
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-phase synthesis and evaluation of tumour-targeting phenylboronate-based MRI contrast agents†
Jonathan
Martinelli
 ab,
Rogelio
Jiménez-Juárez
ac,
Diego
Alberti
d,
Simonetta
Geninatti Crich
d and
Kristina
Djanashvili
*a
ab,
Rogelio
Jiménez-Juárez
ac,
Diego
Alberti
d,
Simonetta
Geninatti Crich
d and
Kristina
Djanashvili
*a
aDepartment of Biotechnology, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: k.djanashvili@tudelft.nl
bDepartment of Science and Technological Innovation, Università del Piemonte Orientale, Viale Michel 11, 15121 Alessandria, Italy
cDepartment of Organic Chemistry, National School of Biological Sciences, National Polytechnical Institute, Prolongación de Carpio y Plan de Ayala S/N, 11340, Mexico D.F., Mexico
dDepartment of Molecular Biotechnology and Health Sciences, University of Torino, via Nizza 52, 10126 Torino, Italy
First published on 21st September 2020
Abstract
Paramagnetic macrocycles functionalized with phenylboronic moieties have proven to be interesting for MRI applications based on their ability to recognize cancer cells and generate local contrast. However, full use of the potential of this class of compounds is hampered by laborious and inefficient synthetic and, especially, purification procedures. The amphiphilic character of water-soluble phenylboronates renders them difficult compounds to be prepared through conventional solution synthesis due to the tendency to aggregate and form adducts with other nucleophiles. The new strategy described herein exploits the advantage of solid-phase synthesis with the application of DEAM-PS resin for anchorage and the subsequent simplified derivatization of boronates. GdDOTA-EN-PBA and its fluorinated analogue GdDOTA-EN-F2PBA were synthesized in a much easier, faster and economically convenient way to achieve good yields and purity. Furthermore, the effect of electron-withdrawing fluorine atoms on the aromatic ring of the latter compound was investigated by comparing the physico-chemical properties of both compounds as well as their binding affinity towards melanoma cancer cells.
Introduction
Boronic acids are a class of compounds that keep attracting increasing interest in many fields. Their applications range from biomedical research to materials science, chemical synthesis and electronics.1 For example, boronates have been successfully employed as enzymatic inhibitors,2 in the synthesis of organogels,3 as chemosensors,4 and as binding sites in the preparation of electroconductive molecules,5 not to mention their use as synthetic partners in the Suzuki–Miyaura coupling reaction.6 Boron-10 is also the nuclide of choice for neutron capture therapy, an emerging anti-cancer treatment based on the use of thermal neutrons to destroy tumour tissues.7Another interesting biomedical application of boron-based compounds lies in their capability for molecular recognition in the design of targeting contrast agents. In particular, phenylboronic acid (PBA) and its derivatives have shown the ability to strongly bind sialic acid, a nine-carbon monosaccharide unit that is overexpressed as the terminal group of glycolipids and glycoproteins on the surface of tumour cells.8 The mechanism of such interaction is based on the reversible formation of five- and six-membered cyclic boronate esters between the boronic group of PBA and the exocyclic polyol chain of sialic acid.9 This property of PBAs has been exploited in the preparation of several diagnostic and/or therapeutic targeting agents,10–12 the most successful of them being GdDOTA-EN-PBA (DOTA = 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, EN = ethylenediamine). Such a Gd(III)-complex bears a PBA moiety conjugated to a ligand (DOTA) through an aminoethylamide linker (EN) (Scheme 1). GdDOTA-EN-PBA has proved to be successful as a magnetic resonance imaging (MRI) contrast agent for in vivo tumour targeting based on the recognition of overexpressed sialic acid.13 In this case, a two-site cooperative binding mechanism is involved: in addition to the previously mentioned formation of cyclic boronates between PBA and the diol of the substrate, the recognition is enhanced by an electrostatic interaction involving the negatively charged carboxylate on the sialic acid residue and the positively charged amino group of the linker between the macrocycle and PBA. The same strategy was applied later with the radioactive Ga-68 complex of DOTA-EN-PBA enabling visualization of tumours by positron emission tomography (PET).11,14 Additionally, Yamamoto et al. have demonstrated the potential of a PBA-containing GdDTPA-based MRI contrast agent (DTPA = diethylenetriaminepentaacetic acid) for simultaneous neutron capture therapy, thus widening the medical applications of such systems.15
 | ||
Scheme 1 Solid-phase synthesis of DOTA-EN-PBA and DOTA-EN-F2PBA. (i) THF, rt, 2 h; (ii) THF, rt, 3 h; (iii) NaBH4, THF, rt, 4 h; and (iv) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DCM/TFA, rt, 15 h. :1 DCM/TFA, rt, 15 h. | ||
Unfortunately, the amphiphilic character of boronates translates into severe difficulties in their preparation and purification, including solubility problems and the inclination to form oligomers or react with alcohols, amines, carboxylates and other nucleophilic species. Also, the pH-dependent equilibrium between a boronic acid and its boronate form represents an additional source of synthetic obstacles.16 For instance, the conventional synthesis of DOTA-EN-PBA was heavily affected in terms of the final yield and the time and effort required to purify the ligand.10 As an example of tedious procedures needed for the purification of PBA derivatives, time-consuming ion-exchange chromatography was also responsible for partial loss of the product.
