
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalized fluorescent terephthalate monomers and their attempted polyester formation†
Yvonne S. L.
Choo
 a,
Marta
Giamberini
b,
José
Antonio
c,
Paul G.
Waddell
d and
Andrew C.
Benniston
*e
a,
Marta
Giamberini
b,
José
Antonio
c,
Paul G.
Waddell
d and
Andrew C.
Benniston
*e
aSchool of Energy and Chemical Engineering, Xiamen University Malaysia, Jalan Sunsuria, Bandar Sunsuria, Sepang 43900, Selangor Darul Ehsan, Malaysia
bDepartament d'Enginyeria Química, Universitat Rovira i Virgili, Av. Països Catalans 26, 43007 Tarragona, Spain
cDepartament de Química Analítica I Química Orgànica, Universitat Rovira I Virgili, Carrer Marcel.lí Domingo s/n, 43007 Tarragona, Spain
dCrystallography Laboratory, Chemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
eMolecular Photonics Laboratory, Chemistry-School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: andrew.benniston@ncl.ac.uk
First published on 13th October 2020
Abstract
The reaction of diethyl 2,5-bis(tert-butyl)phenoxy-3,6-dihydroxyterephthalate (1) with tetraethylene glycol di(p-toluenesulfonate) under high-dilution conditions afforded several isolated products. Two products were identified as macrocycles with one being the 1 + 1 crown ether derivative 3 (10% yield), and the second being the 2 + 2 crown ether compound D3 (19% yield). The X-ray structure for 3 was determined with the asymmetric unit observed to comprise half of the molecule. The small crown ether ring of 3 interacts with K+ or H+ ions in MeOH, but binding is weak and the macrocyclic cavity is too small to fully encapsulate the K+ ion. Transesterification of compounds 1, its methylated version 2 and 3 with diols such as ethylene glycol or 1,4-butandiol produced monomers (M1–M3) which were reacted with terephthaloyl chloride. Short oligomers were produced (PolyM1–PolyM3) rather than extensive polymeric materials and all displayed solid state fluorescence. The absorption and fluorescence properties of M1–M2 and their polymers can be related to subtle structural changes. The Stokes shift for M2 of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 627 cm−1 in DCM is one of the largest observed for a simple organic chromophore in fluid solution.
627 cm−1 in DCM is one of the largest observed for a simple organic chromophore in fluid solution.
Introduction
Terephthalate derivatives are the cornerstone of the manufacture of the widely used polyester plastics,1 which have found numerous commercial applications in areas such as bottles, packaging, cosmetics, thermal insulating materials and car accessories, to name but a few. The preparation of, for example, poly(ethylene terephthalate) (PET) is relatively straightforward, requiring only high temperatures to perform in the first instance, the transesterification of dimethylterephthalate with ethylene glycol.2 The final stage is the driving of polymer formation by vacuum distillation of ethylene glycol under high temperatures. The properties of PET stand it in good stead because of its suitable mechanical features, chemical resistance, thermal stability, high transparency/flexibility and low permittivity to gases.3 Considering all of these positive features of PET it is not surprising that non-commercial applications of the polymer have also been sought in research-biased areas including force-sensing resistor materials,4 nanofiber phototransistors,5 rechargeable polymer batteries,6 solar cells7 and OLED devices.8 One feature from these studies is the consistency in structure of the basic terephthalate core with any additional functionality arising from blending of additional agents. There is clearly no reason, except based on a steric argument, that the actual terephthalate could not support other substituents, which could themselves have additional properties that could be imparted to the final polymer. Features could include, for example, pH sensitivity,9 thermochromic response,10 electrical conductivity,11 ion transport12 and fluorescence-based remote sensing.13 The final two categories are of special interest for potential applications in proton transport membranes14 and explosives detection.15Recently we developed the preparation of highly substituted terephthalate derivatives based on diethyl 2,5-bis(aryl)phenoxy-3,6-dihydroxyterephthalate, where the aryl groups are several disparate aromatic subunits.16 The compounds displayed moderately intense fluorescence both in fluidic solution as well as the solid state; the latter probably a result of the bulky groups which restricted close packing. In addition, the Stokes shift for the compounds was relatively large, supporting major internal structural change upon excitation. Noting the potential for such compounds to be further functionalized at the hydroxyl (e.g., crown ether) or ester groups (e.g., transesterification) we envisaged that new terephthalate-based polymeric materials could also be feasible (Fig. 1). The additional functionality of a crown ether offering a cation-binding site to manipulate the polymer structure and impose alteration to its fluorescence properties. Despite success in preparation of the crown-based monomer, its polymerisation proved challenging, but simpler monomer versions could be produced. The fluorescence properties of these monomers and their polyesters revealed subtle differences, highlighting larger Stokes shifts than reported previously. In addition, it was shown that the internal polymer structure restricted structural motions within the terephthalate core upon photoexcitation; the flexibility of the spacer group between each monomer being an important parameter in this observation.
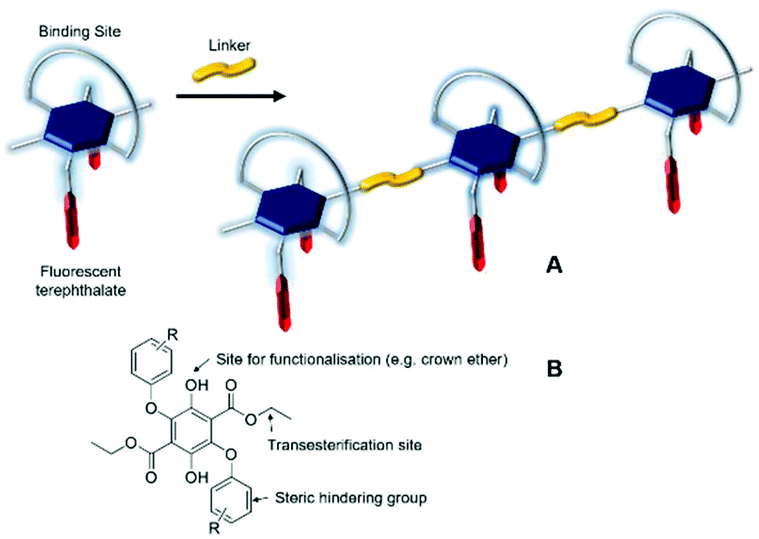 | ||
| Fig. 1 (A) Cartoon representation of polymer formation by linking of a functionalized fluorescent terephthalate subunit. (B) The basic building block for preparation of a terephthalate monomer. | ||
Results and discussion
Synthesis and structure
The synthetic procedure used in the production of the crown ether is illustrated in Scheme 1 starting from the precursor 1 prepared previously using published methods.16 In an attempt to minimise polymerisation reactions, the high dilution method was used in which the diol 1 and the tetraethylene glycol di(p-toluenesulfonate) were added slowly to a refluxing MeCN solution. After 9 days the reaction was worked up and the crude product purified by careful column chromatography on silica gel by using ethyl acetate/petroleum ether (1:5 then 1:4) as the eluent to afford two main products, 3 and D3. The unequivocal identification of the products was performed using a combination of 1H NMR spectroscopy, mass spectrometry and where possible, X-ray crystallography.
 | ||
| Scheme 1 Reagents and conditions (i) MeCN, K2CO3, KPF6, reflux, 9 days. | ||
The 1H NMR spectrum for 3 in CDCl3 is shown in Fig. 2. The prominent multiplet signals in the δ = 3.94–3.45 and 4.73 ppm are readily assigned to the OCH2 protons for the crown ether. By careful inspection of the COSY spectrum a partial assignment of the signals to the methylene units was possible. The first point to note is that the 1H NMR spectrum for 1 is not straightforward because of slowly interconverting conformers which impart inequivalence to the CH2 protons of the ethyl groups. Very noticeable are the downfield signals at δ = 4.73 corresponding to the H1,1′ protons which appear as a doublet of doublet of doublets, confirming that each separate proton of a CH2 unit are diastereotopic. Variable temperature 1H NMR spectra, coupled to COSY assignments, were highly informative of changes to the conformation of 3 in d8-toluene (see ESI S1†). A significant downfield shift for the H2,2′ proton resonances is observed with increasing temperature whereas upfield shifts are witnessed for the remaining CH2 protons. Apart from the shift in peaks, no coalescence of the two methylene multiplets (H5) or the diastereotopic CH2 protons are observed. However, there is a loss in resolution when the temperature is lowered to 183 K.
