes and benzofurans) to afford the γ-arylated products. These were then used to construct 2-(4-(oxazol-2-yl)phenyl)acetonitrile-3-benzothio/furanphens whose fluorescent properties change by the physical act of grinding (Scheme 17).
3. C–H functionalisation of ketones
In comparison to aldehydes, ketones exhibit lower electrophilicity and greater steric and directional constraints, which often requires different designs of transient directing groups. Fewer recent advances have been made on these substrates than their aldehyde analogues.C(sp2)–H functionalisation of ketones
Prabhu described amidation of aromatic ketones (Scheme 18a),14 using the same electron-poor aniline as a monodentate transient directing group as used for aldehyde functionalisation. Further elevated temperatures were required to achieve effective reactivity. A small scope of ketones were demonstrated, where R = Me and Et, which gave the amidated products in low to good yields.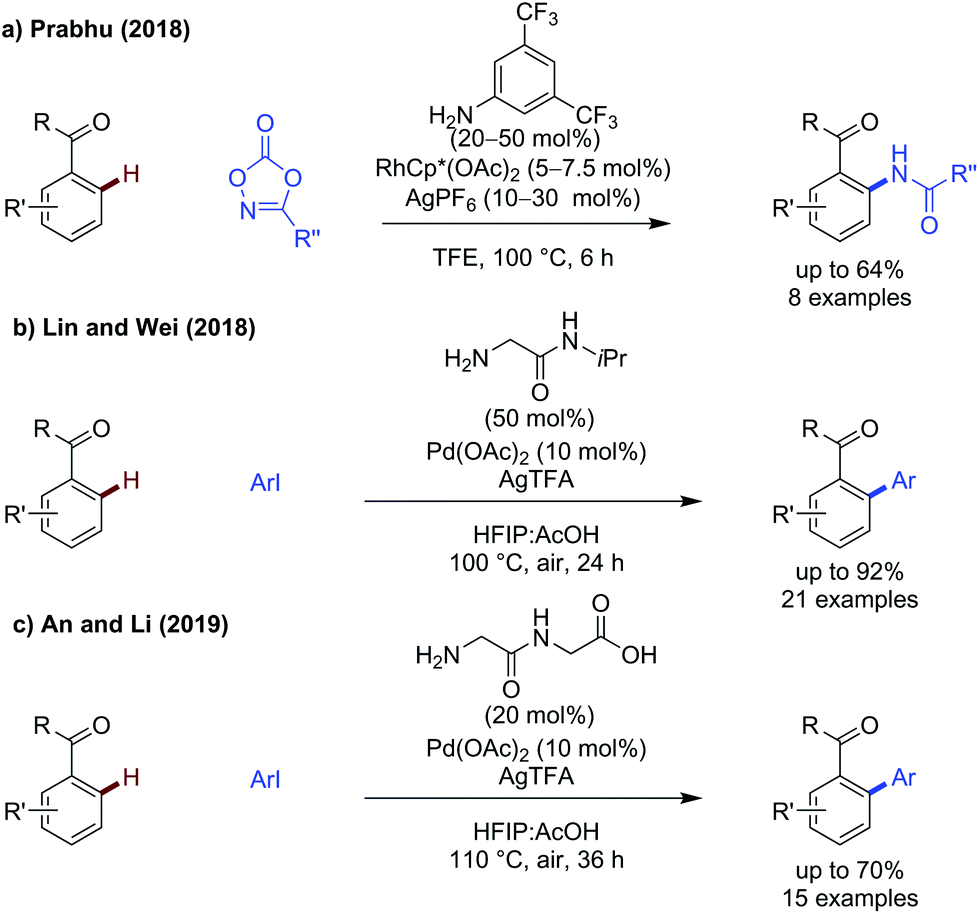 | ||
| Scheme 18 C(sp2) amidation and arylation of aryl ketones. | ||
Lin and Wei showed a glycine derived amino amide could effectively promote arylation of acetophenones (Scheme 18b).60 Several aryl ketones could be functionalised in up to 91% yield. The reaction was tolerant of steric and electronic changes on the aryl iodide, with the yield remaining high in most cases. Use of electron-poor acetophenones bearing p-Cl or p-CO2Me gave reduced yield (54% and 50% respectively) when using methyl 4-iodobenzoate as the coupling partner.
An and Li showed that a dipeptide consisting of two glycine units was an effective transient directing group for the arylation of aryl ketones where R = Me, Et and a cyclic ketone (Scheme 18c).61 This is one of only a few examples of potentially tridentate directing groups being used. This tridentate directing group can be used in relatively low catalytic loadings to promote arylation on a range of substituted acetophenones. The proposed role of the extra binding site is not elaborated on, though it presumably provides additional stabilisation for palladacyclic intermediates/transition states while still being labile enough to prevent catalyst poisoning.
C(sp3)–H functionalisation of ketones
The glycine derived amino amide used by Lin and Wei could also be used for the functionalisation of aliphatic ketones at both methyl and methylene centres (Scheme 19a).60 This shows the versatility of their directing group, as it functions equally well on more challenging C(sp3)–H activation. | ||
| Scheme 19 C(sp3)–H activation of ketones. | ||
The unusual glycylglycine tridentate transient directing group was suitable as a transient directing group for the arylation of aliphatic ketones.61 Arylation of methyl and methylene C(sp3)–H bonds could be achieved in up to 80% yield using the meta-nitro substituted aryl iodide.
Wei showed the β-alanine amide was effective for ketone arylation, using slightly elevated temperatures a broader scope could be achieved (Scheme 19b).56 Electron-poor, neutral and rich aryl iodides were all similarly effective, as was a sterically hindered o-tolyl example. Cyclic and acyclic ketones could be arylated in comparably good yields. Clearly there is a geometric requirement for reactivity, as while secondary ketones were effective, tertiary pinacolone was ineffective. As before, the directing group was proposed to bind in a L,L-binding mode, through the imine and carbonyl lone pairs respectively.
Yu achieved enantioselective arylation of cyclobutyl methyl ketones with the aryl iodide as the limiting reagent (Scheme 19c).62 Valine was found to be the most effective transient directing group, and the addition of a pyridone ligand improved reactivity and chiral induction. The reaction gave modest to excellent yields, with high ees observed for most aryl iodides, with an erosion in yield and ee when heteroaromatic aryl iodides were used. The aliphatic chain on the ketone could be changed readily, with the yield only being significantly reduced when introducing ether substituents which could potentially coordinate competitively. Cyclopentyl methyl ketone and octyl methyl ketone could also be arylated, however a severe reduction in yield and ee were observed. The arylated cyclobutanes could also be resubjected with a different aryl iodide to access diverse arylated cyclobutanes in two consecutive C–H arylation reactions.
Iodide abstraction from the PdIV intermediate prior to reductive elimination was proposed to be rate (and enantio) determining. In the absence of the pyridone ligand, the opposite enantiomer was observed as the major product, implying that under pyridone-free conditions the rate determining step was the C–H activation.
4. C–H functionalisation of amines
Amines represent an important functional group in both organic chemistry and in pharmacologically relevant compounds. Thus, means to quickly, and directly functionalise these sought after compounds in a divergent manner are of high value. Until recently, efforts were directed at modifying the coordinating ability of the amine as part of a larger DG. Increasingly these additional steps are not required by the use of TDGs as well as exploiting the innate directing ability of the amine.C(sp2)–H functionalisation of amines
Advances in amine C–H functionalisation has focused on the development on sp3 systems, though a few advances in C(sp2)–H functionalisation have also been published. Anilines have not been demonstrated as substrates for C–H functionalisation directly on the ring, and there are well established mechanisms for their C–H arylation. Therefore, this has been applied to examples where the amine is separated from the reaction site such as benzylic amines.Kamenecka showed the use of a pyridone aldehyde directing group, previous used by Yu for amine C(sp3)–H arylation,63 was also effective for sp2 arylation of 2-phenyl-α-aminoesters (Scheme 20a).64 Despite the additional coordination site and acid labile ester on the substrate, a wide scope of arylated amino esters could be generated in up to 96% yield. The crude product was treated with SOCl2 and ethanol in a one pot manner, presumably to re-esterify any amino ester which had hydrolysed in the reaction. Silver hexafluoroantimonate was shown to be an effective silver source for this reaction.
 | ||
| Scheme 20 Arylation of benzylamines. | ||
Young used CO2 as a transient directing group for several C–H functionalisation reactions,65 including C(sp2)–H arylation of primary and secondary benzylamines (Scheme 20b).66 Based on preliminary mechanistic investigation, the role of CO2 was primarily to promote the formation of a carbamic acid in situ as both a protecting and directing group. Additionally, this process may prevent the formation of inactive palladium diamine complexes which is a hurdle all palladium catalysed amine functionalisation strategies must overcome.
Oxidative addition with ArI followed by reductive elimination installed the aromatic group. A high loading of CO2 (in the form of dry ice) was required, which poses safety concerns running reactions at high pressures and high temperatures. The reaction displayed a broad substrate scope, encompassing primary and secondary benzylamines, even benzylamines possessing secondary binding sites could be arylated, in spite of the potential for competitive coordination. The reaction tolerated a wide range of different aryl iodides as coupling partners, including an impressive, highly complex, strychnine analogue.
For substrates biased to mono-arylation, high yields of arylated benzylamines were accessed. The proposed in situ formed carbamic acid directing group also limited α-oxidation of the amine, allowing benzylamines without α-substitution to be arylated in high yields.