In order to prevent the boronic acid moiety from affecting the efficacy of the various synthetic steps and the purification of the intermediate products, the boronate can be “protected” by binding it to a solid support. This would also allow the exploitation of the well-known advantages of solid-phase synthesis, such as the easy removal of excess reagents and by-products through simple filtration and washing. A few cases have been already described where conventional solid-phase peptide synthesis was exploited to build molecular parts of contrast agents.17–20 Several solid supports are reported in the literature to specifically bind boronic acids, including citronellic acid attached to Rink's amide resin, polystyrene derivatised with 2-methyl-2-(hydroxymethyl)-1,3-propanediol,21 catechol pendant polystyrene polymer,22 and more recently, 1-glycerol Merrifield resin.23 Among these, polystyrene functionalized with diethanolamine groups (DEAM-PS resin) seems to be endowed with more effective binding properties, and requires mild conditions to release the boronic acids after their derivatization.24–26
Herein, we report a new way to prepare DOTA-EN-PBA derivatives based on solid-phase synthesis. We were also interested in investigating the effect of the increased acidity of the boronate moiety on the binding to sialic acid. Therefore, an additional fluorinated ligand (DOTA-EN-F2PBA) was synthesised via the same successful procedure. Such a strategy allowed both compounds to be obtained in a more straightforward way and with higher yields compared to those reported previously for DOTA-EN-PBA. Gd(III)-complexes of both species were then prepared and their relaxometric properties were determined through standard procedures. Finally, the targeting ability of novel GdDOTA-EN-F2PBA towards sialic acid was studied on suitable cell cultures and compared to that of GdDOTA-EN-PBA.
Results and discussion
Synthetic procedures
DEAM-PS resin was synthesized according to the procedure described by Gravel et al.25 starting from chloromethylated polystyrene (Merrifield) resin and diethanolamine. Generally, a functionalized boronic acid can be anchored to the resin by simply dissolving it in a suspension of DEAM-PS and shaking the reaction vessel for a short time at room temperature.25,26 The synthesis can then be continued in a divergent way analogously to common solid-phase peptide synthesis until the final cleavage is performed. The novel preparation of DOTA-EN-PBA in a few simple steps was therefore carried out accordingly, as shown in Scheme 1.After swelling the DEAM-PS resin in dry tetrahydrofuran for 30 min, 3-formylphenylboronic acid (1a) dissolved in anhydrous THF was first immobilized onto the resin (2a) by agitation in a filter syringe fixed onto an orbital shaker (50 rpm) for 2 hours at room temperature. This step was followed by reductive amination under analogous conditions (anhydrous THF, ambient temperature) between the aldehyde moiety and the primary amine of DOTA(OtBu)3-EN to form the anchored and protected precursor (3a) of the aimed ligand. This was obtained in one step by shaking the functionalized resin in dichloromethane and trifluoroacetic acid to achieve deprotection of the carboxylates and cleavage from the resin at the same time.
The yield calculated (as the trifluoroacetate form) with respect to DOTA(OtBu)3-EN was 62%, which definitely represents an improvement when compared to that reported in the literature for the synthesis in solution (33%).11 Following this protocol, all the solubility problems typical of PBA derivatives were avoided. The identity of DOTA-EN-PBA was confirmed by ESI mass spectrometry and full NMR characterization (see the ESI†) by comparison with the data previously reported.11 It is noteworthy that the main species observable in the mass spectrum of the ligand were the mono- and bis-dehydrated forms (Fig. S3 and S4†). By recording mass spectra at different values of cone voltage, these have been proved to be the consequence of fragmentation happening in the spectrometer and not of degradation prior to analysis; by decreasing the ionization voltage, the bis-dehydrated form progressively disappears while the mono-hydrated species increases (Fig. S4†).
Prompted by the positive outcome, the novel procedure was exploited to prepare a derivative of DOTA-EN-PBA bearing two electron-withdrawing fluorine atoms on the aromatic ring. Such a modification is expected to increase the acidity of the boronic acid,16 thus strengthening the binding to the sialic acid residues on the targeted tumour cells. Preliminarily, the effect of the F–atoms in different positions on the acidity of the boronic acid was tested by 11B NMR titration measurements (see the ESI†) on four commercial model compounds: one resembling the position of DOTA-EN-PBA (namely, 3-methylphenylboronic acid) and three bearing the halogens and the B(OH)2 groups in various sites of the aromatic ring (2,3-difluoro-4-methylphenylboronic acid [2,3-F-4-Me-PBA], 2,4-difluoro-5-methylphenylboronic acid [2,4-F-5-Me-PBA], 2,6-difluoro-5-methylphenylboronic acid [2,6-F-5-Me-PBA]). The measurements (Fig. S1†) revealed a pKa value of 7.2 for 2,3-F-4-Me-PBA, compared to 8.8 for 3-Me-PBA, 8.6 for 2,6-F-5-Me-PBA and 8.0 for 2,4-F-5-Me-PBA. Expecting an analogous two-orders-of-magnitude increase in pKa, a novel ligand (DOTA-EN-F2PBA) was designed with two fluorine atoms in the ortho- and meta-positions with respect to the attachment site of the pendant arm, while the boronic moiety was switched to the para-position. This would also allow the testing of the importance of the formation of the cyclic boronate in the recognition mechanism regardless of the additional contribution of the amine-carboxylate electrostatic interaction mentioned previously.