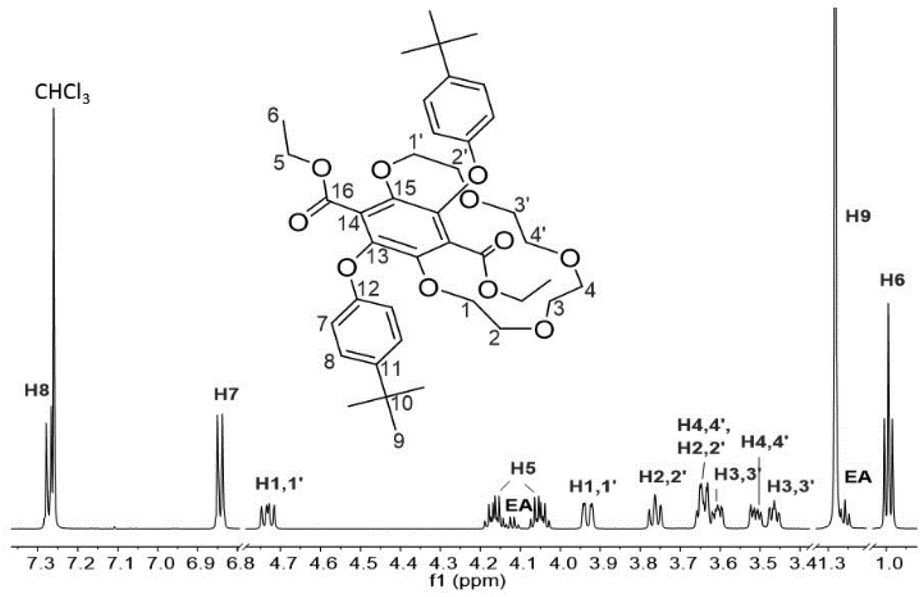 | ||
| Fig. 2 1H NMR spectrum (700 MHz) of 3 in CDCl3 showing the proton signals assignment. EA = trace of ethyl acetate solvent. | ||
Further corroboration that the solid was in fact the 1 + 1 product was obtained by positive nanoESI (DCM/MeOH + KOAc) which displayed a prominent peak at m/z = 747.3126 corresponding interestingly to the [M + K]+ ion and a peak at m/z = 726.3848 corresponding to the [M + NH4]+ ion (DCM/MeOH + NH4OAc). However, the unequivocal identification of 3 was obtained by the use of single-crystal X-ray crystallography, and the molecular structure is illustrated in Fig. 3. Selected bond lengths and angles are collected in Table 1. The two bulky tert-butyl aryl groups are syn to each other and point away from the crown ether moiety which encloses the central benzene ring. The effective size of the cavity can be assessed by considering the atom of the crown ether moiety closest to the benzene ring centroid (Fig. 3). This centroid⋯O2 distance was observed to be 3.59(1) Å, a relatively short contact. It is conceivable however that the conformational flexibility of the ring may allow it to expand further.
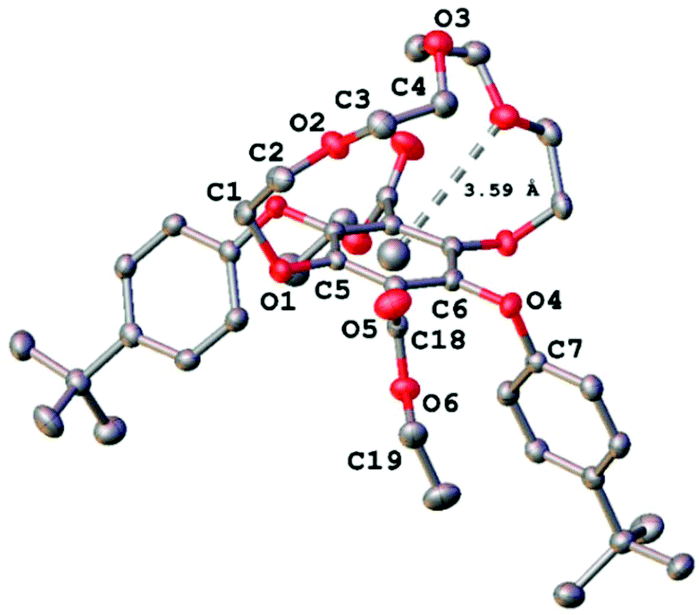 | ||
| Fig. 3 X-ray crystal structure of 3 with the centroid⋯O2 distance highlighted. Ellipsoids are drawn at the 50% probability level and hydrogen atoms have been omitted for clarity. | ||
| Atoms | Bond length/Å | Atoms | Angle/° |
|---|---|---|---|
| O1–C1 | 1.4459(18) | C5–O1–C1 | 116.84(10) |
| O1–C5 | 1.3760(16) | C3–O2–C2 | 113.70(11) |
| O2–C2 | 1.4211(18) | C4–O3–C4 | 115.93(17) |
| O2–C3 | 1.421(2) | C6–O4–C7 | 117.64(10) |
| O3–C4 | 1.4240(18) | C18–O6–C19 | 117.35(11) |
| O4–C6 | 1.3892(17) | O1–C1–C2 | 112.25(13) |
| O4–C7 | 1.3932(17) | O2–C2–C1 | 107.81(12) |
| O5–C18 | 1.1982(18) | O2–C3–C4 | 109.09(12) |
| O6–C18 | 1.3349(18) | O3–C4–C3 | 113.32(13) |
| O6–C19 | 1.4553(17) | O5–C18–O6 | 124.87(13) |
Noting the conformational flexibility of the compound in solution, and that the jellyfish-like shape of the molecule in the crystal structure was conceivably not the lowest energy conformation, complementary molecular modelling calculations were performed. Using the molecular geometry of the crystal structure as the starting point an energy minimisation calculation was performed using DFT (B3LYP) and a 6-311G basis set. There was only a minor structural alteration which suggested the solid state structure actually represented the lowest energy conformation. Basic molecular dynamics simulations (MM+) were far more informative (see ESI S2†) and several different energy conformations were observed. In full agreement with the solid-state structure and the DFT calculation, the syn tert-butyl aryl arrangement would appear to be the lowest energy conformation; the anti structure is slightly higher in energy by ca. 5 kcal mol−1. Two other higher energy conformations sampled are associated with variations in the crown ether geometry.
Cation binding studies of 3
Based on the crystal structure of 3 the crown ether moiety appears unobstructed by the two bulky tert-butyl aryl groups as they are orientated away from the cavity and are arranged syn to one another. However, the relatively short centroid⋯O2 distance and the potential steric hindrance of the carbonyl groups for the tert-butyl aryl groups either side of the ring raises the question of whether the cavity would be large enough for any cation to fit in it. Upon performing a structural database search on ConQuest,17 no X-ray crystal structures of compounds with similar features to 3 could be found, let alone examples with a cation bound within the cavity.To our surprise, the 1H NMR spectra of 3 recorded in CD3OD in the absence and presence of K+ ions (KPF6) did show subtle differences as shown in Fig. 4. Small downfield shifts and changes in splitting patterns were observed from the OCH2 protons on the alkoxy chain but the aromatic protons (H7 and H8) and the CH3 protons of the ethyl ester groups (H6) remained unchanged. This may potentially be attributed to some cation-crown interaction whereby the K+ ion does not necessarily bind within the cavity but may reside at the periphery of the crown and interact with the oxygen atoms on the alkoxy chain. This would explain the positive nanoESI mass spectrometry results of 3 for the presence of [M + K]+ species. The unique interaction of a K+ ion with 3 in CD3OD was found to be selective as all the other cation salts (LiPF6, NaPF6 and NH4PF6) revealed no changes or shifts in the 1H NMR signals. Since all the salts used were hexafluorophosphates, any anion interaction with the crown ether can be ruled out.