C(sp3)–H functionalisation of amines
Several important advances have been described recently in the advancement of amine directed C(sp3)–H activation at β-, γ- and δ-positions enabled by a growing number of bespoke directing groups and ligands.Young first reported the use of CO2 as a transient directing group in 2018, to promote the β-arylation of primary and secondary amines (Scheme 21a).65 A broad scope of α-branched amines could be arylated in good yields. Of particular note were a valine-derived ethyl ester which was functionalised without any reported hydrolysis, and a more structurally complex bornylamine, functionalised in 62% yield. The scope of the aryl iodides encompassed a diverse set of substitution patterns in good to excellent yields. Lower yields were generally observed with more sterically hindered examples, or those bearing highly electron-withdrawing substituents.
 | ||
| Scheme 21 γ-C(sp3)–H arylation of amines. | ||
Unlike other amine functionalisation methodologies using a transient directing group, this work could also functionalise secondary amines, though a large excess of CO2 was required (30 equiv.), raising some safety concerns related to the maximum admitted pressure in a closed reaction flask.
Kamenecka presented a method for the C(sp3)–H arylation of amino esters using the versatile pyridone aldehyde directing group (Scheme 21b).64 As with the previous method, one pot treatment of the crude product with thionyl chloride and ethanol was necessary. The reaction proceeded in good yields with electronically and sterically diverse iodoarenes, including four heteroaromatic examples including indole and substituted pyridines. The utility of this strategy was exemplified by its use in the formal synthesis of the immunomodulator Fingolimod.
We (Bull) demonstrated that bench stable alkyl acetals could be used as directing groups for the arylation of amines (Scheme 21c).67 Under the reaction conditions, the acetal was cleaved to the aliphatic aldehyde which was proposed to form the directing group in the reaction. Several acetal directing groups were evaluated, and those bearing ether secondary binding sites proved promising. Aryl ether secondary binding sites were preferred with 57% of the arylated product observed when using 4-trifluoromethylphenoxyacetaldehyde dimethyl acetal as the transient directing group. Commercially available phenoxyacetaldehyde dimethyl acetal was effective for most aryl iodides giving arylated products in the range of 13–59%. For challenging examples, higher yields could be obtained using 4-trifluoromethylphenoxyacetaldehyde dimethyl acetal.
Several α-branched amines of varying chain length could be functionalised, including a THP containing example. For amines bearing no α-substituents, slightly modified conditions were developed to avoid α-oxidation. Kinetic studies and deuteration studies were conducted, revealing C–H activation was likely rate determining due to an observed KIE = 2.26. No deuterium incorporation was observed at γ-position of the starting material in deuterated solvent implying an irreversible C–H activation. Same excess experiments,68 implied that product inhibition was occurring; an inactive 7-membered palladacycle was proposed as the source of inhibition, which was observed by 1H NMR. Moreover, ε-deuteration of the product supported this as a valid off-cycle intermediate.
In the same year, Yu developed a transient methylene C–H functionalisation protocol for the heteroarylation of cyclic amines, accelerated by pyridone ligands as carboxylate surrogates (Scheme 21d).58 2-Chlorosalicylaldehyde was the most effective directing group for this transformation, giving high yields of heteroarylated product with a high degree of selectivity for the γ-position. Forcing reaction conditions, including high temperatures (150 °C) that are significantly above the solvent boiling point, as well as superstoichiometric silver trifluoroacetate were required for effective reactivity. A vast scope of electron-poor heteroaromatic iodides was demonstrated, including unsubstituted pyridines which are intrinsically challenging for palladium catalysis. All amines were isolated as the Boc protected derivatives after in situ Boc-protection. There was is a single example with an electron-rich thiophene. Impressively, activated heteroaryl bromides can be used as coupling partners, without significant loss in yield. In addition to cyclohexylamine, amines of varying ring size (5- and 8-membered rings, 47% and 64% respectively), complexity (adamantylamine and norbornylamine in 40% and 60% respectively) and also acyclic substrates could all be arylated in modest to good yields, up to 61%.
Liu and Wang demonstrated an effective method for the arylation of β-alanine derived amino esters using 2-hydroxynicotinaldehyde as a transient directing group (Scheme 21e).69 Unusually, this method used a Pd/Pd co-catalytic system which was found to be most effective to promote the reaction. The use of the amino ester was key to reactivity, preventing potential competitive coordination from the substrate/product and catalyst poisoning. The reaction was treated with an acid, then the amine was Boc protected in a one pot manner.
A reasonably broad scope of aryl iodides was reported in yields up to 74%, with electron-neutral p-tolyl and electron-poor p-NO2 substrates being particularly effective in 70% and 67% respectively. More sterically encumbered o-tolyl example showed reduced reactivity, giving product in 40% yield. Heteroaromatic indole and chloropyridine could be installed in 74% and 45% respectively showing some compatibility with heteroaromatic substrates.
Yu reported a strategy of γ-oxygenation of aliphatic amines, using a 2-hydroxynicotinaldehyde transient directing group, an N-fluoropyridinium salt as a bystanding oxidant and either acid or alcohol nucleophilic coupling partners (Scheme 22a).70 This is related to an earlier work by Shi,71 however the work presented by Yu has a wider scope, encompassing α-secondary and challenging fully unsubstituted propylamine as substrates. All amines were isolated as the benzoyl protected derivatives after a one-pot benzoyl protection. Acetoxylation proceeded in 22–91% yield, with 2-aminobutylamine being a particularly effective substrate for acetoxylation. Aromatic benzoic acids were also used.
 | ||
| Scheme 22 C(sp3)–H oxidative C–O and C–F bond forming reactions using 2-hydroxynicotinaldehyde transient directing groups. | ||
Changes in the substitution of the acid coupling partner were well tolerated by the reaction, with more acidic benzoic acids being more effective (p-NO2 in 91% and m-Cl in 81%). Sterically bulky 2,4,6-triisopropylbenzoic acid could also be coupled in 74%. Potentially coordinating pyridine carboxylic acids could also be used without erosion in yield, however 75 mol% of the TDG is required. In addition to simple carboxylic acids, the late stage functionalisation of more complex carboxylic acid drugs and natural products was demonstrated in 53–73% yield, including: ibuprofen, isozepac, fenbufen and lithocholic acid.
A modified set of conditions was used to enable alkoxylation, in which the alcohol was used as a solvent, the directing group was used in stochiometric quantities and longer times were employed. While a narrower scope of alcohols was demonstrated than with the acids, primary, secondary and tertiary alcohols could all be used to give oxygenated product (primary: 37–65% yield, secondary: 40–55% yield and tertiary: 18–23% yield). A pyridone ligand was necessary to promote reactivity when using methanol or cyclohexanemethanol as coupling partners. This method was limited to the oxygenation of methyl positions.
Sunoj and Yu disclosed the use of F+ oxidants for both the methylene and methyl fluorination of aliphatic substrates using a subtly different methodology for each substrate class (Scheme 22b).72 Both sets of conditions use 2-hydroxylnicotinaldehyde directing group and a pyridone ligand. However, for methylene substrates, N-fluorosuccinimide (NFSI) was found to be the most effective oxidant and fluorine source, which was used in combination with silver trifluoroacetate, pyridone A in superstoichiometric quantities and a mixed HFIP:PhCl solvent system. The product amines were isolated as the benzoyl protected derivatives.
Fluorination could be achieved at the γ-methylene position of several cyclic and acyclic aliphatic amines in 23–82% yield. The use of a catalytic pyridone ligand (50 mol%) in a mixed HFIP:PhCl solvent system with cyclohexylamine gave 70% yield. In all other cases the ligand was used in 2 equivalents in only HFIP as solvent. Substituted analogs of cyclohexylamine bearing ester and carboxylic acid functional groups at varying positions on the ring were also tolerated. Linear aliphatic 2-aminopentane could be fluorinated in 61%; other longer chain analogs were also reactive. With two potential reactive methylene sites, as in the case of 4-aminoheptane, the difluorinated product is observed exclusively (36% yield).
For selective functionalisation of methyl substrates, a set of silver-free conditions was developed, which allowed the use of catalytic directing group and ligands. This work also studied the underlying mechanism of these transformations using DFT analysis to understand the origin of the silver dependence in the methylene conditions and the underlying roles of the pyridone ligand.
For methylene substrates, oxidative addition was proposed to be rate determining, 2-hydroxynicotinaldehyde additive acts as both directing group and carboxylate surrogate promoting C–H activation (hence high loading required) and the pyridone ligand additive was proposed to aid oxidative addition by assisting the formation of a bimetallic Pd/Ag species. In contrast, the C–H activation was found to be rate limiting for methyl C–H activation and the inclusion of silver salts led to an increase in the energy barrier to reaction, consistent with experimental observations.
Distal δ-C–H functionalisation was demonstrated by Yu in 2018, using a keto-acid and a modified pyridone ligand (Scheme 23a).58 Slightly milder reaction conditions and a similarly broad scope of (het)aryl iodides could be used, giving the arylated products in up to 82%. This method showed an impressive functional group tolerance, with good yields observed for the majority of substrates, and the reaction struggling only with very electron-poor heteroaromatic substrates. A high selectivity for δ-arylation was observed for substrates where γ-arylation was possible, conveying the robust site selectivity of this process.