An analogous synthetic pathway was followed starting from 2,3-difluoro-4-formylphenylboronic acid (1b), which was first immobilized onto the DEAM-PS resin (2b), then conjugated to DOTA(OtBu)3-EN (3b) and finally deprotected/cleaved. The newly formed DOTA-EN-F2PBA ligand (trifluoroacetate form) was obtained in 52% yield and fully characterized by ESI MS and NMR spectroscopy (Fig. S2 and S8–S11†). The water solubility of DOTA-EN-PBA and DOTA-EN-F2PBA represents an important extension with respect to the compounds prepared by Gravel et al. via solid-phase synthesis on the DEAM-PS resin, especially when the amphiphilic behaviour of boronates is considered.
In order to prove the effectiveness of such a protocol, the literature procedure for the preparation of DOTA-EN-PBA was applied to prepare DOTA-EN-F2PBA without a solid-phase synthetic strategy (see the Synthetic procedures section). In this case, the product was achieved in 24% yield, less than half compared to the new procedure.
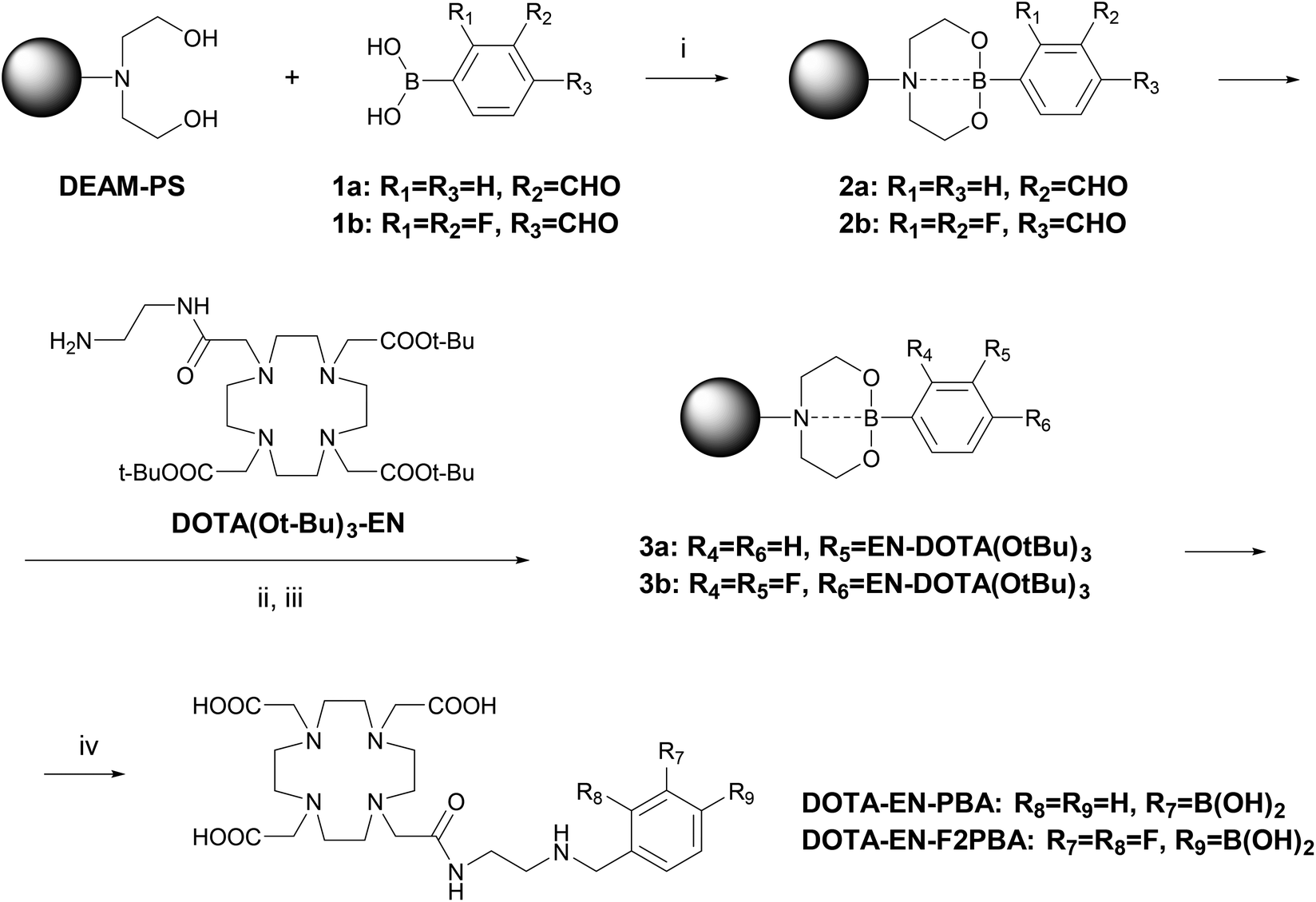
Besides the final yield, another advantage of the resin-binding approach is clear when the purity of the compound after the deprotection step is considered: the 19F NMR spectrum of DOTA-EN-F2PBA prepared in solution (Fig. 1 and S12†) revealed the coexistence of two species bearing a fluorinated arene, which could not be observed by 1H, 13C and 11B NMR spectroscopy. In particular, in addition to the two double doublets at −132 and −142 ppm from the expected product, two multiplets were also detected at −138 and −141 ppm, which furthermore gave cross-peaks in the 19F NMR COSY spectrum of the liquid-synthesis product (Fig. S13†). In contrast, this was not the case for DOTA-EN-F2PBA prepared through the novel procedure, whose 19F NMR spectrum shows only one set of signals (Fig. 1 and S11†). The presence of contaminants in the commercial reactant (2,3-difluoro-4-formylphenylboronic acid) was excluded by checking its NMR spectra.