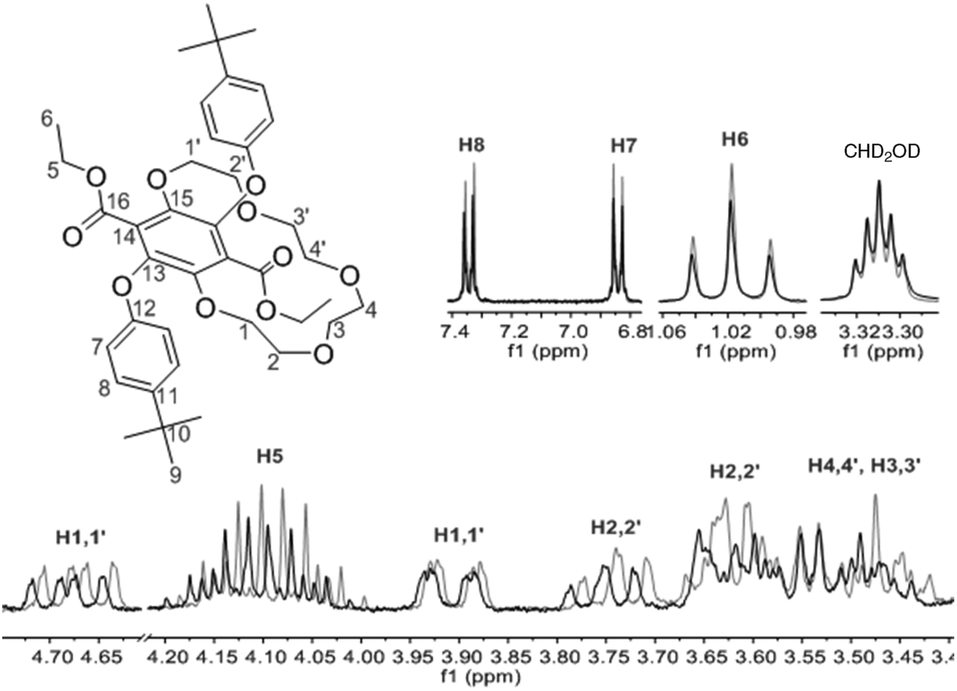 | ||
| Fig. 4 1H NMR spectra (300 MHz, CD3OD) of 3 before (grey) and after (bold) addition of K+ ions. | ||
Absorption and emission spectra of 3 in MeOH were also recorded in the absence and presence of cation salts. Unfortunately, no significant change was observed. As for the crown-ether-cation complex crystallisations attempted with sodium, potassium and ammonium hexafluorophosphate salts, only free 3 (uncomplexed) was crystallised in all of the cases. Since all attempts to grow crystals of the K+ adduct with 3 failed we turned our attention to DFT calculations to try and ascertain a probable structure. Starting from the solid state structure a K+ ion was placed in several positions within bonding distances to disparate oxygen atoms of the crown, and the structures were energy minimised in Gaussian 09 using B3LYP and a 6-311G basis set. From several calculations two structures, as shown in Fig. 5, emerged as conceivable adducts of 3 with a K+ ion; differing only slightly in energy because of small structural variations. Several key aspects are apparent from examination of the structures. The first to note is that only three oxygen atoms of the crown ether bind to the potassium ion and that the two crown-based phenoxyl atoms do not. The latter observation is perhaps not too surprising considering the severe structural distortion that would be required to fully accommodate the potassium ion. The K–O bond distances relating to the oxygen atoms of the crown ether are in good agreement with those found in several X-ray structures of relevant macrocyclic complexes.18 Additional oxygen donor interactions to the K+ appear to be via the ethyl ester, using the ethyl oxygen in structure A and the carbonyl oxygen for structure B; the latter structure being ca. 4 kcal mol−1 more stable. For both structures there is the possibility of a K-O5 interaction although the actual distances are rather long. The K-C1 and K-C2 distances are potentially just within the crystallographically determined range (2.990–3.117 Å) to be considered an η2 interaction to the aryl ring.19 It is worth noting that the analogues structures calculated for 3 with the smaller Li+ ion (see ESI S3†) showed that only two oxygen atoms of the crown bind to the metal ion, but with the ester still able to participate in ion binding. The Li–O5 distance is also far too long to be considered significant. The combination of weaker interactions is likely one reason for the observed lack of binding of 3 with Li+. The structure of 3 with Na+ (see ESI S4†) is more akin to the K+ structure shown.
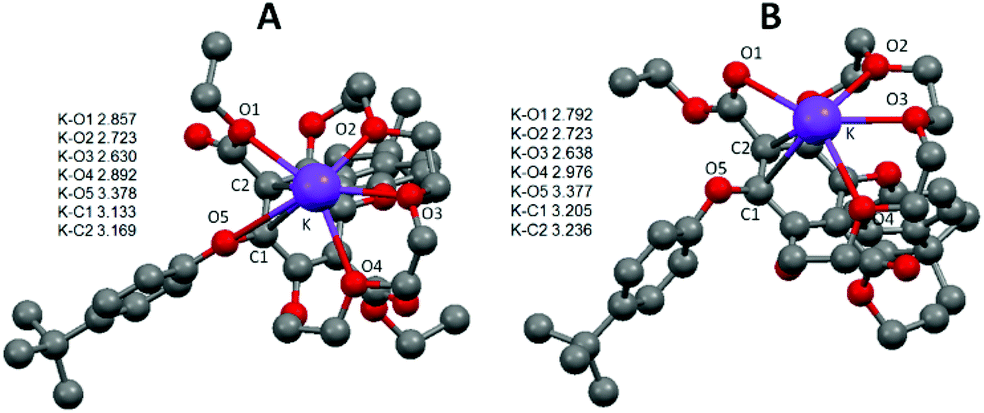 | ||
| Fig. 5 Gaussian calculated energy-minimised gas phase structures for a K+ adduct of 3 using B3LYP and a 6-311G basis set. Distances shown as the insert are in Å and hydrogens are omitted for clarity. | ||
Proton binding studies of 3
Since 3 was designed with the intention of its being polymerised to create a polymer containing proton binding sites, it was deemed reasonable to first test its ability to coordinate with protons. To this end, several NMR titration experiments were conducted in CDCl3 using trifluoroacetic acid as the proton source. Interestingly, no chemical shift changes were observed throughout the titration experiment for most of the proton peaks of 3 aside from H3,3′ and H4,4′ (protons from the crown ether chain) and H5 (protons from the ester side groups). By focusing on the region between 3.90–3.40 ppm in the stacked 1H NMR spectra (see ESI S74–76(a)†) the chemical shift data obtained from peaks corresponding to H3,3′, H3,3′ and H4,4′ were non-linearly fitted to a binding equation (see ESI S74–76(b)†). The average binding constant logKa was calculated to be 2.2 M−1 which is a relatively small value. There are two potential binding sites: the ester group and the crown cavity. Based on literature, the association constant for the protonation of an ester is found to be very small with logKa values between −15 and −1 M−1.20 The binding constants of cation complexes fall within the logKa range of 1.0 and 2.1 M−1 while proton complexes have logKa value of around 2.9 M−1 and ΔG° (300 K) of −15.9 kJ mol−1 (15-crown-5).20 Therefore, to a certain degree of certainty, the values obtained were indeed the binding constants of 3 with H+ (logKa of 2.2 M−1 and ΔG° of −13 kJ mol−1). Several attempts to crystallise the protonated complex of 3 with TFA were to no avail. Likewise, no [M + H]+ ion could be detected in the positive nanoESI spectrum of 3. Given the results it is speculated that the proton interacts weakly with oxygen atoms of the crown moiety and is not buried deep within the cavity.