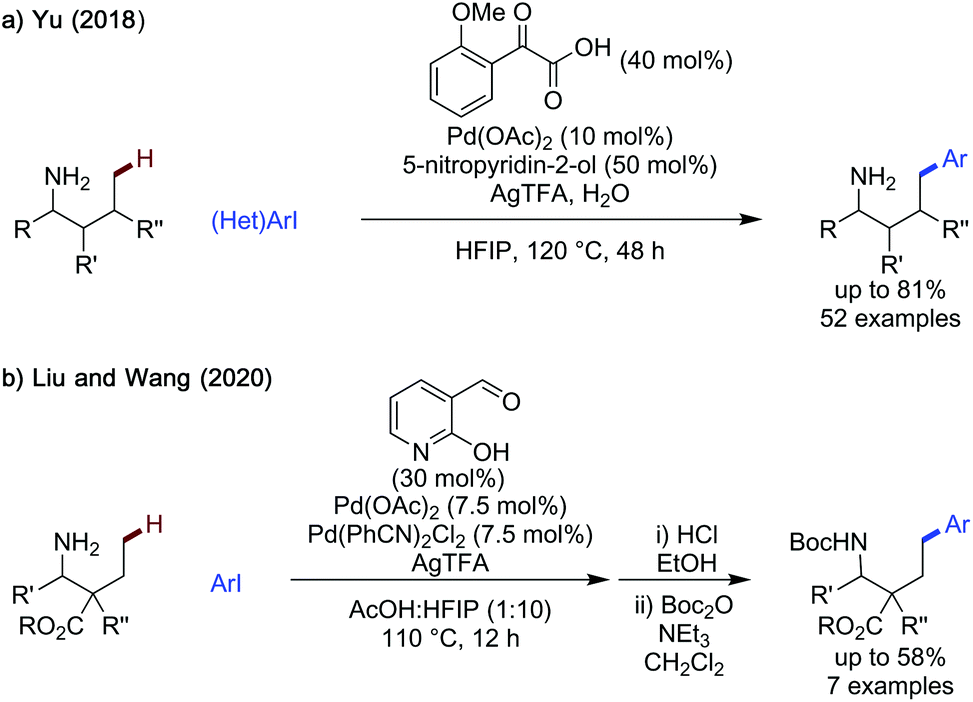 | ||
| Scheme 23 δ-C(sp3)–H arylation of amines. | ||
Liu and Wang also demonstrated the method used previously for γ-functionalisation of β-alanines could also be used for δ-C–H arylation, using 2-hydroxynicotinaldehyde as the transient directing group (Scheme 23b).69 This would result from the reaction going by a rare 6,6 palladacyclic intermediate.
Hartwig published a mechanistically intriguing method to achieve β-acetoxylation of imines via a strained 4-membered palladacyclic intermediate (Scheme 24).73 While the reaction required a preformed imine to be effective, hence not transient, it was impressive that such a strained intermediate can be accessed. Salicylaldehyde was found to be an effective directing group along with N-Boc-alanine as a ligand. Treatment of the imine with HCl cleaved both the directing group, revealing the amine functionality, and the acetoxy group, leaving an unprotected β-amino alcohol. Overall, the reaction accessed β-acetoxylated products in low to modest yields.
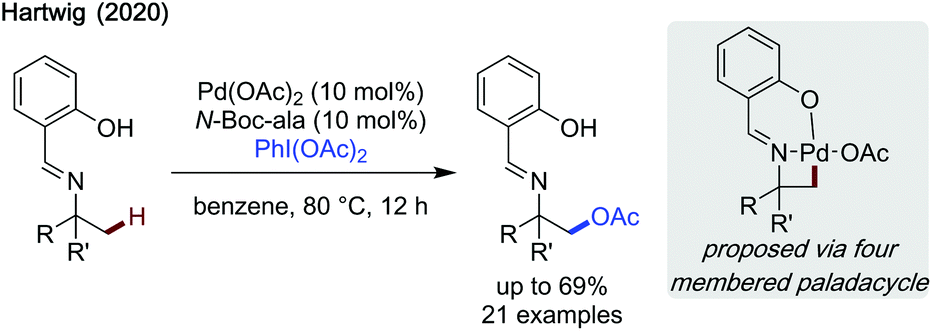 | ||
| Scheme 24 β-C(sp3)–H acetoxylation. | ||
Directing group ‘free’ approaches to amine functionalisation
In addition to developments in directing group design, several groups have published works in which the free amine acts as an innate directing group, and auxiliary ligands are employed to realise directing group free approaches to amine C(sp3)–H activation. We have chosen to also include this topic here to demonstrate recent advances in this related emerging field.Yao showed amine directed arylation of challenging amino ester substrates (Scheme 25a).74 Aryliodonium salts were shown to be required as coupling partners for this transformation, enabling arylation in up to 72% yield using silver oxide as an additive. The reaction was effective fully α-substituted alkyl amines, but ceased to function when less substituted amines were used.
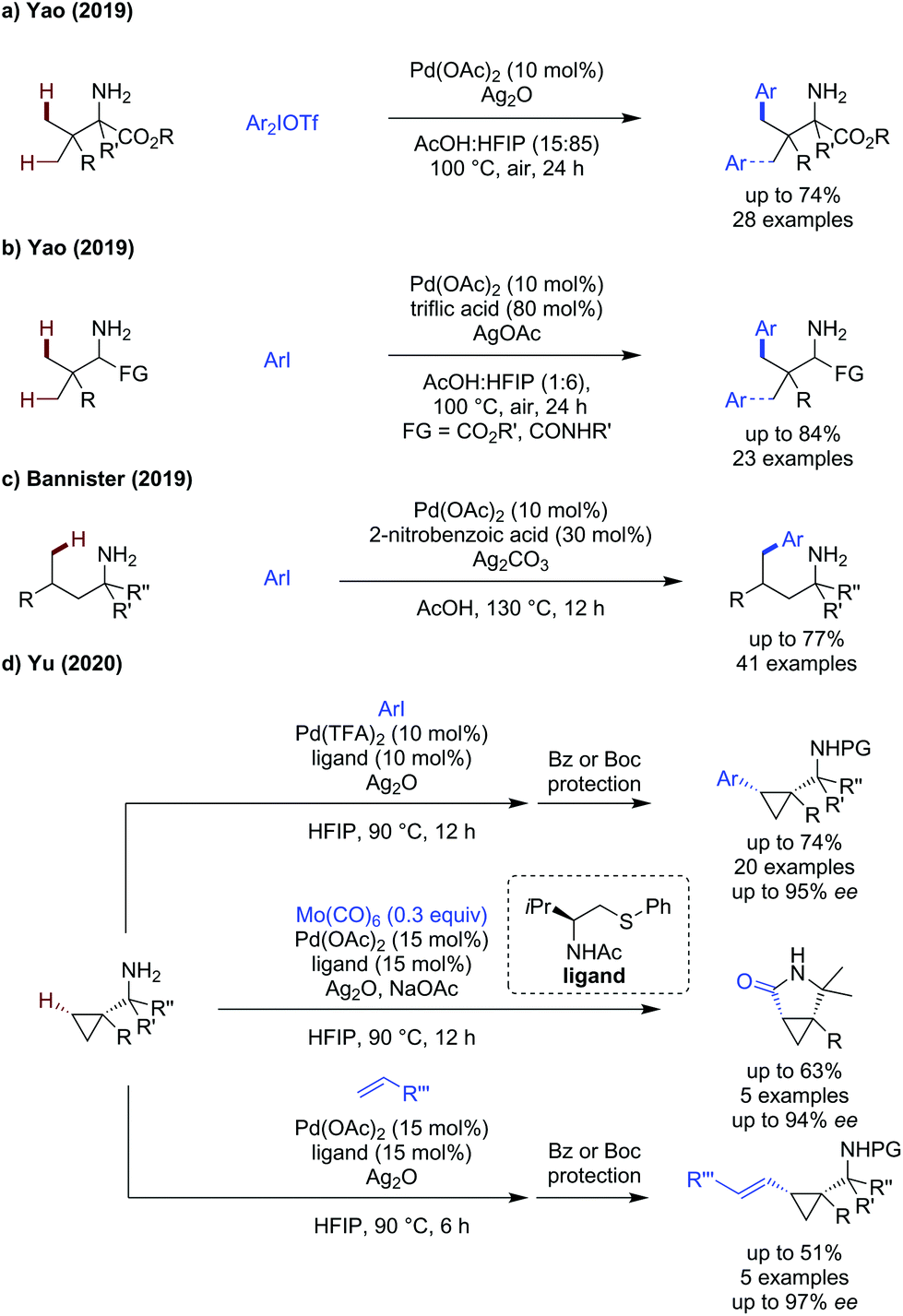 | ||
| Scheme 25 External transient directing group free approaches to primary amine C(sp3)–H functionalisation. | ||
In a follow up paper, Yao developed a directing group free approach using aryl iodides (Scheme 25b).75 The amine scope included complex oligopeptide substrates possessing many competing coordination sites for palladium. Triflic acid was identified as an effective additive for this transformation, both its high acidity and ligand effect being proposed to improve the reaction. A modest scope of aryl iodides was presented, all reacting in high yields, the compatibility of this method with di and tripeptides was truly impressive.