 | ||
| Fig. 1 Comparison between the 19F NMR spectra of DOTA-EN-F2PBA obtained after cleavage from the solid support (top) and recovered after the final deprotection step of the conventional preparation in solution (bottom). | ||
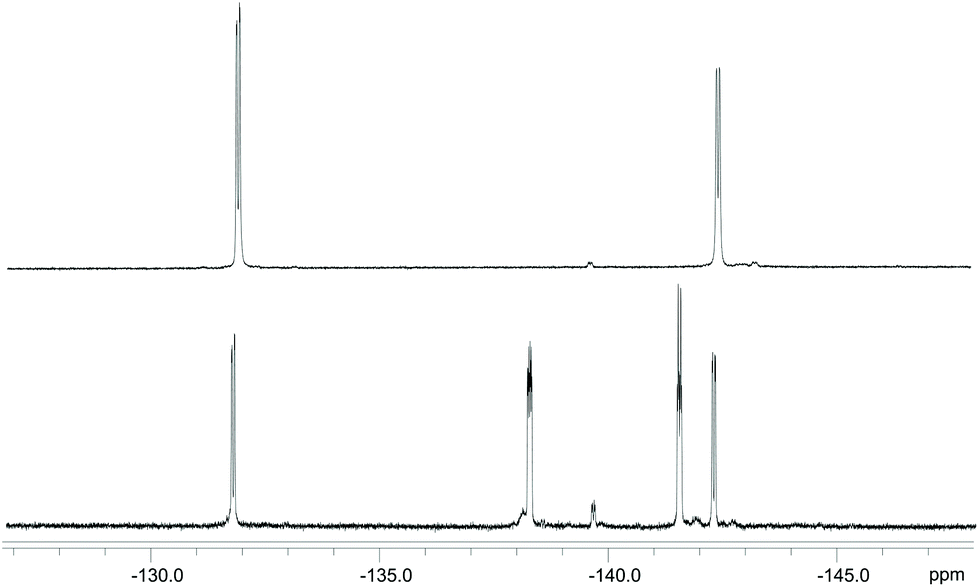
Since the 11B NMR spectrum of the ligand prepared in solution only reports one resonance from the boronic moiety of the ligand (as compared to the solid-phase synthesis product) without additional signals from side-products (Fig. 2), it was concluded that the extra emerged species is the result of protodeboronation typical of phenylboronic acids with a fluorine in the ortho-position, as reported by Cox et al.27
 | ||
| Fig. 2 Comparison between the 11B NMR spectra of DOTA-EN-F2PBA prepared on the solid support (top) and in solution (bottom). | ||
The presence of such a by-product was confirmed by both positive and negative ESI MS (Fig. S3†). The cleaner output further emphasizes the benefits of our novel solid-phase synthesis for the preparation of this kind of ligand: should such a side-reaction take place, the by-product would be released from the solid support and eliminated through successive washings.
Relaxometric characterization
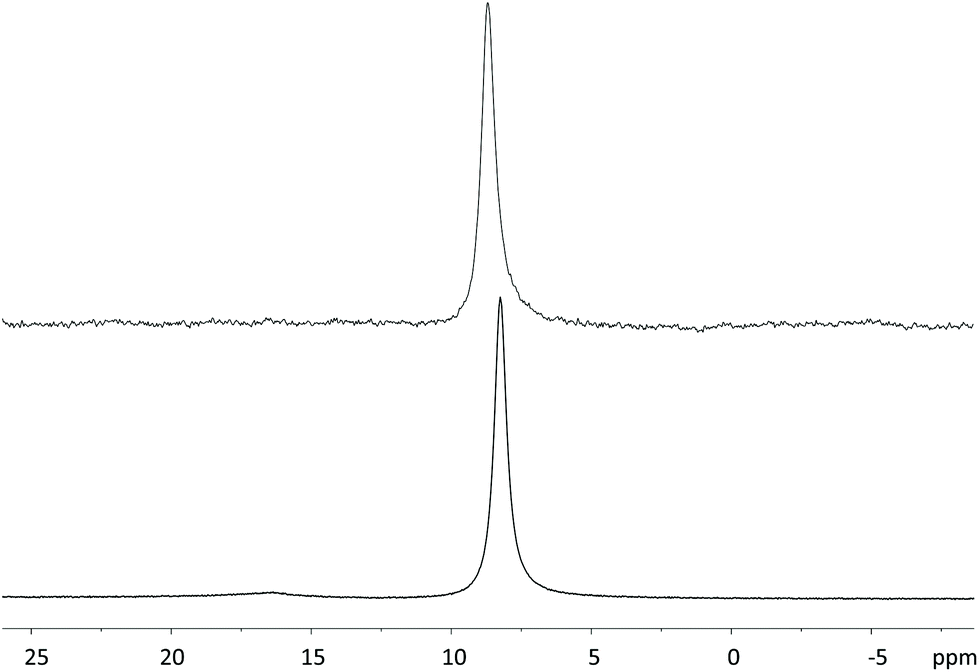
Although GdDOTA-EN-PBA is reported in the literature, its relaxometric parameters have never been evaluated. Therefore, full relaxometric characterization of both GdDOTA-EN-PBA and GdDOTA-EN-F2PBA chelates was carried out. The complexes were prepared according to a standard procedure and their formation was preliminarily confirmed by mass spectrometry, including the distinctive Gd isotopic pattern (Fig. S5 and S6†). Then the 1H nuclear magnetic relaxation dispersion (NMRD) profiles, in the proton Larmor frequency range of 0.01 to 70 MHz, were recorded at 25 and 37 °C (Fig. 3). The exact Gd(III) concentrations were determined according to the Evans method.28 | ||
| Fig. 3 1H NMRD profiles acquired at pH 7 and 25 or 37 °C for aqueous solutions of GdDOTA-EN-PBA (top) and GdDOTA-EN-F2PBA (bottom). | ||
The NMRD profiles measured for the two complexes at 25 °C and 37 °C were analysed using the conventional Solomon–Bloembergen–Morgan theory,29 leading to the determination of a set of characteristic parameters, such as the rotational correlation time (τR) and the exchange time (τM) of water coordinated to the metal centre with the bulk (Table 1). The longitudinal relaxivity values (r1p), representing the proton relaxation rate enhancement per 1 mM Gd(III) ion, were determined at 25 °C and 20 MHz to be 6.24 and 5.80 mM−1 s−1 for GdDOTA-EN-PBA and GdDOTA-EN-F2PBA, respectively. These values, slightly higher than those of typical monomeric GdDOTA complexes, can be attributed to the bigger molecular size due to the additional PBA pendant arm, which results in slower tumbling times as compared to GdDOTA (τR = 90 ps).30 The increased relaxivity values are expected to translate into an enhanced MRI contrast effect.
| Parameter | GdDOTA-EN-PBA | GdDOTA-EN-F2PBA |
|---|---|---|
| a The following parameters were fixed to common values during the fitting procedure: rGd-H = 3.0 Å, D = 2.24 × 10−5 cm2 s−1, q = 1, a = 4.0 Å. | ||
| 20 MHz r 1p [mM−1 s−1] | 6.24 ± 0.21 | 5.80 ± 0.35 |
| τ M [ns] | 69 ± 19 | 100 ± 23 |
| τ R [ps] | 115 ± 5 | 112 ± 6 |
| τ v [ps] | 36 ± 5 | 40 ± 7 |
| Δ2 [1019 s−2] | 1.48 ± 0.35 | 1.14 ± 0.36 |