Polymerisation results
Polymerization of terephthalate derivatives is often performed using high temperature transesterification in the presence of an appropriate diol and a Lewis acid catalyst.21 To aid in the polymerization studies control compounds were employed to ascertain appropriate conditions and identify any problems (Scheme 2). The attempted polymerization of 1 with stoichiometric ethylene glycol at high temperature immediately identified a drawback in that the O-aryl group tended to be cleaved off. The design protocol was altered slightly to introduce two terminal alcohol groups so that simple esterification with a suitable diacid chloride would form a polyester. The reaction of 1 with excess ethylene glycol containing zinc(II) acetate at 175 °C produced the monomer diol M1 in 24% yield, but still showed signs of some degradation in the purification products. Given this problem a different catalyst system was identified and the two OH groups were protected as their methyl ether (2). To also extend the spacer length the reaction of 2 in excess butanol but using Ti(OnBn)4 as the catalyst was employed and afforded both M2 and M4 after column chromatography.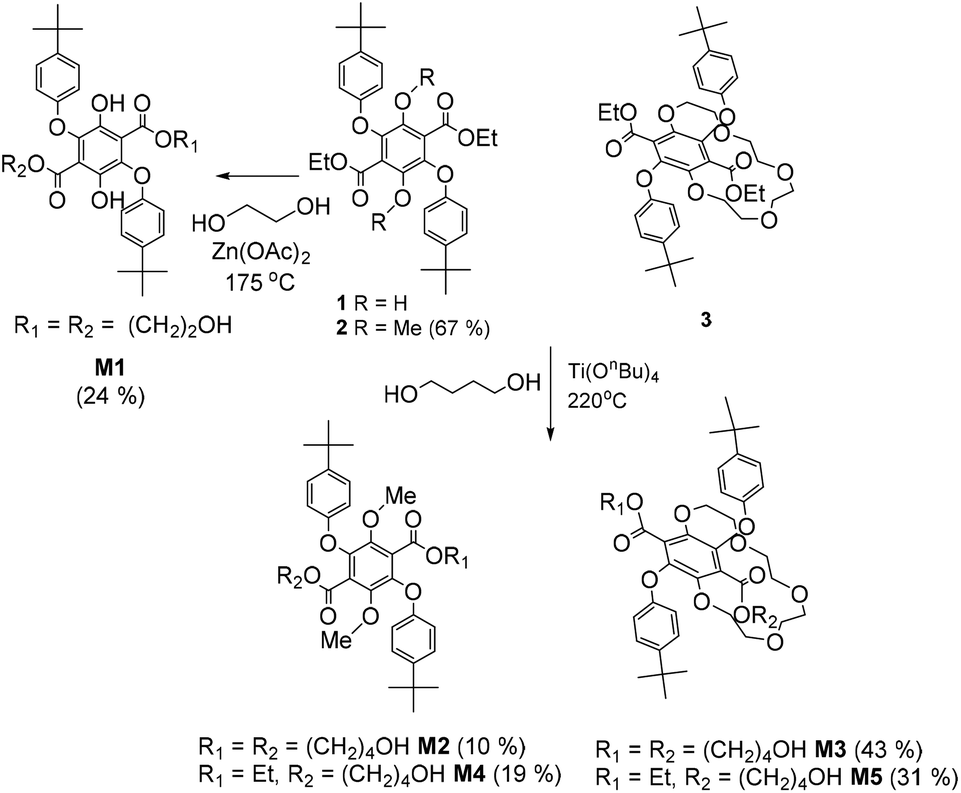 | ||
| Scheme 2 Procedures used in the preparation of the diol monomers M1–M3 and the side products M4–M5. | ||
Given the information gleaned by the test procedures the reaction of 3 in excess butanol using Ti(OnBn)4 as the catalyst worked well and afforded M3 and the side product M5 after purification. In the 1H NMR spectrum of M3 (see ESI S5†), peaks attributed to the CH2 protons on the newly attached butyl chains are seen as two sets of mirroring multiplets (at 4.20 and 4.07 ppm – H5), an overlapped multiplet at 3.48 ppm (H18) and additional multiplets at 1.50 ppm (H6) and 1.35 ppm (H17). The two protons from the terminal OH groups (H19) could also be seen as a broad peak at 1.59 ppm. The diastereotropic nature of the OCH2 protons of the crown ether characteristic of 3 was unaltered by transesterification, and was observed as prominent multiplet signals around 3.93–3.42 ppm and 4.69 ppm. The 1H NMR spectrum of asymmetrical M5 (see ESI S56†) appeared to be much more complicated that its symmetrical counterpart with two sets of peaks each in addition to overlapping ones.
Polyester formation
The attempted polyester formation reactions are illustrated in Scheme 3 and for simplicity terephthaloyl chloride was used as the linker unit. In all the reactions a slight excess of the monomer was used to ensure that any polymer would be end capped with a hydroxyl group. An excess of the base triethylamine was used to mop up the HCl generated by the ester formation reaction. The 1H NMR spectrum of PolyM1 (see ESI S10†) showed several broad peaks which are generally observed in the spectrum of a polymeric material. In addition, by comparing the 13C NMR spectrum of PolyM1 with that of its monomer (see ESI S13†), the stronger intensity carbon peaks could be attributed to the repeating unit of the polymer and the weaker intensity carbon peaks to the end groups.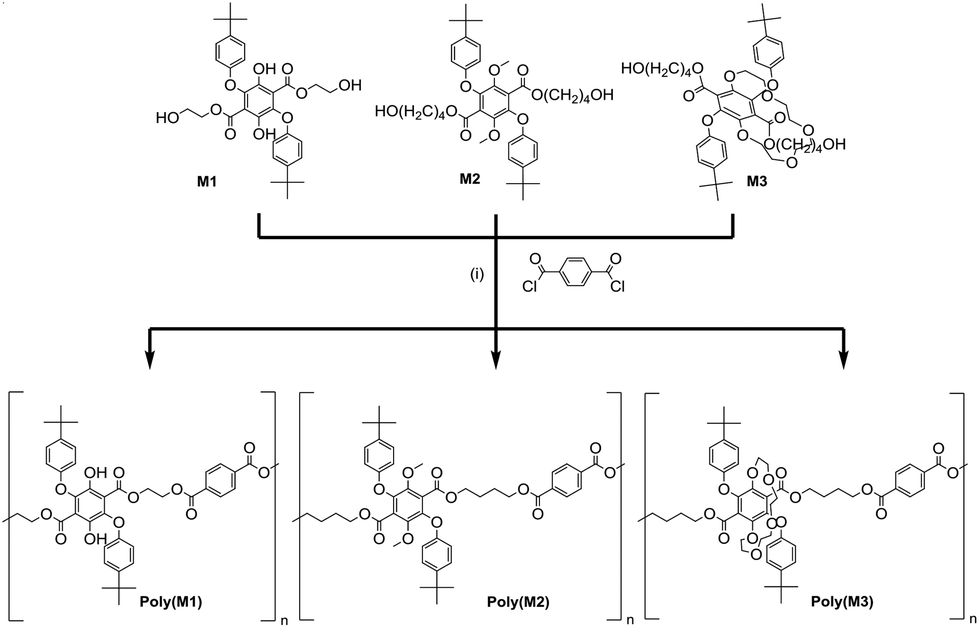 | ||
| Scheme 3 Reagents and conditions: (i) Triethylamine, THF, 0 °C then room temperature, 3 days. | ||
DOSY NMR spectroscopy was employed for the determination of the diffusion coefficient (D = 9.06 × 10−11 m2 s−1), and the identification of proton peaks of a polymer from a mixture of compounds. The diffusion coefficient was used to estimate the molecular weight (Mw) of PolyM1 (23360 g mol−1), which is relatively close to the Mw value obtained from a GPC experiment (25047 g mol−1). In contrast, the calculated Mn from the 1H NMR spectrum (8423 g mol−1) is much lower reflecting the difference in the methods. It is important to note that the difference may be attributed to the fact that GPC operates with the assumption that the polymer introduced in the system behaves like that of PS in THF/toluene. The copolymer in this case could have had very different hydrodynamic volume and behaviour in THF/toluene, resulting in an under/overestimation of the Mw and Mn. The glass transition temperature, Tg of the polymer, as determined from the second heating curve in the DSC thermogram, was found to be 91 °C. This value is consistent with its brittle nature of the polymer as observed at room temperature.