In 2019, Bannister disclosed an amine directed δ-functionalisation method, using 2-nitrobenzoic acid as a ligand additive (Scheme 25c).76 Under their standard conditions, in the absence of an external ligand, the amine is intrinsically very reactive to arylation, with 60% of the δ-arylated product observed. An improved yield was obtained by the use of a carboxylic or sulfonic acid additive, o-nitrobenzoic acid was shown to be the most effective, giving 73% yield. The reaction was reasonably tolerant to changes to the aryl iodide used, though electron-poor examples generally gave lower yields. The chain length of the aliphatic substituents could be changed without major changes in the yield, though in substrates with multiple reactive sites, low selectivity for mono, di or tri arylated products were observed. This method was also effective for γ-and δ-C(sp2)–H arylation.
Yu published an enantioselective arylation, carbonylation and alkenylation of cyclopropylamines (Scheme 25d).77 A bidentate NAc thioether ligand was most effective at promoting: arylation, carbonylation and alkenylation of cyclopropyl amines in moderate to good yields (30–74%) and excellent enantioselectivity (up to 97% ee). A one pot benzoyl protection allowed isolation of the benzoylated product. The scope of the arylation was broad, with electron-rich and electron-poor aryl iodides tolerated in the reaction without a significant change in yield (p-tolyl, 68% vs. p-CF3, 72%). Heteroaromatic coupling partners could also be used including substituted pyridines where the nitrogen was suitable blocked by an adjacent substituent, however use of 4-iodopyridine was not tolerated. Molybdenum hexacarbonyl could be used as a source of CO to achieve carbonylation, which proceeded in 33–63% to form the γ-lactam products in high ees, with 5 examples reported. A similarly sized scope of alkenylated products were disclosed. The reaction was less tolerant of styrenes as coupling partners as either low to moderate yields were observed in all cases, however an improved enantioselectivity could be obtained.
Gaunt reported the use of 2-bromobenzoic acids as coupling partners for the γ-C(sp3)–H arylation of secondary amines.78 The extra carboxylate coordination site on the aromatic coupling partner improved reactivity, and a decarboxylation event gave the arylated product (Scheme 26).79 An amine directed cyclopalladation is proposed to give palladacycle A. Ligand exchange with the bromo carboxylic acid gave intermediate B in which the C–Br bond is forced close to the palladium centre, initiating oxidative addition to give PdIV intermediate C. The silver source was proposed to abstract the halide, giving intermediate D which was active to a concerted decarboxylative 1,2-Pd migration/reductive elimination sequence to give 7-membered palladacyclic intermediate E (isolated and characterised by X-ray). Protodemetalation of E gives the arylated product, while regenerating the PdII catalyst.
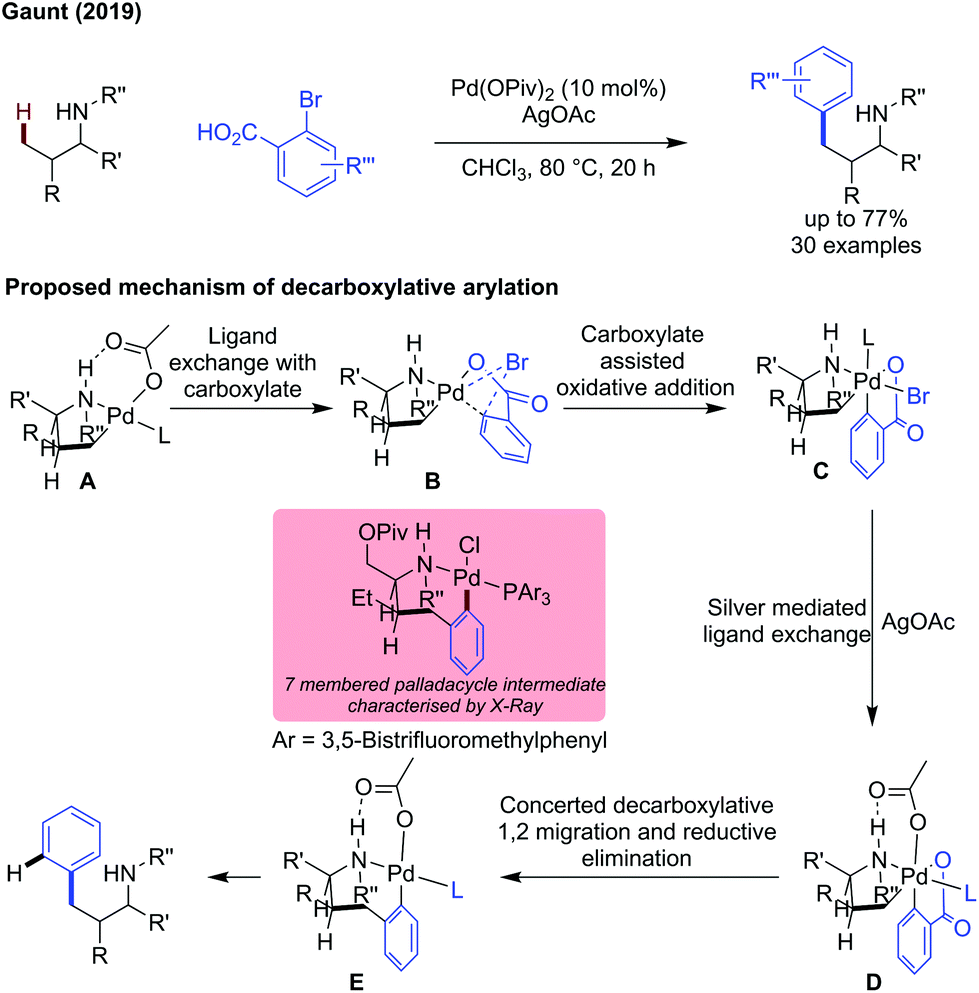 | ||
| Scheme 26 External directing group free decarboxylative arylation of secondary amines with 2-bromobenzoic acids. Proposed mechanism is shown. | ||
An ortho-methyl substituent was poorly tolerated, as the increased steric hinderance prevented oxidative addition. The amine substituent could be readily changed without significant decrease in yield. Both α and β positions of the amine's aliphatic backbone must be substituted for reactivity.
Gaunt also demonstrated the first method allowing direct C–H functionalisation of tertiary alkylamines (Scheme 27).80 The use of N-Ac-tert-leucine as a ligand additive limited competitive β-hydride elimination. Silver carbonate and benzoquinone were used as oxidants (also a ligand in the case of benzoquinone). The reaction proceeded under mild conditions requiring neither an inert atmosphere, nor steric activation. While a slight reduction in yield was observed when using substituted alkyl amines, the scope was broad and included potentially coordinating ethers, esters and pyridines, installed on either the alkyl chain, or on the cyclic portion of the substrate. The scope of arylboronic acids was similarly broad, with high yields, up to 93% when using 3-fluorophenylboronic acid and N,N-dimethylbutan-2-amine as coupling partners. The late stage arylation of tertiary amine containing drug derivatives was also demonstrated, in moderate to excellent yields. An additional point of utility is this strategy's ability to desymmetrise alkylamines possessing enantiotopic methyl groups in good yields and moderate ees. Finally, a single example of methylene C–H activation of a tertiary amine containing THP substrate was shown, though a lower yield was reported. One main issue with this method, however, is the need to use the amine in excess, which may limit its feasibility in reactions with more valuable alkylamines.
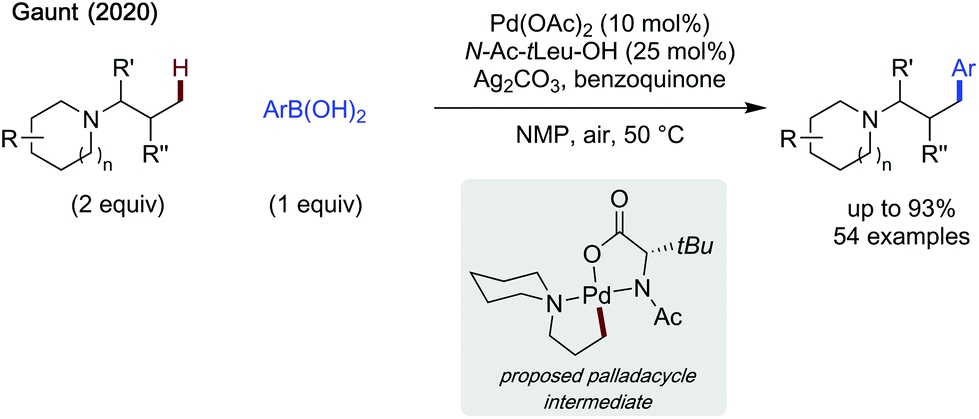 | ||
| Scheme 27 Oxidative C(sp3)–H arylation of tertiary alkylamines. | ||
In 2018, Li and Li demonstrated a palladium catalysed, copper promoted, free amine directed C(sp2)–H carbonylation of benzylamines to the respective lactam products (Scheme 28a).81 This method could be applied to the carbonylation of primary and secondary benzylamines bearing diverse functionality on the aromatic ring. This carbonylation/lactamisation could also be used to generate 6-membered δ-lactams by using the appropriate homologated starting material. Li later showed a rhodium catalysed carboxylation/lactamisation of biaryl amines to afford δ-lactams (Scheme 28b).82 A broad scope of biaryl amines were effective in the reaction, including some pyridine containing heteroaromatics. Secondary arylamines were not demonstrated to be reactive.