As expected, the values for the two complexes are similar, as there is no particular difference of relaxometric interest in their molecular structures. The slightly lower relaxivity of the fluorinated analogue suggests the influence of the rather remote aromatic group on the first coordination sphere of the complex, whereby a relative decrease in hydrophilicity induced by the fluorine atoms affects the water exchange rate at the Gd(III) ion (100 vs. 69 ns) and consequently the r1p value.
Determination of the effect of pKa on binding efficacy
As previously mentioned, one of the objectives of this work was to assess the importance of the acidity of the boronic group towards the binding strength regardless of the additional contribution arising from the electrostatic interaction between the amine of the linker and the carboxylate of sialic acid residues overexpressed by cancerous cells. In this perspective, switching the boronate from the meta- to para-position of the aromatic ring allows the effects of the lower pKa to be highlighted (see above and the ESI†) in the recognition mechanism while at the same time reducing or removing the electrostatic influence. Since the expression of sialic acid is directly correlated to melanogenesis, the recognition ability of GdDOTA-EN-PBA and GdDOTA-EN-F2PBA was evaluated on murine melanoma B16-F10 cells. In fact, there is a direct correlation between sialic acid expression and the melanogenesis process occurring in these melanoma cells. Upon incubation of the cells in the presence of both Gd(III)-complexes, when the amount of the Gd(III) ion per cell was determined, comparable values were obtained (Fig. 4).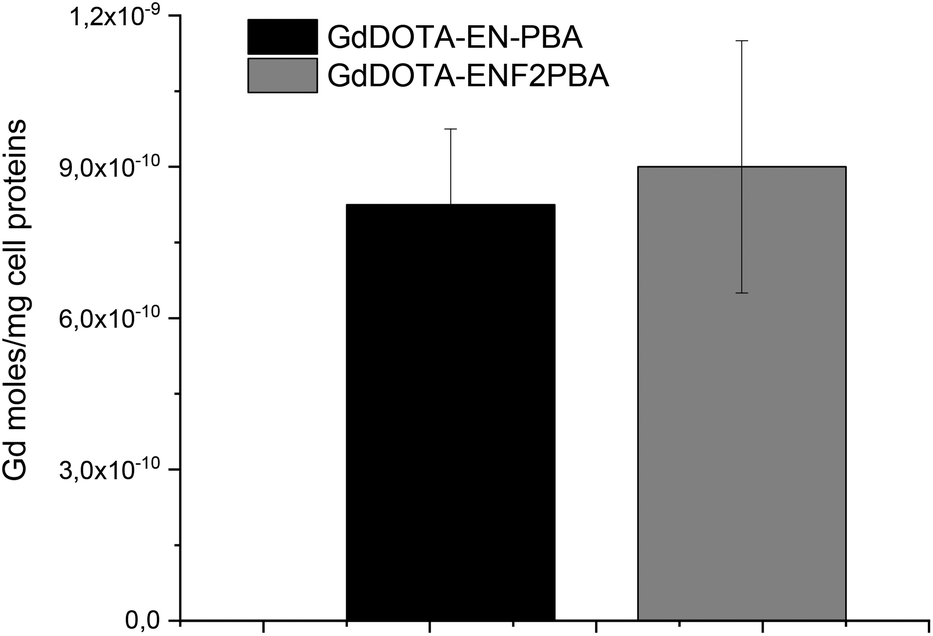 | ||
| Fig. 4 Evaluation of the bound Gd(III)-complexes upon incubation with melanoma cells: B16-F10 cells, 4 h of incubation in EBSS buffer w/o glucose at a Gd concentration of 0.6 mM. | ||
This seems to suggest that the advantageous effect of the electronegative substituents on increasing the acidity of the boronic group was counterbalanced by the shift of the latter from the meta-position in GdDOTA-EN-PBA to the para-position in the fluorinated derivative. As previously mentioned, because the amino group of DOTA-EN-PBA at physiological pH facilitates the recognition and binding to sialic acid residues by electrostatic interaction with the carboxylate of the glycan chain, when moving from a meta- to a para-substituted boronic acid, this interaction can be much less effective or even missed completely, thus weakening the targeting ability of the system. The similar results obtained for GdDOTA-EN-PBA and GdDOTA-EN-F2PBA indicate that the contribution of the electrostatic interaction is basically comparable to the enhanced binding strength achieved by increasing the PBA acidity. This confirms therefore that, as we aimed to prove, playing with the acidity of the vector, e.g. by introducing electron-withdrawing groups on the aromatic ring, is a good strategy to improve the targeting abilities of this type of ligand.
Conclusions
The advantages of solid-phase synthesis were successfully exploited to prepare DOTA-EN-PBA and the novel DOTA-EN-F2PBA ligands, the latter bearing two fluorine substituents on the aromatic ring to increase the acidity of the boronic moiety. The positions of the electron-withdrawing groups and the B(OH)2 groups were chosen upon investigation of the pKa values of several model compounds by 11B NMR titration, where the strongest acidity was assessed for a PBA derivative with two fluorines in the 2,3-positions and the boronate in the 4-position relatively to the linker. With work-up procedures mostly