The copolymer, PolyM2 was synthesised in a similar manner to PolyM1 and characterised using a similar method (see ESI S14–18†). Collected in Table 2 are data comparing the two polymers. The Tg for PolyM2 of 48 °C is significantly lower than that of PolyM1 (91 °C). This difference is likely a result of the longer aliphatic chain (butylene spacer instead of ethylene spacer) and a smaller number of repeating units (n = 8). Given the test conditions the attempted polymerisation of M3 was carried out and afforded an opaque off-white material. The 1H NMR spectrum of the material (see ESI S19†) appeared only slightly broadened with respect to the starting material. Most of the peaks observed in the 1H NMR spectrum could be readily assigned to the main parts of the chemical structure for PolyM3, and suggested that the compound only contained a few repeating units. The lack of polymerisation of M3 was disappointing and seemed be related to the presence of the crown group, but the reason is not immediately obvious. Molecular dynamics simulations for the oligomers (n = 2, 4, 8) show that all the structures tend to fold in on themselves the greater the number of repeat units, and hence start to enclose and hinder the terminal alcohol groups. The crown ether of M3 unlike the other two monomers provides multiple hydrogen bonding sites which may interact with the terminal hydroxyl groups to retard the esterification reaction.
| Polymer | 1H and DOSY NMR | GPCd | |||||
|---|---|---|---|---|---|---|---|
|
M
na (g mol−1) |
M
wb (g mol−1) |
PDIc | D (m2 s−1) | M n (g mol−1) | M w (g mol−1) | PDI | |
|
a Number average molecular weight (Mn) was calculated from the 1H NMR spectrum.
b Weight average molecular weight (Mw) was calculated from diffusion coefficient, D obtained from the 2D DOSY NMR spectrum together with the equation of PS calibration curve by Grubbs et al. logD = −0.537 logMw − 7.697 (R2 = 0.9991).
c PDI = Mw/Mn.
d Monodispersed PS standard as the calibrant, THF/toluene as the eluent.
|
|||||||
| PolyM1 | 8423 | 23360 |
2.77 | 9.06 × 10−11 | 12806 |
25047 |
1.96 |
| PolyM2 | 7839 | 10670 |
1.36 | 1.38 × 10−10 | 1809 | 3471 | 1.92 |
Absorption and emission
All three polymers exhibited solid-state fluorescence as shown in Fig. 6, but with slightly different colours as observed by eye. The subtle colour difference between PolyM1 and PolyM2 appears to arise from the more structured emission in the blue-end of the spectrum (see ESI S20†). Although there are several polymers that emit in the blue region,22 the unique features of the two materials are revealed by careful inspection of the absorption and emission/excitation profiles for M1, M2 and the starting material 1. The X-ray crystal structure and molecular modelling calculations (DFT, B3LYP, 6-311G) for 1 show that the ground-state molecule is planar with the hydroxyl group forming an intramolecular hydrogen bond to the ester. The structure also remains planar in the first-excited singlet state, but there is a rearrangement manifest in changes to the bond lengths within the terephthalate core (see ESI S21†). The absorption maximum (λABS) in DCM is located at 392 nm (25510 cm−1) nm and the broad emission profile has a maximum (λEM) at 488 nm (20492 cm−1). The fluorescence excitation spectrum is a good match to the absorption profile (see ESI S23†). The large Stokes shift (SS) of 5018 cm−1 is a prominent feature of this type of structure. It is noted that an absorption spectrum calculated using time-dependent DFT (TD-DFT) and using a planar structure as the starting geometry is consistent with the experimental spectrum, but it is considerably blue-shifted if both ester groups are twisted out of the plane (see ESI S22†). This observation is important when comparing the absorption and emission properties of M1, M2 and their polymers. The ethylene glycol transesterification of 1 has little effect on the absorption and emission properties; the λABS and λEM for M1 are located at 393 nm and 488 nm, respectively (see ESI 25†). The absorption and fluorescence spectra for M2 (Fig. 7) are significantly different and must be related to the methylation of the two hydroxyl groups. The absorption profile is slightly structured and considerably blue-shifted compared to M1 (λABS = 281 nm). Again, the fluorescence profile is broad with λEM located at 501 nm, and red-shifted with respect to M1. Even though the fluorescence excitation spectrum matches well to the absorption profile in most parts, there is an additional feature (marked) which is significant and is related to the structure of the compound. The high-energy absorption profile is consistent with a ground-state structure in which the ester groups are twisted out of the aromatic plane (cf. 1). Further evidence for this can be observed in the calculated structure (DFT, B3LYP, 6-311G) of the ground-state for 2 (simplified version of M2) (see ESI S26†). The two ester groups adopt a more co-planar arrangement with the central aromatic ring as illustrated in the TD-DFT calculated structure of the first-excited singlet state (see ESI S26†). Such a large-scale structural alteration accompanying the ground to relaxed excited-state transition is the reason for the large SS. It should be noted that the apparent SS of 15627 cm−1 for M2 is one of the largest observed for a simple organic chromophore in fluid solution.23 This large SS is certainly conducive for the application of M2 (or a derivative) as a bio-fluorescent tag, but with the drawback of requiring excitation in the UV region.
 | ||
| Fig. 6 Side-by-side comparison of PolyM1, PolyM2 and PolyM3 in (a) normal light conditions and (b) under 312 nm UV irradiation. | ||
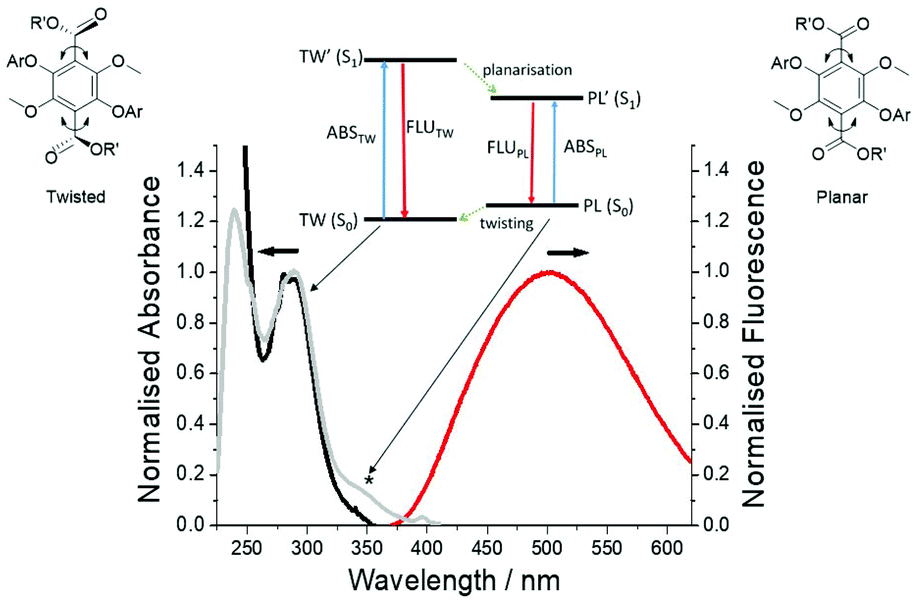 | ||
| Fig. 7 Absorption (black), excitation (grey) and fluorescence (red) spectra of M2 in dilute DCM. λex = 320 nm. The asterisk notes the additional feature seen in the excitation spectrum to longer wavelength. Insert shows a simple picture of the absorption and emission from the two different conformers. PL = planar, TW = twisted and that the potential energy surfaces TW′ ≠ TW and PL ≠ PL. | ||
Given that the absorption profile for a planar structure is more red-shifted, the additional feature seen in the excitation spectrum is assigned to a planar conformation, which is formed following deactivation of the planar S1 state. This planar conformation must relax readily to the lower energy twisted conformation under normal conditions. The low-energy absorption feature is much more prominent (λabs = 365 nm) in the spectrum for PolyM2 (see ESI S20†). In addition, the emission profile contains a slight shoulder at ca. 428 nm, which is not observed for the monomer (Fig. 7). As the absorption spectrum for M2 and PolyM2 are remarkably similar, the ground-state structures of the aromatic core are likely comparable and hence exhibit the twisted ester subunits. The vertical Franck–Condon transition in both cases produces the twisted S1 conformation. However, in PolyM2 the twisting of the ester groups to generate the planar conformation is more hindered and emission is also observed from this state, explaining the high-energy shoulder in the emission profile. This type of dual emission is not uncommon in molecular systems that can twist and is observed in porphyrin dimers.24
Major perturbation of the ground-state structure is observed for PolyM1 by inspection of its absorption spectrum; λABS is located at 300 nm but the profile is broad stretching to around 375 nm (Fig. 8). By comparison to the absorption profile of Fig. 7, the major conformer comprises ester groups twisted out of the plane, implying that the intramolecular hydrogen bonding is disrupted when the monomer M1 is polymerised. The broadness of the absorption profile does suggest that there is still a small distribution of the hydrogen bonded planar conformer in the ground state. The excitation spectrum however clearly indicates that this conformer is populated after deactivation of the first-excited state using the model discussed for PolyM2. That the excitation profile is more intense and the high-energy structured emission band is far more prominent suggests that the short ethylene glycol spacer severely restricts molecular rearrangement after excitation.