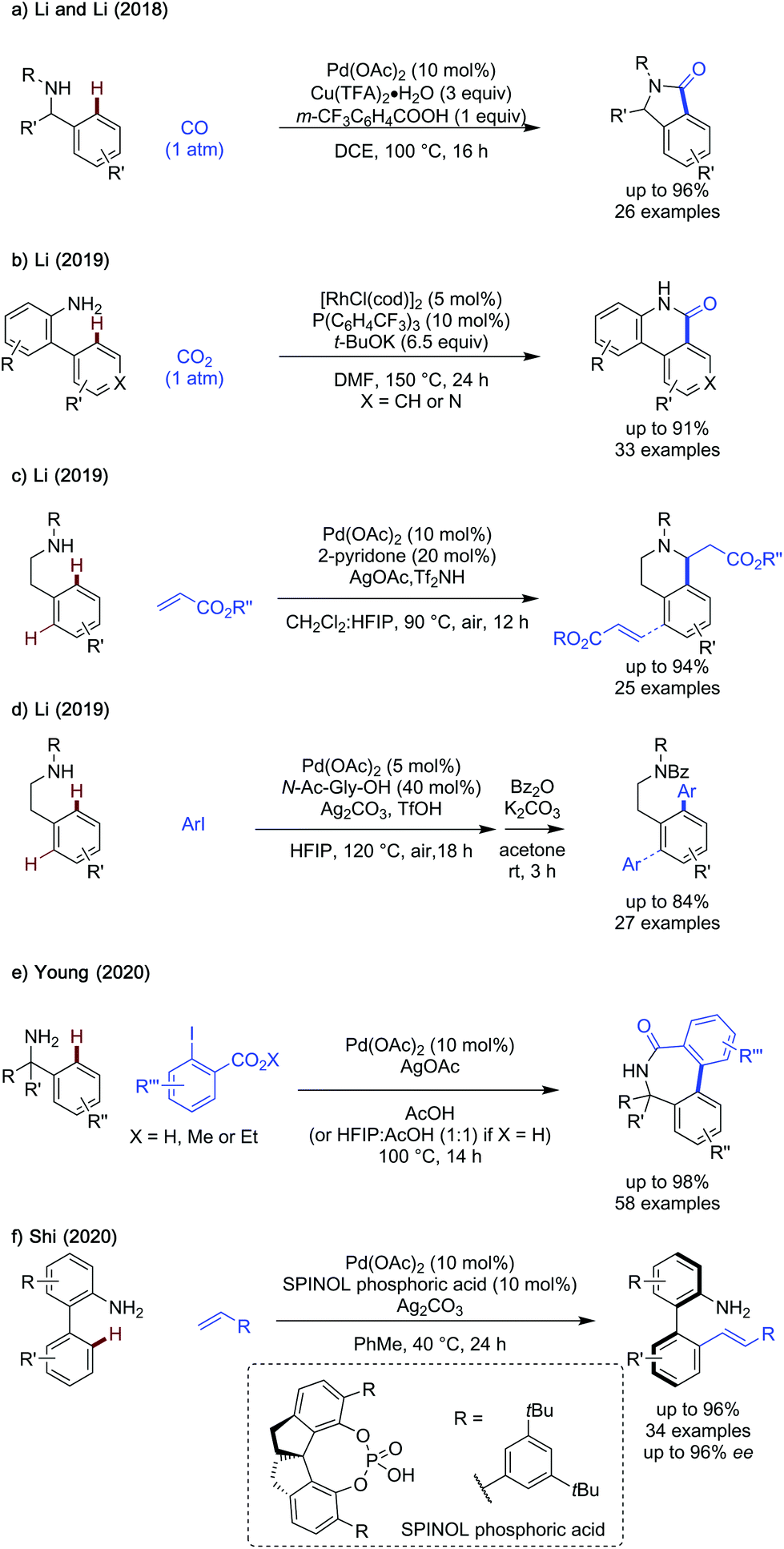 | ||
| Scheme 28 Free amine directed C–H arylation and alkenylation. | ||
The innate directing ability of primary and secondary 2-phenylethylamines was demonstrated in 2019 by reports of ‘transient directing group free’ methods of alkenylation83 and arylation84 of these scaffolds. Alkenylation was achieved by palladium catalysis using a pyridone ligand, silver oxidant and highly acidic trifluoromethanesulfonimide additive (Scheme 28c). The alkenylated intermediate spontaneously cyclised to the tetrahydroisoquinoline in up to 94%. Difunctionalisation was observed in cases where there was no ortho- or meta-substituent guiding the selectivity. The scope of aryl amines was broad for reaction with ethyl and n-butyl acrylate.
Arylation could be achieved under conditions modified from the alkenylation conditions, using a bidentate amino acid derived ligand instead of the pyridone additive and silver carbonate and triflic acid. Aromatic substrates bearing electron rich (o-OMe), electron neutral (o-Me, m-Me) and electron poor (o-Cl, m-Cl) substituents were be arylated in good to excellent yields (>50%), however in cases where the aromatic amine bore no ortho- or meta-substituent, diarylation was the major product. A phenylalanine derived ester and amphetamine could both be effectively arylated under these conditions.
In 2020, Young reported a procedure for the o-arylation/cyclisation cascade for the formation of biaryl lactams from benzylamines and 2-iodobenzoates (Scheme 28a).85 While related to an earlier work using CO2 as a transient directing group, this work does not require the inclusion of CO2 and is directed by the free amine. An extensive investigation of benzylamines was conducted including a variety of substituents on the aromatic moiety of the benzylamine, with excellent yields observed in the majority of cases (up to 98%). The reaction is relatively insensitive to changes on the electronic and steric properties of the aromatic component. HFIP was required as a co-solvent when using 2-iodobenzoic acid coupling partners, though lower yields were observed compared to the case of an ester. The exact reason for why a directing group free approach is feasible when using 2-iodobenzoate coupling partners is unclear at the moment.
In addition to numerous atroposelective methodologies, Shi reported a ‘transient directing group free’ approach to stereoselective alkenylation of biaryl amines via an oxidative palladium catalysed approach (Scheme 28b).86 The use of a chiral phosphonic acid with a spirocyclic backbone was key for effective stereoselectivity, with SPINOL derived ligands being ineffective. High ees were observed throughout in excess of 90%. Acrylate coupling partners gave particularly high yields, up to 96% when using butyl acrylate, and less activated styrene substrates could also be coupled in similar yields. Impressively, 2-substituted bi-aryl substrates were compatible and afforded alkenylated products in high ees despite the lower energy barrier to racemisation. Further derivatisation of the alkenylated product could be performed without erosion of ee.
5. Transient imine directing groups in other transition metal catalysed transformations
Transient imine directing groups have begun to find applications in transformations other than C–H activation. Liu and Engle showed hydroarylation of styrenes can be achieved enantioselectivly using L-tert-leucine (Scheme 29).87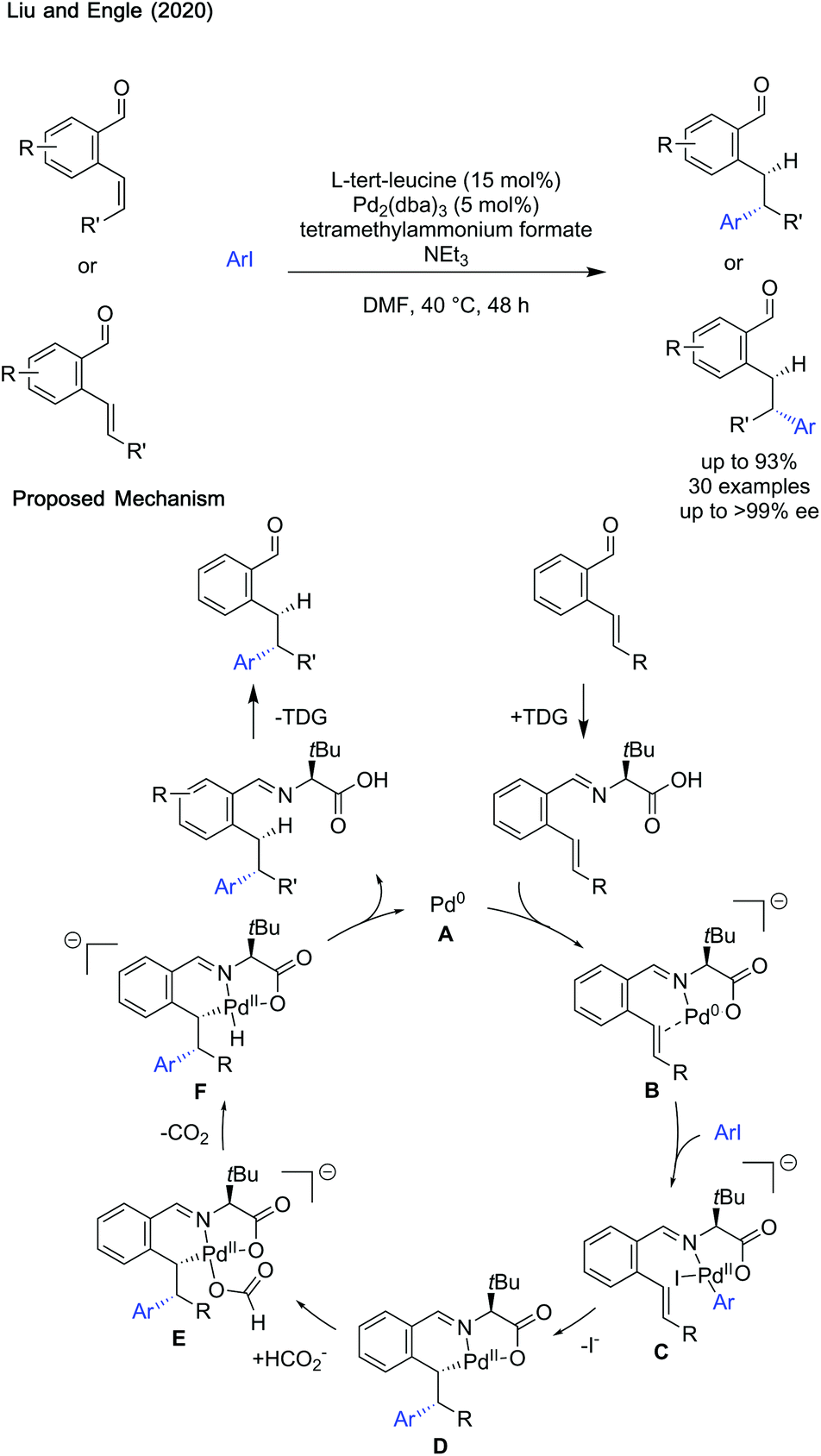 | ||
| Scheme 29 Enantioselective hydroarylation of alkene enabled by a transient directing group. | ||
The reaction was proposed to proceed by coordination of Pd0 with the imine formed in situ to form intermediate B. Oxidative addition with ArI leads to the formation of tethered aryl palladium(II) complex C which can then undergo an enantiodetermining carbopalladation with loss of an iodide ligand to give D. Association of formate with the palladium centre forms anionic complex E. Decarboxylation and reductive elimination reforms palladium(0) and affords the product imine. Hydrolysis afforded the aldehyde product and reformed the transient directing group.