consisting of washing only, the yields of both compounds were doubled compared to the respective syntheses in solution. Moreover, the products appeared to be cleaner without the final by-products to separate. The relaxometric characterization of the GdDOTA-EN-PBA and GdDOTA-EN-F2PBA complexes allowed the determination of the longitudinal relaxivity values to be 6.24 and 5.80 mM−1 s−1, respectively; such r1p values are higher than those commonly obtained for simple monomeric GdDOTA chelates and are the consequence of a bigger size (due to the PBA functionality) and fast water exchange rates (τM < 100 ns). These relatively high relaxivities render GdDOTA-EN-PBA and GdDOTA-EN-F2PBA very promising for applications as targeting MRI contrast agents, as already reported.13 When the targeting abilities of the Gd(III)-complexes of the two ligands were tested towards sialic acid residues overexpressed by melanoma cells, a satisfactory response was obtained. However, GdDOTA-EN-F2PBA appeared to bind cancer cells similarly to GdDOTA-EN-PBA; this is due to the different position of the PBA group in the fluorinated agent (para) with respect to the non-fluorinated system (meta), which affects the secondary interaction between the amine of the ligand and the carboxylate of sialic acid, thus reducing the binding strength. This aspect, being one of the objectives of this work, has to be taken into consideration in the future design of this promising kind of MRI targeting contrast agent. The solid-state method proved to be suitable for the preparation of the targeted species with good yield and purity for applications as theranostic (therapeutic and diagnostic) agents.Experimental
Materials and methods
DO3AtBu-N-(2-aminoethyl)ethanamide (DOTA(OtBu)3-EN) was purchased from CheMatech. All chemicals were used as supplied from commercial sources. “H2O” refers to high-purity water with a conductivity of 0.04 μS cm−1, obtained from a Milli-Q purification system. Solid-phase reactions were performed by orbital shaking (50 rpm) in polypropylene vessels (filter syringes) purchased from MultiSynTech GmbH. Resin-washing operations were carried out on a vortexer. Anhydrous THF was obtained by distillation of a commercial solvent over sodium/benzophenone. 1H, 13C, 11B and 19F NMR was performed on an Agilent MR400DD2 spectrometer operating at 9.4 T. Samples were prepared in 5 mm NMR tubes by dissolving the compounds in D2O. Chemical shifts are reported in ppm relative to tert-BuOH (1.2 and 31.2 ppm for 1H and 13C NMR, respectively), H3BO3 (0 ppm for 11B NMR) and CF3COOH (−74.4 ppm for 19F NMR) as the internal standards. For 11B NMR experiments, a quartz tube was used. Coupling constants are reported in Hz. Splitting patterns are described as singlet (s), broad singlet (bs), double doublet (dd), multiplet (m) or broad multiplet (bm). Electrospray mass spectra were obtained on an ESI Waters SQD 3100 spectrometer for the low-resolution mass spectra and on a QSTAR Pulsar (AB/MDS Sciex) spectrometer for the high-resolution mass spectra (HR MS).Synthetic procedures
:1 DCM/TFA (6 mL) and shaken at room temperature for 15 h. The suspension was filtered and the resin was washed with 1:1 DCM/TFA (3 × 3 mL, 1 min). The combined filtrates were evaporated under reduced pressure and the residue was dissolved again in TFA (1 mL) and added to Et2O (10 mL). The resulting suspension was centrifuged (4000 rpm, 30 min, 10 °C) and the solid residue was washed/centrifuged with Et2O (3 × 10 mL) and dried under vacuum.