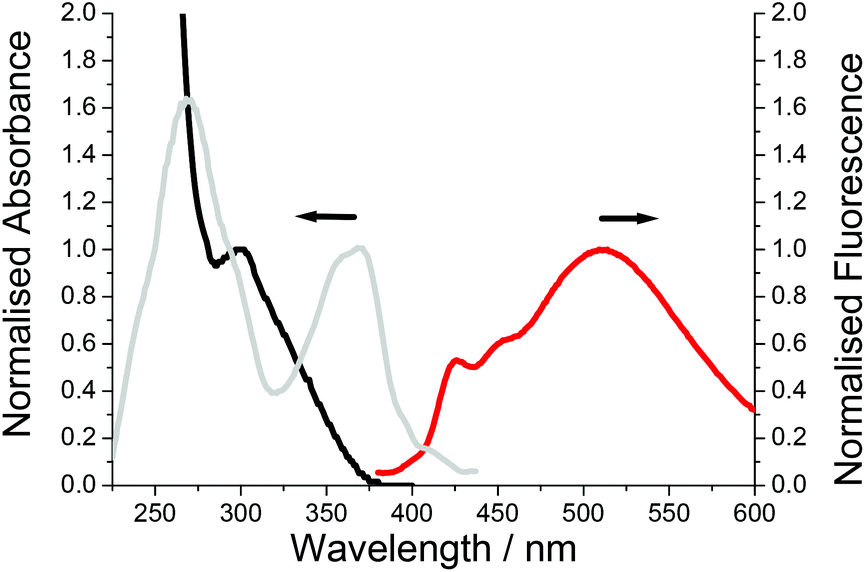 | ||
| Fig. 8 Absorption (black), excitation (grey) and fluorescence (red) spectra of PolyM1 in dilute DCM. λex = 320 nm. | ||
Conclusion
The two hydroxyl groups of diethyl 2,5-bis(tert-butyl)phenoxy-3,6-dihydroxyterephthalate are readily functionalised, and are ideal sites for crown ether formation. The ring size for the first example produced (3) is too small for complete cation encapsulation, but the methodology could be readily adapted to increase the number of ethylene glycol spacer units. However, as observed in the macrocycle forming reaction the 2 + 2 adduct may also be formed, and dominate the mixture of products. The 2 + 2 adduct (D3) is interesting as its core resembles the well-known 34-Crown-10 macrocycle.25 This compound has found wide application in the binding of electron deficient moieties such a paraquat (N,N′-dimethyl-4,4-bipyridinium chloride).26 Preliminary results support the same type of self-assembly with D3 despite the steric demands imposed by the additional ester and aromatic groups.The highly substituted phthalate unit is also a prime candidate for the production of fluorescent polymeric materials, but direct transesterification is not appropriate due to problems caused by degradation. A better approach appears to be the attachment of short spacer groups under milder transesterification conditions, which contain an end group that facilitates polymerization. Even so, using the esterification method detailed in this work only short oligomers are produced. A RAFT approach may be more conducive to control the growth of the polymer.
Experimental
Materials
Reagents and solvents were purchased from Sigma Aldrich at the highest purity possible and were used without further purification unless otherwise stated. Standard solvents were dried by literature methods before being distilled and stored under nitrogen over 4 Å molecular sieves. Thin layer chromatography (TLC) was performed with silica gel TLC plates. The spots were visualized under UV light at 312 nm and 365 nm to monitor progress of the reaction.1H and 13C NMR spectra were recorded on either the Bruker Avance III 300 MHz, Bruker Avance III HD 500 MHz or Bruker Avance III 700 MHz. 2D NMR such as 1H–1H correlation (COSY), 1H–13C heteronuclear single quantum correlation (HSQC), 1H–13C heteronuclear multiple bond correlation (HMBC) were used to substantiate 1H and 13C NMR assignments of the compounds. Deuterated chloroform (CDCl3), methanol (d4-MeOH) and dimethylsulphoxide (DMSO-d6) were used as the solvents and TMS as the internal standard. FTIR spectra were obtained using Varian 800 FTIR instrument (Varian Inc.) fitted with a diamond crystal plate ATR unit (Pike Technologies). Melting points of intermediates were measured using a Stuart™ melting point apparatus SMP3. Absorption spectra were recorded using UV-1800 UV-Vis Spectrophotometer. Matrix-assisted laser desorption ionization (MALDI) mass spectrometry was carried out using the Applied BioSystems Voyager DE-STR and Bruker ultrafleXtreme mass spectrometers at the EPSRC UK National Mass Spectrometry Facility at Swansea. Thermogravimetric analysis experiments (TGA) were carried out in a Mettler TGA SDTA 851e thermobalance. Approximately 8 mg of samples were heated from 30 to 650 °C at 10 °C min−1 in a nitrogen atmosphere (100 mL min−1). Calorimetric studies were carried out in a Mettler DSC822e Differential Scanning Calorimeter. Approximately 5 mg of samples were weighed into 40 μL aluminium crucibles covered with a pierced lid. The analyses were performed in dynamic mode at a heating and cooling rate of 10 °C min−1 using nitrogen as a purge gas (100 mL min−1). The number average (Mn) and weight average (Mw) molecular weight of the polymers were determined by the use of an Agilent 1200 Series GPC-SEC system consisting of three columns in series (PLgel 20 μm MIXED-A, PLgel 5 μm MIXED-D and PLgel 3 μm MIXED-E) and a refractive index detector. Monodispersed polystyrene (PS) standards were used for calibration and the mobile phase (THF with toluene as internal standard) was eluted at a flow rate of 1.0 mL min−1. The sample concentrations used were 5–10 mg mL−1.
Crystal structure data for 3 were collected at 150 K on an Xcalibur, Atlas, Gemini ultra diffractometer equipped with a fine-focus sealed X-ray tube (λCuKα = 1.54184 Å) and an Oxford Cryosystems CryoStreamPlus open-flow N2 cooling device. Cell refinement, data collection and data reduction were undertaken via the software CrysAlisPro.27 Intensities were corrected for absorption empirically using spherical harmonics. All structures were solved using XT28 and refined using XL.29 All non-hydrogen atoms were refined as anisotropic. Hydrogen atoms were positioned with idealised geometry with the exception of those bound to heteroatoms the locations of which were identified using peaks in the Fourier difference map. The atomic displacement parameters of the hydrogen atoms were set to be an appropriate multiple of those of the parent atom.
Computational calculations were performed using a 32-bit version of Gaussian0930 on a quadruple-core Intel Xeon system with 4GB RAM. The calculations were run in parallel, fully utilising they multi-core processor. To reduce computational time low-level calculations were carried out to minimise structures using Hartree–Fock and a low basis set. Energy-minimised structures were then used to feed high-level DFT calculations starting firstly with B3LYP and the 3-21G basis set. The complexity of the basis set was then increased and the results compared to the previous calculations. The 6-311G basis set was deemed sufficient for comparison purposes. Simple MM+ calculations were performed using Chem3D and conformations were sampled using the dihedral angle driver and minimization function. Dynamic simulations used a step interval of 2 fs and a frame interval of 10 fs. The heating/cooling rate was set at 1 kcal/atom/ps and a target temperature of 300 K.