The mild reaction conditions aided a wide functional group tolerance for both the aryl iodide and aldehyde coupling partner. The stereochemistry of the starting aldehyde was conserved in the product, with the chirality of the directing group transferred to give exceptional enantioselectivity (99% ee in most cases). Good to excellent yields are reported, though there are a few limitations to the current methodology, especially compatibility with aniline and imidazole containing aryl iodides. Additionally, alkynyl halides could not be used as coupling partners. The mild conditions, and vast scope, makes this a powerful method of directed hydroarylation and the use of a transient directing group marks an improvement on other directed hydroarylation methodologies, avoiding discrete directing group installation and cleavage steps.
6. Advances in mechanistic understanding
Several advances in mechanistic understanding have been discussed in earlier sections of this review, including possible modes of catalyst deactivation,67 and computational investigations of additional role of pyridone ligands.72There have also been several publications purely dealing with gaining a greater understanding of the mechanism underlying these impressive catalytic transformations, including uncovering potential roles of specific reagents, the mechanisms of C–H activation and the origins of regioselectivity for different substrates.
Wang and Dang studied arylation reactions previously reported by Yu in 2016,7 investigating the mechanistic origins for the different regioselectivities observed for 2-alkylbenzaldehydes and 3-methylpentan-2-one when using glycine as an amino acid directing group with palladium catalysis (Scheme 30).88 Namely why is a 6-membered palladacycle favoured in the former case, and a 5-membered palladacycle in the latter. For both substrates, acetate mediated CMD was determined to have the highest energetic barrier in the process, and so was regiodetermining in both cases. For 2-methylbenzaldehyde, there was the choice of forming a 5-membered palladacycle, cleaving the C(sp2)–H bond, or forming a 6-membered palladacycle cleaving the benzylic C(sp3)–H bond. It was found that the rigid conformation of the substrate makes a CMD forming the 6-membered palladacycle more energetically favourable. This was ascribed to the lower ring strain, the intrinsically lower bond strength of the benzylic C–H bond and better orbital overlap between the palladium d orbitals and C–H bond. Calculations were also performed with chiral L-tert-leucine as the directing group, showing a key steric clash between the directing group side chain and the alkyl substituent on the benzylic site was responsible for the enantioselectivity.
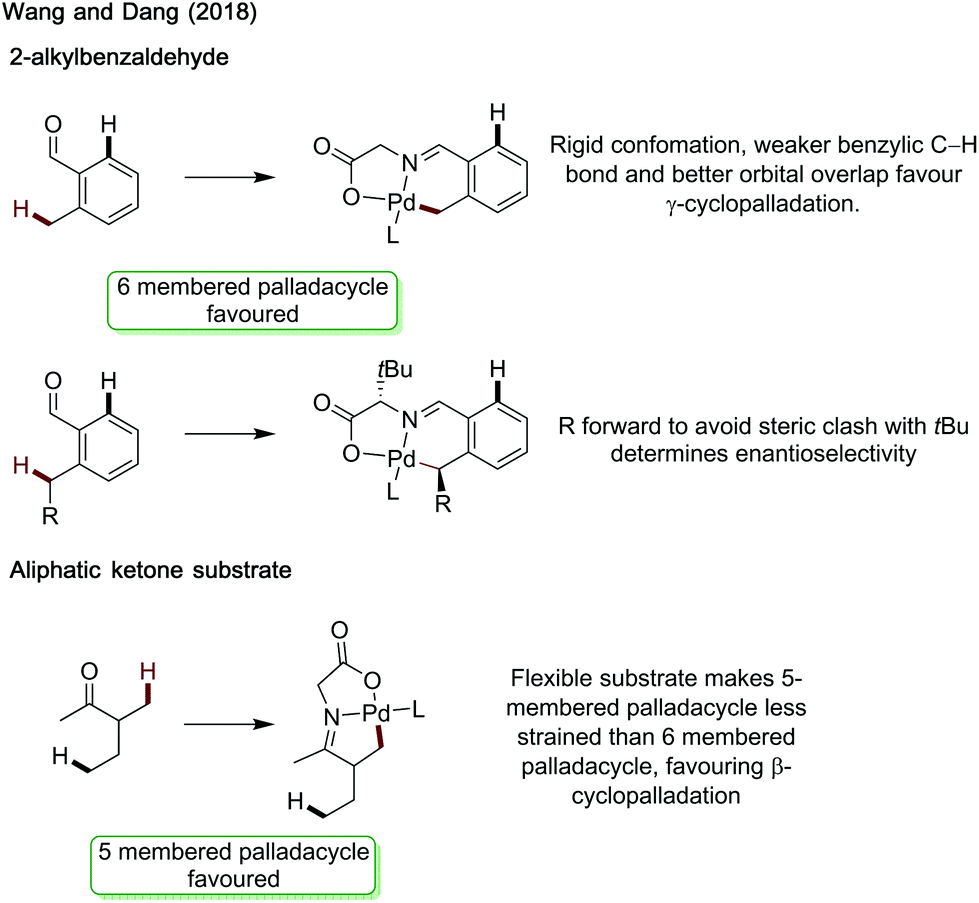 | ||
| Scheme 30 Origins of regioselectivity with amino acid directing group. | ||
For the aliphatic substrate, the increased flexibility leads to a less strained 5-membered palladacycle and so β-cyclopalladation is more energetically preferred.
Chen reported an in-depth computational study on the γ-arylation of amines as reported by Dong,52a specifically to understand the stabilising effect of the imine transient directing group (Scheme 31).89 Computationally, the CMD step was determined to have the highest energetic barrier, with an outer sphere deprotonation by pivalate being energetically preferred to a more traditional inner sphere CMD. A comparison of the activation energy of the CMD, both with and without the directing group, reveals that the activation energy of the C–H activation step is significantly lower when the palladium is bound in a bidentate manner with the imine directing group, compared to being bound in a monodentate manner to the free amine. Hence the role of the directing group was to lower the activation barrier of the rate limiting step. The pyridine additive is likely to act as a non-coordinating base to neutralise HBF4 formed as part of the reaction.