DOTA-EN-PBA: 62% yield (calculated as the trifluoroacetate form with respect to DOTA(OtBu)3-EN). The NMR characterization is in agreement with that reported in the literature (Fig. S3†).11
1H NMR (400 MHz, 25 °C, D2O): 7.75 (m, 2H), 7.48 (m, 2H), 4.24 (s, 2H), 3.7–3.3 (bm, 8H), 3.3–2.5 (bm, 20H). 13C NMR (100 MHz, 25 °C, D2O): 178.4, 173.0, 169.7, 151.5, 146.9, 129.7, 128.9, 126.8, 123.9, 61.3, 57.1, 56.4, 56.0, 52.0, 50.4, 48.7, 48.3, 46.7, 43.9, 35.6, 20.6. 11B NMR (128 MHz, 25 °C, D2O): −19.2 (bs). HR MS (ESI): [M − H2O + H]+m/z calculated for C25H40BN6O8 563.2995, observed 563.2989.
DOTA-EN-F2PBA: 52% yield (calculated as the trifluoroacetate form with respect to DOTA(OtBu)3-EN). 1H NMR (400 MHz, 25 °C, D2O[CD3CN]): 7.28 (m), 7.17 (m), 4.37 (bs), 3.81 (bm), 3.57 (m), 3.6–3.3 (bm), 3.39 (bm), 3.33 (bm), 3.23 (m), 3.2–2.8 (bm). 13C NMR (100 MHz, 25 °C, D2O[CD3CN]): 173–169 (CO), 155.3, 153.5, 151.3, 149.3, 131.3, 128.5, 61.3, 56–50, 34.9, 29.4, 20.7. 11B NMR (128 MHz, 25 °C, D2O[CD3CN]): 8.22 (bs). 19F NMR (376 MHz, 25 °C, D2O[CD3CN]): −131.79 (dd, 3JFF = 23.7 Hz, 4JFH = 4.5 Hz, C(F)CCH2), −141.54 (dd, 3JFF = 23.7 Hz, 4JFH = 3.0 Hz, C(F)C(B)). HR MS (ESI): [M − H2O + H]+m/z calculated for C25H38BF2N6O8 599.2807, observed 599.2803.
:47.5:47.5). After evaporation of the solvents under reduced pressure, the residue was dissolved again in H2O and lyophilized (0.12 g, 24% yield calculated from the initial step). The NMR spectra are discussed in the text.
GdDOTA-EN-PBA: 96% yield. ESI+ MS: [M + H]+m/z calculated for C25H39BGdN6O9+ 736.2, observed 735.8.
GdDOTA-EN-F2PBA: 93% yield. ESI+ MS: [M + H]+m/z calculated for C25H37BF2GdN6O9+ 772.2, observed 771.8.
Relaxometric measurements
The water-proton longitudinal relaxation rates as a function of the magnetic field strength were measured in non-deuterated aqueous solutions on a fast field-cycling Stelar SmarTracer relaxometer (Stelar s.r.l., Mede (PV), Italy) over a continuum of magnetic-field strengths from 0.00024 to 0.25 T (corresponding to 0.01–10 MHz proton Larmor frequencies). The relaxometer operates under computer control with an absolute uncertainty in 1/T1 of ±1%. Additional longitudinal and transverse relaxation data in the range of 15–70 MHz were obtained on a Stelar relaxometer connected to a Bruker WP80 NMR electromagnet adapted to variable-field measurements (15–80 MHz proton Larmor frequency). The exact concentration of Gd(III) ions was determined by measuring the bulk magnetic susceptibility shifts of a tBuOH signal. The 1H T1 relaxation times were acquired by the standard inversion recovery method with a typical pulse width (90°) of 3.5 ms and 16 experiments of 4 scans. The temperature was controlled with a Stelar VTC-91 airflow heater equipped with a calibrated copper-constantan thermocouple (uncertainty of ±0.1 °C).Cell lines
Mouse melanoma (B16-F10) cell lines were purchased from American Type Culture Collection. Melanogenic B16-F10m cells were obtained by growing cells in standard DMEM (Lonza) medium supplemented with sodium bicarbonate, and glutamine. The cells were incubated at 37 °C under a humidified atmosphere of 5% CO2. The medium was supplemented with 10% (v/v) FBS, 2 mM glutamine, 100 U ml−1 penicillin, and 100 U ml−1 streptomycin. Melanogenesis was evaluated by measuring the absorbance of the cell lysate and medium (dissolved in 1 M NaOH) at 490 nm observing its linear dependency on the number of cell passages. The sialic acid expression on B16-F10 cells was determined using a commercially available AbCAM assay.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (NWO VENI grant-722.012.009) and the Instituto Politécnico National (COFAA, EDI). The authors thank Prof. Mauro Botta (Università del Piemonte Orientale, Alessandria, Italy) for the availability of the relaxometery equipment and the data fitting.Notes and references
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS.
- R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS.
- L. Xu, Y. Hu, M. Liu, J. Chen, X. Huang, W. Gao and H. Wu, Tetrahedron, 2015, 71, 2079–2088 CrossRef CAS.
- S. D. Bull, M. G. Davidson, J. M. H. Van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y. B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Z. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS.
- T. Miyahara and K. Kurihara, J. Am. Chem. Soc., 2004, 126, 5684–5685 CrossRef CAS.
- I. Maluenda and O. Navarro, Molecules, 2015, 20, 7528–7557 CrossRef CAS.
- A. H. Soloway, W. Tjarks, B. A. Barnum, F. G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS.
- R. Schauer, Glycoconjugate J., 2000, 17, 485–499 CrossRef CAS.
- K. Djanashvili, L. Frullano and J. A. Peters, Chem. – Eur. J., 2005, 11, 4010–4018 CrossRef CAS.
- K. Djanashvili, G. A. Koning, A. van der Meer, H. T. Wolterbeek and J. A. Peters, Contrast Media Mol. Imaging, 2007, 2, 35–41 CrossRef CAS.
- K. Djanashvili, T. L. M. ten Hagen, R. Blange, D. Schipper, J. A. Peters and G. A. Koning, Bioorg. Med. Chem., 2011, 19, 1123–1130 CrossRef CAS.
- L. Frullano, J. Rohovec, S. Aime, T. Maschmeyer, M. I. Prata, J. J. P. de Lima, C. Geraldes and J. A. Peters, Chem. – Eur. J., 2004, 10, 5205–5217 CrossRef CAS.
- S. G. Crich, D. Alberti, I. Szabo, S. Aime and K. Djanashvili, Angew. Chem., Int. Ed., 2013, 52, 1161–1164 CrossRef.
- C. Tsoukalas, S. Geninatti-Crich, A. Gaitanis, T. Tsotakos, M. Paravatou-Petsotas, S. Aime, R. Jimenez-Juarez, C. D. Anagnostopoulos, K. Djanashvili and P. Bouziotis, Mol. Imaging Biol., 2018, 20, 798–807 CrossRef CAS.
- K. Takahashi, H. Nakamura, S. Furumoto, K. Yamamoto, H. Fukuda, A. Matsumura and Y. Yamamoto, Bioorg. Med. Chem., 2005, 13, 735–743 CrossRef CAS.
- J. Yan, G. Springsteen, S. Deeter and B. H. Wang, Tetrahedron, 2004, 60, 11205–11209 CrossRef CAS.
- L. Connah, R. Joshi, S. Vibhute, G. Gambino, J. D. G. Correia and G. Angelovski, Org. Lett., 2019, 21, 5378–5382 CrossRef CAS.
- J. Singh, V. Rustagi, S. R. Zhang, A. D. Sherry and D. G. Udugamasooriya, Magn. Reson. Chem., 2017, 55, 747–753 CrossRef CAS.
- M. Tripepi, F. Capuana, E. Gianolio, F. V. C. Kock, A. Pagoto, R. Stefania, G. Digilio and S. Aime, Bioconjugate Chem., 2018, 29, 1428–1437 CrossRef CAS.
- W. R. Li and Y. C. Yo, Tetrahedron Lett., 1999, 40, 9085–9089 CrossRef CAS.
- B. Carboni, C. Pourbaix, F. Carreaux, H. Deleuze and B. Maillard, Tetrahedron Lett., 1999, 40, 7979–7983 CrossRef CAS.
- W. Q. Yang, X. M. Gao, G. Springsteen and B. H. Wang, Tetrahedron Lett., 2002, 43, 6339–6342 CrossRef CAS.
- M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Org. Lett., 2016, 18, 2016–2019 CrossRef CAS.
- S. Arimori, J. H. Hartley, M. L. Bell, C. S. Oh and T. D. James, Tetrahedron Lett., 2000, 41, 10291–10294 CrossRef CAS.
- M. Gravel, K. A. Thompson, M. Zak, C. Berube and D. G. Hall, J. Org. Chem., 2002, 67, 3–15 CrossRef CAS.
- D. G. Hall, J. Tailor and M. Gravel, Angew. Chem., Int. Ed., 1999, 38, 3064–3067 CrossRef CAS.
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS.
- D. M. Corsi, C. Platas-Iglesias, H. Van Bekkum and J. A. Peters, Magn. Reson. Chem., 2001, 39, 723–726 CrossRef CAS.
- The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and E. Toth, John Wiley & Sons Ltd., 2013 Search PubMed.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, J. Am. Chem. Soc., 1996, 118, 9333–9346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01552k |
| This journal is © The Royal Society of Chemistry 2020 |