Synthesis of compounds 3 and D3
Anhydrous DCM (150 mL) was added to a flask (F1) containing 1 (6.2 g, 11.3 mmol) and TetraEGdiOTs (5.7 g, 11.3 mmol) under N2. In a separate flask (F2), anhydrous MeCN (50 mL) was added to K2CO3 (4.7 g, 34.0 mmol) and KPF6 (3.1 g, 16.8 mmol). F2 was heated to reflux and was kept under N2. The solution in F1 was then added dropwise to F2 via a syringe pump over 3 days. The reaction mixture was vigorously stirred for another 6 days under N2 and reflux conditions. The flasks were both covered in aluminium foil throughout the 9-days-long reaction with TLC monitoring. At day 9, the reaction mixture was cooled to RT and the suspension was filtered and washed with DCM. The filtrate was dried under reduced pressure. The resulting residue was re-dissolved with DCM and washed with H2O. The combined organic layer was rapidly flushed through a silica plug with DCM as eluent and gradually switched to ethyl acetate (EA). All of the flushes were combined and dried under reduce pressure to yield a viscous oil. The crude material was then dry packed into a silica column with DCM and was eluted with petroleum ether:EA (5:1), slowly increasing the ratio to 4:1. From the viscous oil, several products (3 and D3) were isolated and identified. The isolated 3 was recrystallized from hot MeOH (1 mg 3 in 0.4 mL hot MeOH) to yield pure 3 (0.80 g, 1.1 mmol, 10%) in the form of sugar-like rectangular crystals while D3 (1.60 g, 1.1 mmol, 19%) was isolated as a viscous oil. Crystals of 3 suitable for X-ray crystallography were grown from hot MeOH upon slow cooling.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1179 (aromatic ester C–O stretching vibrations), 838, 811 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + K]+ 747.3126, calcd for C40H52O11K: 221.0608 and [M + NH4]+ 726.3834, calcd for C40H56O11N: 726.3848.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1177 (aromatic ester C–O stretching vibrations), 836, 815 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + NH4]+ 1434.7349, calcd for C80H108O22N: 1434.7358.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1179 (aromatic ester C–O stretching vibrations), 838, 811 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + K]+ 747.3126, calcd for C40H52O11K: 221.0608 and [M + NH4]+ 726.3834, calcd for C40H56O11N: 726.3848.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1177 (aromatic ester C–O stretching vibrations), 836, 815 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + NH4]+ 1434.7349, calcd for C80H108O22N: 1434.7358.
Synthesis of M1
Ethylene glycol (26 mL, 0.5 mol, excess) was added to a flask containing PI (1.0 g, 1.8 mmol) and Zn(OAc)2·2H2O (50.0 mg, 0.2 mmol). The distillation set-up was covered in aluminium foil and was heated to 175 °C with a sand bath for 17 hours with TLC monitoring. The reaction mixture was cooled to RT. The suspension was suction filtered, the resulting solid was rinsed with H2O and left to dry. The solid was dissolved in DCM and quickly flushed through a silica plug with DCM/EA. The flushes were collected and dried under reduced pressure. The resulting solid was dry packed into a silica column with DCM:petroleum ether (1:1) and was eluted with the same solvent gradually increasing to DCM and finally DCM with 5% MeOH. Note that this column chromatography had to be done quickly as the compound seemed to decompose on silica. Alternative purification attempts with alumina column and recrystallisation were both unsuccessful. The purification yielded greenish yellow crystals (0.25 g, 0.4 mmol, 24%). 1H NMR (400 MHz, CDCl3) δ 10.27 (s, 2H, H6), 7.41–7.34 (m, 4H, H4), 6.88–6.81 (m, 4H, H3), 4.30–4.25 (m, 4H, H1), 3.55–3.49 (m, 4H, H2), 1.31 (s, 18H, H5). 13C NMR (101 MHz, CDCl3) δ 168.59 (C8), 155.96 (C12), 147.41 (C11), 145.46 (C13), 138.69 (C10), 126.88 (C4), 114.79 (C9), 113.68 (C3), 68.53 (C1), 60.45 (C2), 34.40 (C14), 31.59 (C5). FT-IR (vmax, cm−1) 3574 (alcohol O–H stretching vibration), 2960 (asymmetric C–H stretching vibration from –CH2–, –CH3), 1676 (aromatic ester CO stretching vibration), 1507 (aromatic CC stretching vibration), 1227, 1174 (aromatic ester C–O stretching vibrations), 826, 721 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + H]+ 583.2538, calcd for C32H39O10: 583.2538 and [M + NH4]+ 600.2798, calcd for C32H42O10N: 600.2803. Crystals suitable for X-ray crystallography were grown in the fridge via vapour diffusion from chloroform/hexane. X-ray crystal structure and refinement data are available in Table 1 of ESI.†
Synthesis of 2
CH3I (0.8 mL, 12.9 mmol) was added dropwise into a flask containing 1 (1.5 g, 2.7 mmol) and K2CO3 (1.9 g, 13.7 mmol) pre-dissolved in acetone (106 mL) at reflux. The reaction mixture was left to stir for 1 hour with TLC monitoring. It was then cooled to RT. The suspension was filtered and the filtrate was dried under reduced pressure. The crude was recrystallized with hot hexane to form colourless sugar-like tiny cube crystals (1.03 g, 1.8 mmol, 67%). 1H NMR (300 MHz, CDCl3) δ 7.34–7.22 (m, 4H, H4), 6.86–6.80 (m, 4H, H3), 4.16 (q, J = 7.1 Hz, 4H, H1), 3.81 (s, 6H, H6), 1.29 (s, 18H, H5), 1.08 (t, J = 7.1 Hz, 6H, H2). 13C NMR (75 MHz, CDCl3) δ 164.00 (C10), 155.58 (C11), 146.48 (C7), 145.40 (C12), 142.04 (C8), 126.84 (C9), 126.34 (C4), 115.11 (C3), 62.08 (C6), 61.89 (C1), 34.34 (C13), 31.63 (C5), 13.96 (C2). FT-IR (vmax, cm−1) 2959 (asymmetric C–H stretching vibration from –CH2–, –CH3), 1736 (aromatic ester CO stretching vibration), 1507 (aromatic CC stretching vibration), 1205, 1170 (aromatic ester C–O stretching vibrations), 821 (aromatic C–H out of plane bending vibration). (+)nanoESI-FTMS (m/z): found [M + H]+ 579.2946, calcd for C34H43O8: 579.2952. Crystals suitable for X-ray crystallography were grown from acetone at RT. X-ray crystal structure and refinement data are available in Table 2 of ESI.†
Synthesis of M2
1,4-Butandiol (35 mL, 0.4 mol, excess) was added to a flask containing 2 (1.8 g, 3.1 mmol) and Ti(OnBu)4 (0.3 mL, 0.9 mmol). The distillation set-up was covered in aluminium foil and was heated to 220 °C with a sand bath for 76 hours with TLC monitoring. The reaction mixture was cooled to RT. H2O was added to the reaction mixture until a suspension formed before it was suction filtered. The residue was allowed to dry and was dissolved in DCM. The solution was quickly flushed through silica plug with DCM/EA. The first flush containing unreacted 2 was discarded while the subsequent flushes were combined and dried under reduced pressure. The resulting viscous oil was wet packed into a silica column with minimum amount of DCM and was eluted with DCM:EA (19:1) gradually increasing the polarity to 4:1. The fractions were collected in vials and were combined based on TLC. From the crude, several products were isolated and identified as follows: M2 (0.21 g, 0.3 mmol, 10%) and its asymmetric form M4 (0.35 g, 0.6 mmol, 19%). Both products were isolated in the form of a brown oil.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1213, 1170 (aromatic ester C–O stretching vibrations), 830 (aromatic C–H out of plane bending vibration), 733 (C–C skeletal rocking vibration of –(CH2)2– and –(CH2)3–). (+)nanoESI-FTMS (m/z): found [M + Na]+ 689.3285, calcd for C38H50O10Na: 689.3296 and [M + NH4]+ 684.3737, calcd for C38H54O10N: 684.3742.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1213, 1170 (aromatic ester C–O stretching vibrations), 830 (aromatic C–H out of plane bending vibration), 733 (C–C skeletal rocking vibration of –(CH2)2– and –(CH2)3–). (+)nanoESI-FTMS (m/z): found [M + H]+ 623.3206, calcd for C36H47O9: 623.3215.
Synthesis of M3
1,4-Butandiol (7.3 mL, 0.1 mol, excess) was added to a flask containing 3 (0.4 g, 0.6 mmol) and Ti(OnBu)4 (80 μL, 0.2 mmol). The distillation set-up was covered in aluminium foil and was heated to 220 °C with a sand bath for 18 hours with TLC monitoring. The reaction mixture was cooled to RT. H2O was added to the reaction mixture until a suspension formed before it was suction filtered. The residue was allowed to dry and was wet packed into a silica column with a minimum amount of DCM and was eluted with DCM with 0.5% MeOH gradually increasing polarity to DCM with 3.5% MeOH. The fractions were collected in vials and were combined based on TLC. From the crude, several products were isolated and identified as follows: M3 (0.20 g, 0.3 mmol, 43%) and its asymmetric form M5 (0.13 g, 0.2 mmol, 31%).O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1179 (aromatic ester C–O stretching vibrations), 837, 811 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + NH4]+ 814.4367, calcd for C44H64O13N: 814.4372.