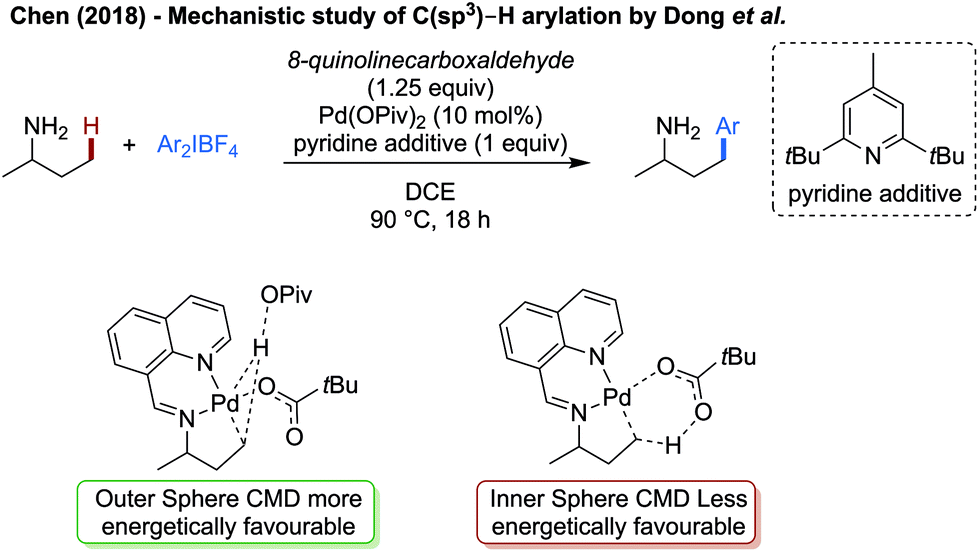 | ||
| Scheme 31 Mechanistic work elucidating the role of 8-formylquinoline as a transient directing group. | ||
Silver additives are commonly used in palladium catalysed transient C–H arylation strategies and are necessary for reactivity. While a common role of silver(I) is generally proposed to be the abstraction of halide from the catalyst to ensure turnover of palladium, several other roles have also been proposed including, an activator for palladium, aiding transmetalation by the formation of silver aryl species and as a reoxidant in oxidative couplings.90 In addition to these roles, Wang and Dang published a mechanistic investigation in 2019, detailing another crucial role of silver beyond simple halide abstraction (Scheme 32).91
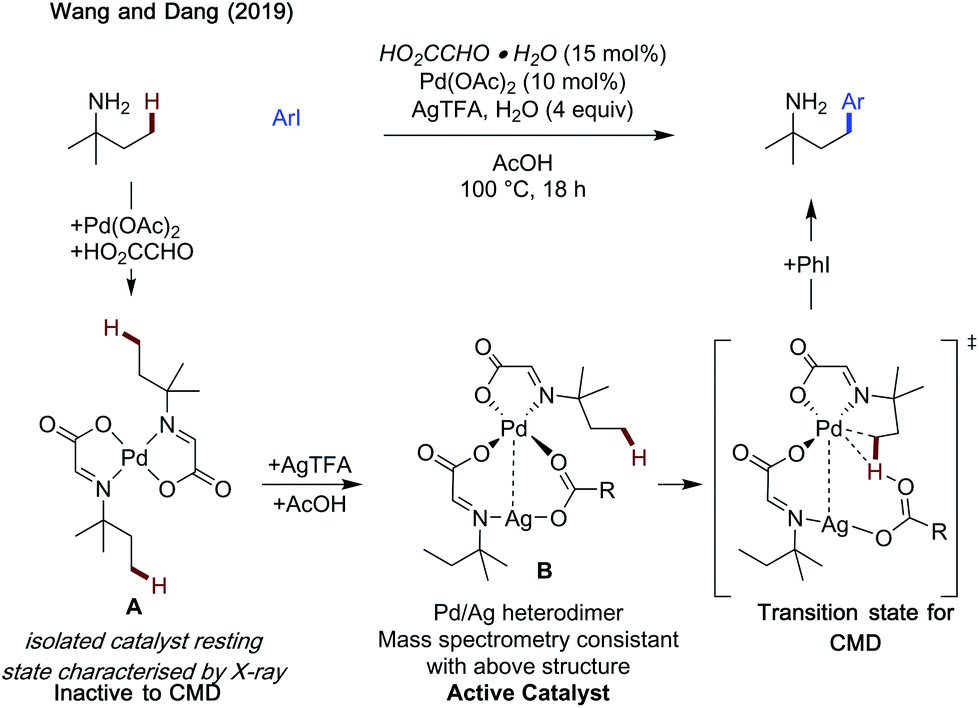 | ||
| Scheme 32 Mechanistic investigation of the role of silver(I) salts for transient C–H activations. | ||
A combination of computational and experimental investigations of the γ-C(sp3)–H arylation of t-amylamine were performed. Palladium species A was identified computationally as the catalyst resting state. This was supported experimentally as when the amine substrate was treated with stoichiometric directing group and palladium acetate, A could be isolated and characterised by X-ray crystallography. A could also be used as the catalyst in the reaction instead of palladium acetate without any reduction in yield of the arylated product, implying it is a competent intermediate in the reaction.
The CMD was determined to be the rate limiting step in the process, and a direct CMD occurring directing from intermediate A was determined computationally to be energetically unfavourable. The silver trifluoroacetate can instead break up the catalyst resting state to form bimetallic intermediate B which allows access to a lower energy CMD transition state in which the carboxylate is bound through silver. This heterodimeric species was observed experimentally via MALDI-TOF mass spectrometry, supporting its role in this reaction. This work has identified an important additional role of silver additives in this C–H activation. It is likely that other C–H arylation strategies proceed by similar bimetallic Pd/Ag intermediates, a potential reason for the dependence on the use of silver trifluoroacetate as an additive.
7. Conclusion
This review has detailed the progress made in this fast moving, and highly competitive field since 2018. The sheer number of publications is an accurate reflection of the interest in the continuing development of these remarkable mechanistically interesting, step efficient processes. Powerful advances in new C(sp2)–H and C(sp3)–H activation methodologies, enabled by new directing group design, has unlocked many potential avenues for future research in both C–C and C–X bond forming reactions.While palladium catalysis is by far the most popular in these transient C–H activation methodologies, advances in the use of other metals has enabled the development of new C–C and C–X bond forming methods and novel modes of attaining enantioselective reactions.
Advances in the use of more benign solvents, electrosynthetic methods, and identifying ligands to render reactions milder have all demonstrated a transient C–H activation can be made more sustainable, which will see further development in the future. High TDG loadings are often required (40 to >100 mol%) which we expect will be greatly reduced in the future as new and more efficacious transient directing groups are discovered. Additionally, the advances in mechanistic understanding through experimental and computational methods have uncovered important insight which will undoubtedly aid in the development in future methods, extending to the late stage functionalisation of complex substrates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
For financial support, we gratefully acknowledge The Royal Society [University Research Fellowship (UF140161, to J. A. B.) and Research Grants (RG150444 and RGF\EA\180031)].Notes and references
- For general reviews on C–H activation, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS; (b) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS; (e) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (f) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS.
- M. Wasa, K. S. L. Chan, X. G. Zhang, J. He, M. Miura and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570–18572 CrossRef CAS.
- A. Uttry and M. Van Gemmeren, Synthesis, 2020, 52, 479–488 CrossRef CAS.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- C. H. Jun, D. Y. Lee and J. B. Hong, Tetrahedron Lett., 1997, 38, 6673–6676 CrossRef CAS.
- F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS.
- For previous reviews of C–H activation using a transient directing group, see: (a) Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 49, 4808–4826 CrossRef CAS; (b) O. K. Rasheed and B. Sun, ChemistrySelect, 2018, 3, 5689–5708 CrossRef CAS; (c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (d) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC; (e) S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC; (f) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955–2969 CrossRef CAS. For review of enantioselective strategies see: (g) K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 68, 42–61 Search PubMed; (h) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202008437; (i) D. Bag, P. K. Verma and S. D. Sawant, Chem. – Asian J., 2020 DOI:10.1002/asia.202000657. For amine functionalisation, see: (j) H. Ha, J. Lee, M. H. Park, B. Jung and M. Kim, Bull. Korean Chem. Soc., 2020, 41, 582–587 CrossRef CAS.
- M. Y. Zhang and R. A. Barrow, J. Org. Chem., 2018, 83, 6776–6782 CrossRef CAS.
- G. Li, J. Jiang, H. Xie and J. Wang, Chem. – Eur. J., 2019, 25, 4688–4694 CrossRef CAS.
- P. W. Tan, N. A. B. Juwaini and J. Seayad, Org. Lett., 2013, 15, 5166–5169 CrossRef CAS.
- Z. Guan, S. Chen, Y. Huang and H. Yao, Org. Lett., 2019, 21, 3959–3962 CrossRef CAS.
- D. Sun, J. Du, H. Fang, J. Lan, D. Wu and J. You, Chem. Commun., 2019, 55, 7518–7521 RSC.
- A. E. Hande, V. B. Ramesh and K. R. Prabhu, Chem. Commun., 2018, 54, 12113–12116 RSC.
- X. Wang, S. Song and N. Jiao, Chin. J. Chem., 2018, 36, 213–216 CrossRef CAS.
- J. Shen, X. Liu, L. Wang, Q. Chen and M. He, Synth. Commun., 2018, 48, 1354–1362 CrossRef CAS.
- S. Kim, S. H. Han, N. K. Mishra, R. Chun, Y. H. Jung, H. S. Kim, J. S. Park and I. S. Kim, Org. Lett., 2018, 20, 4010–4014 CrossRef CAS.
- Z.-Y. Li, H. H. C. Lakmal, X. Qian, Z. Zhu, B. Donnadieu, S. J. McClain, X. Xu and X. Cui, J. Am. Chem. Soc., 2019, 141, 15730–15736 CrossRef CAS.
- G. Li, Q. Liu, L. Vasamsetty, W. Guo and J. Wang, Angew. Chem., 2020, 59, 3475–3479 CrossRef CAS.
- X.-Y. Chen and E. J. Sorensen, Chem. Sci., 2018, 9, 8951–8956 RSC.
- X.-Y. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS.
- J. Huang, J. Ding, T.-M. Ding, S. Zhang, Y. Wang, F. Sha, S.-Y. Zhang, X.-Y. Wu and Q. Li, Org. Lett., 2019, 21, 7342–7345 CrossRef CAS.
- B. Khan, V. Dwivedi and B. Sundararaju, Adv. Synth. Catal., 2020, 362, 1195–1200 CrossRef CAS.
- D. Mu, G. He and G. Chen, Chem. – Asian J., 2018, 13, 2423–2426 CrossRef CAS.
- R. Shi, F. Liao, H. Niu and A. Lei, Org. Chem. Front., 2018, 5, 1957–1961 RSC.
- Y.-F. Wang, W.-G. Xu, B. Sun, Q.-Q. Yu, T.-J. Li and F.-L. Zhang, J. Org. Chem., 2019, 84, 13104–13111 CrossRef CAS.
- J. Fan, L. Li, J. Zhang and M. Xie, Chem. Commun., 2020, 56, 2775–2778 RSC.