O stretching vibration), 1507 (aromatic CC stretching vibration), 1212, 1179 (aromatic ester C–O stretching vibrations), 837, 811 (aromatic C–H out of plane bending vibrations). (+)nanoESI-FTMS (m/z): found [M + NH4]+ 770.4103, calcd for C42H60O12N: 770.4110.
Synthesis of PolyM1
M1 (250.0 mg, 0.4 mmol) was dissolved in anhydrous THF (5 mL) at RT. Upon complete solubilisation, the solution was cooled to 0 °C in an ice bath. TEA (180 μL, 1.3 mmol) was added. In a separate flask, terephthaloyl chloride (87.0 mg, 0.4 mmol) was dissolved in anhydrous THF (5 mL). The solution was then added dropwise into the reaction mixture via a syringe pump over the course of 1 hour. The reaction mixture was allowed to react at RT over 3 days covered in aluminium foil. The reaction mixture was then suction filtered and the filtrate was dried under reduced pressure. The residue was purified by re-dissolving in CHCl3 and added to Et2O. The precipitate was dissolved in small amounts of CHCl3 and allowed to dry to yield a semi-opaque pale yellow film (61.0 mg, 7.2 μmol, 2%).Synthesis of PolyM2
M2 (250.0 mg, 0.4 mmol) was dissolved in anhydrous THF (5 mL) at RT. Upon complete solubilisation, the solution was cooled to 0 °C in an ice bath. TEA (160 μL, 1.1 mmol) was added. In a separate flask, terephthaloyl chloride (76.0 mg, 0.4 mmol) was dissolved in anhydrous THF (4 mL). The solution was then added dropwise into the reaction mixture via a syringe pump over the course of 1 hour. The reaction mixture was allowed to react at RT over 4 days covered in aluminium foil. The reaction mixture was then suction filtered and the filtrate was dried under reduced pressure. The residue was purified by re-dissolving in CHCl3 and added to Et2O. The precipitate was dissolved in small amounts of CHCl3 and allowed to dry to yield an off-white opaque brittle film (28.00 mg) which was found to be a mixture. The filtrate from the purification was dried under reduced pressure, re-dissolved in CHCl3 and re-precipitated in hexane twice to yield PolyM2, similarly being off-white, opaque and brittle (263.00 mg, 33.6 μmol, 8%).Synthesis of PolyM3
M3 (115.0 mg, 0.1 mmol) was dissolved in anhydrous THF (2 mL) at RT. Upon complete solubilisation, the solution was cooled to 0 °C in an ice bath. TEA (60 μL, 0.4 mmol) pre-dissolved in anhydrous THF (0.8 mL) was added. In a separate flask, terephthaloyl chloride (29.0 mg, 0.1 mmol) was dissolved in anhydrous THF (1 mL). The solution was then added dropwise into the reaction mixture via a syringe over the course of 1 hour. The reaction mixture was allowed to react at RT over 6 days covered in aluminium foil. The reaction mixture was then filtered through a pipette cotton plug and the filtrate was dried under reduced pressure. The residue was purified by dissolving in THF and added to CHCl3. The filtrate was dried under reduced pressure to yield PolyM3 as an off-white opaque polymer film (95.8 mg).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the mass spectrometry service at Swansea University for collecting mass spectra and Newcastle University for financial support. Dr C. Wills is thanked for collecting the DOSY spectra. Financial support from the Spanish Ministerio de Ciencia, Innovación y Universidades (ENE2017-86711-C3-3-R) is gratefully acknowledged.Notes and references
- B. Lepoittevin and P. Roger, Handbook of Engineering and Speciality Thermoplastics: Polyethers and Polyesters, ed. S. Thomas and P. M. Visakh, Wiley, Hoboken, 2011, p. 97 Search PubMed.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 342 RSC.
- K. Ravindranath and R. A. Mashelka, Chem. Eng. Sci., 1986, 41, 2197 CrossRef CAS.
- D. Giovanelli and E. Farella, J. Sens., 2016, 9391850 Search PubMed.
- M. Y. Lee, J. Hong, E. K. Lee, H. Yu, H. Kim, J. U. Lee, W. Lee and J. H. Oh, Adv. Chem. Mater., 2016, 26, 1445 CAS.
- K. Peng, B. Wang and C. Ji, J. Appl. Polym. Sci., 2017, 134, 44907 Search PubMed.
- T. Kuwabara, T. Nakashima, T. Yamaguchi and K. Takahashi, Org. Electron., 2012, 13, 1136 CrossRef CAS.
- K. Hong and J.-L. Leez, J. Electrochem. Soc., 2007, 154, H782 CrossRef CAS.
- G. Kocak, C. Tuncera and V. Bütün, J. Polym. Chem., 2017, 8, 144 RSC.
- A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037 CrossRef CAS.
- (a) G. Wegner, Angew. Chem., Int. Ed., 1981, 20, 361 CrossRef; (b) G. Inzelt, J. Electrochem. Sci. Eng., 2018, 8, 3 CAS.
- P. Wang, X. Wang, Y. Ling, M. Wang, S. Ding, W. Shen, Z. Wang, Y. Wang and F. Liu, Radiat. Meas., 2018, 119, 80 CrossRef CAS.
- D. Gopalakrishnan and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14853 CrossRef.
- S. J. Peighambardoust, S. Rowshanzamir and M. Amjadi, Int. J. Hydrogen Energy, 2010, 35, 9349 CrossRef CAS.
- Y. Salinas, R. Martínez-Mánez, M. D. Marcos, F. Sancenón, A. M. Costero, M. Parraad and S. Gilad, Chem. Soc. Rev., 2012, 41, 1261 RSC.
- A. C. Benniston, T. P. L. Winstanley, H. Lemmetyinen, N. V. Tkachenko, R. W. Harrington and C. Wills, Org. Lett., 2012, 14, 1374 CrossRef CAS.
- Software used in the Cambridge Crystallographic Data Centre.
- P. Liebing, A. Zaeni, F. Olbrich and F. T. Edelmanna, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 1757 CrossRef CAS.
- D. L. Clark, J. C. Gordon, J. C. Huffman, R. L. Vincent-Hollis, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 5903 CrossRef CAS.
- (a) Y. Chiang, J. Kresge and J. Jones, J. Am. Chem. Soc., 1994, 116, 8358–8359 CrossRef CAS; (b) J. M. A. Spiteri, C. J. Mallia, G. J. Scerri and D. C. Magri, Org. Biomol. Chem., 2017, 15, 10116–10121 RSC; (c) T. Terashima, M. Kawabe, Y. Miyabara, H. Yoda and M. Sawamoto, Nat. Commun., 2013, 4, 2321 CrossRef; (d) J. B. Smith, S. H. Kerr, P. S. White and A. J. M. Miller, Organometallics, 2017, 36, 3094–3103 CrossRef CAS; (e) F. Arnaud-Neu, R. Delgado and S. Chaves, Pure Appl. Chem., 2003, 75, 71–102 CAS; (f) R. B. Sharma, P. Kebarle and A. T. Blades, J. Am. Chem. Soc., 1984, 106, 510–516 CrossRef CAS; (g) J. C. Chambron and M. Meyer, Chem. Soc. Rev., 2009, 38, 1663–1673 RSC.
- J. Lui, S.-G. Bian, M. Xiao, S.-J. Wang and Y.-Z. Meng, J. Appl. Polym. Sci., 2010, 115, 3401 CrossRef.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913 RSC.
- (a) A. Marchesi, S. Brenna and G. A. Ardizzoia, Dyes Pigm., 2019, 161, 457 CrossRef CAS; (b) A. K. Gupta, A. Kumar, R. Singh, M. Devi, A. Dhir and C. P. Pradeep, ACS Omega, 2018, 3, 14341 CrossRef CAS.
- F. Hammerer, G. Garcia, P. Charles, A. Sourdon, S. Achelle, M.-P. Teulade-Fichou and P. Maillard, Chem. Commun., 2014, 50, 9529 RSC.
- B. L. Allwood, N. Spencer, H. Shahriari-Zavareh, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1061 RSC.
- B. L. Allwood, N. Spencer, H. Shahriari-Zavareh, J. F. Stoddart and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 1064 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compounds, DFT calculation results, binding data and absorption spectra. CCDC 2018846. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01533d |
| This journal is © The Royal Society of Chemistry 2020 |