- F. Li, Y. Zhou, H. Yang, Z. Wang, Q. Yu and F.-L. Zhang, Org. Lett., 2019, 21, 3692–3695 CrossRef CAS.
- H. Qiao, B. Sun, Q. Yu, Y.-Y. Huang, Y. Zhou and F.-L. Zhang, Org. Lett., 2019, 21, 6914–6918 CrossRef CAS.
- Q. Yong, B. Sun and F.-L. Zhang, Tetrahedron Lett., 2019, 60, 151263 CrossRef CAS.
- X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS.
- Y.-J. Wu, Q.-J. Yao, H.-M. Chen, G. Liao and B.-F. Shi, Sci. China: Chem., 2020, 63, 875–880 CrossRef CAS.
- N. Thrimurtulu, A. Dey, A. Singh, K. Pal, D. Maiti and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 1441–1446 CrossRef CAS.
- G. Liao, B. Li, H.-M. Chen, Q.-J. Yao, Y.-N. Xia, J. Luo and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151–17155 CrossRef CAS.
- G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 11464–11468 CrossRef CAS.
- S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956–1961 CrossRef CAS.
- H.-M. Chen, S. Zhang, G. Liao, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Organometallics, 2019, 38, 4022–4028 CrossRef CAS.
- J. Zhang, Q. Xu, J. Wu, J. Fan and M. Xie, Org. Lett., 2019, 21, 6361–6365 CrossRef CAS.
- H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., 2020, 59, 6576–6580 CrossRef CAS.
- U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Angew. Chem., 2020, 59, 13451–13457 CrossRef CAS.
- S. Xie, S. Li, W. Ma, X. Xu and Z. Jin, Chem. Commun., 2019, 55, 12408–12411 RSC.
- S. Bag, S. Jana, S. Pradhan, S. Bhowmick, N. Goswami, S. Kumar Sinha and D. Maiti, ChemRxiv, 2020, DOI:10.26434/chemrxiv.11521827.v1.
- M. Tang, Q. Yu, Z. Wang, C. Zhang, B. Sun, Y. Yi and F.-L. Zhang, Org. Lett., 2018, 20, 7620–7623 CrossRef CAS.
- H. Park, K. Yoo, B. Jung and M. Kim, Tetrahedron, 2018, 74, 2048–2055 CrossRef CAS.
- Z. Wang, W. Dong, B. Sun, Q. Yu and F. L. Zhang, Tetrahedron, 2019, 75, 4031–4041 CrossRef CAS.
- F. Wen and Z. Li, Adv. Synth. Catal., 2020, 362, 133–138 CrossRef CAS.
- J. Chen, C. Bai, H. Ma, D. Liu and Y.-S. Bao, Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2020.02.055.
- C. Reddy, J. Y. Shaikh and R. G. Bhat, J. Org. Chem., 2020, 85, 6924–6934 CrossRef CAS.
- H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755–762 CrossRef CAS.
- J. Chen, C. Bai, X. Tong, D. Liu and Y.-S. Bao, RSC Adv., 2020, 10, 12192–12196 RSC.
- S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC.
- For reports of amine directed C(sp3)–H activation prior to this review, see: (a) Y. Xu, M. C. Young, C. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084–9087 CrossRef CAS; (b) Y. Liu and H. Ge, Nat. Chem., 2017, 9, 26–32 CrossRef CAS; (c) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS; (d) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073–1076 CrossRef CAS.
- S. St John-Campbell and J. A. Bull, Chem. Commun., 2019, 55, 9172–9175 RSC.
- B.-B. Gou, H.-F. Liu, J. Chen and L. Zhou, Org. Lett., 2019, 21, 7084–7088 CrossRef CAS.
- X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, R.-L. Guo, Y.-R. Gao and Y.-Q. Wang, Org. Lett., 2019, 21, 2731–2735 CrossRef CAS.
- C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085–2089 CrossRef CAS.
- S. St John-Campbell, A. J. P. White and J. A. Bull, Org. Lett., 2020, 22, 1807–1812 CrossRef CAS.
- Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS.
- B. Li, B. Lawrence, G. Li and H. Ge, Angew. Chem., 2020, 59, 3078–3082 CrossRef CAS.
- J. Wang, C. Dong, L. Wu, M. Xu, J. Lin and K. Wei, Adv. Synth. Catal., 2018, 360, 3709–3715 CrossRef CAS.
- Y. Cheng, J. Zheng, C. Tian, Y. He, C. Zhang, Q. Tan, G. An and G. Li, Asian J. Org. Chem., 2019, 8, 526–531 CrossRef CAS.
- L.-J. Xiao, K. Hong, F. Luo, L. Hu, W. R. Ewing, K.-S. Yeung and J.-Q. Yu, Angew. Chem., 2020, 59, 9594–9600 CrossRef CAS.
- Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS.
- H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Chem. – Eur. J., 2018, 24, 9535–9541 CrossRef CAS.
- M. Kapoor, D. Liu and M. C. Young, J. Am. Chem. Soc., 2018, 140, 6818–6822 CrossRef CAS.
- M. Kapoor, P. Chand-Thakuri and M. C. Young, J. Am. Chem. Soc., 2019, 141, 7980–7989 CrossRef CAS.
- S. St John-Campbell, A. K. Ou and J. A. Bull, Chem. – Eur. J., 2018, 24, 17838–17843 CrossRef CAS.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS.
- Z. Wang, Y. Fu, Q. Zhang, H. Liu and J. Wang, J. Org. Chem., 2020, 85, 7683–7693 CrossRef CAS.
- Y.-Q. Chen, Y. Wu, Z. Wang, J. X. Qiao and J.-Q. Yu, ACS Catal., 2020, 10, 5657–5662 CrossRef.
- K. Chen, D. Wang, Z.-W. Li, Z. Liu, F. Pan, Y.-F. Zhang and Z.-J. Shi, Org. Chem. Front., 2017, 4, 2097–2101 RSC.
- Y.-Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS.
- B. Su, A. Bunescu, Y. Qiu, S. J. Zuend, M. Ernst and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 7912–7919 CrossRef CAS.
- P. K. Pramanick, Z. Zhou, Z.-L. Hou and B. Yao, J. Org. Chem., 2019, 84, 5684–5694 CrossRef CAS.
- F. Yuan, Z.-L. Hou, P. K. Pramanick and B. Yao, Org. Lett., 2019, 21, 9381–9385 CrossRef CAS.
- H. Lin, X. Pan, A. L. Barsamian, T. M. Kamenecka and T. D. Bannister, ACS Catal., 2019, 9, 4887–4891 CrossRef CAS.
- Z. Zhuang and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 12015–12019 CrossRef CAS.
- W. G. Whitehurst, J. H. Blackwell, G. N. Hermann and M. J. Gaunt, Angew. Chem., Int. Ed., 2019, 58, 9054–9059 CrossRef CAS.
- This is reminiscent of carboxylate directed C–H activation, decarboxylative functionalisation reactions. For example, see: (a) J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS; (b) A. Biafora and L. J. Gooßen, Synlett, 2017, 28, 1885–1890 CrossRef CAS. For carboxylic acids as a directing group for C–H functionalisation, see: (c) M. Pichette Drapeau and L. J. Gooßen, Chem. – Eur. J., 2016, 22, 18654–18677 CrossRef CAS.
- J. Rodrigalvarez, M. Nappi, H. Azuma, N. J. Flodén, M. E. Burns and M. J. Gaunt, Nat. Chem., 2020, 12, 76–81 CrossRef CAS.
- C. Zhang, Y. Ding, Y. Gao, S. Li and G. Li, Org. Lett., 2018, 20, 2595–2598 CrossRef CAS.
- Y. Gao, Z. Cai, S. Li and G. Li, Org. Lett., 2019, 21, 3663–3669 CrossRef CAS.
- S. Fan, Y. Ding, X. Chen, Y. Gao, L. Fu, S. Li and G. Li, J. Org. Chem., 2019, 84, 13003–13012 CrossRef CAS.
- Y. Ding, S. Fan, X. Chen, Y. Gao, S. Li and G. Li, Org. Lett., 2019, 21, 4224–4228 CrossRef CAS.
- P. Chand-Thakuri, V. G. Landge, M. Kapoor and M. C. Young, J. Org. Chem., 2020, 85, 6626–6644 CrossRef CAS.
- B.-B. Zhan, L. Wang, J. Luo, X.-F. Lin and B.-F. Shi, Angew. Chem., 2020, 59, 3568–3572 CrossRef CAS.
- L. J. Oxtoby, Z.-Q. Li, V. T. Tran, T. G. Erbay, R. Deng, P. Liu and K. M. Engle, Angew. Chem., 2020, 59, 8885–8890 CrossRef CAS.
- W. Liu, J. Zheng, Z. Liu, W. Hu, X. Wang and Y. Dang, ACS Catal., 2018, 8, 7698–7709 CrossRef CAS.
- X.-X. Hu, J.-B. Liu, L.-L. Wang, F. Huang, C.-Z. Sun and D.-Z. Chen, Org. Chem. Front., 2018, 5, 1670–1678 RSC.
- K. L. Bay, Y.-F. Yang and K. N. Houk, J. Organomet. Chem., 2018, 864, 19–25 CrossRef CAS.
- W. Feng, T. Wang, D. Liu, X. Wang and Y. Dang, ACS Catal., 2019, 9, 6672–6680 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |