
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An amide hydrogen bond templated [1]rotaxane displaying a peptide motif – demonstrating an expedient route to synthetic mimics of lasso peptides†‡
Matthew J.
Young§
 ,
Geoffrey R.
Akien
and
Nicholas H.
Evans
*
,
Geoffrey R.
Akien
and
Nicholas H.
Evans
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: n.h.evans@lancaster.ac.uk
First published on 24th June 2020
Abstract
The rapid synthesis of an amide hydrogen bond templated [1]rotaxane is reported – demonstrating a potential pathway to synthetic analogues of lasso peptides. The structures of the [1]rotaxane and its unthreaded isomer have been characterized by NMR spectroscopy and modelled using DFT calculations.
Introduction
Lasso peptides are a fascinating class of structurally unusual peptides consisting of a polypeptide chain that threads its “tail” through a macrolactam “ring” (Fig. 1).1 Due to their entangled structure, lasso peptides have been found to evade degradation by enzymes, and examples have been shown to possess a number of useful bioactive characteristics, including antimicrobial activity,2 receptor antagonism3 and enzyme inhibition.4 The total synthesis of lasso peptides is in its infancy, with the first chemical synthesis of a lasso peptide having only very recently been reported.5 However, it has been established that the conformationally constrained amino acids displayed in the “loop region” (see Fig. 1) are important in determining the biological activity of a lasso peptide.6 Synthetic [1]rotaxanes7 are structurally equivalent to lasso peptides, but examples that display peptide sequences are rare.8,9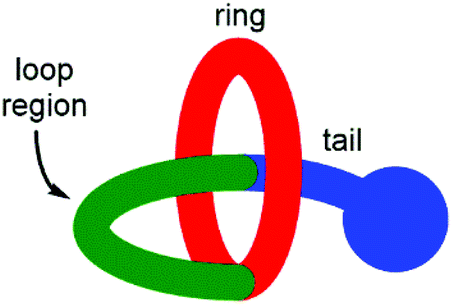 | ||
| Fig. 1 Schematic representation of structure of a lasso peptide and [1]rotaxane. | ||
Recently we have rapidly prepared [2]rotaxanes10–12 by use of hydrogen bond templation13 and CuAAC click chemistry to stopper a simple amide half-axle component threaded through an isophthalamide macrocycle.14 Here we are reporting an adaptation of this method to rapidly prepare a [1]rotaxane displaying a Gly–Gly–Gly–Gly peptide sequence within the key loop region, by appending and then cyclizing a peptide chain on a novel “scaffold” [2]rotaxane. Successful preparation of the target [1]rotaxane was confirmed by NMR spectroscopy and mass spectrometry. Structural comparison with the non-interlocked isomer of the [1]rotaxane has also been undertaken by NMR spectroscopy and DFT calculations.
When compared to prior work from the laboratories of Coutrot8 and Bode,9 the synthetic route reported here has the potential to generate better mimics of naturally occurring lasso peptides. In the previous cases, the entangled structure arises from the non-amide templation of a protonated ammonium passing through a 24-crown-8 macrocycle. Here, amide motifs (used to template formation of the interlocked structure through the carbonyl O atom of the axle to the isophthalamide N–Hs of the macrocycle) are to be found in both the axle and ring components – not just the peptide sequence in the loop region.
Results and discussion
The synthesis of [1]rotaxane 6 is presented in Scheme 1. All novel compounds were characterized by NMR spectroscopy (1H, 13C and where applicable 19F), IR spectroscopy and high resolution mass spectrometry. Scaffold [2]rotaxane 4 was prepared using azide 1, alkyne 2 and amino macrocycle 3. Azide 1 was synthesized according to our previously reported procedures in two chromatography-free steps.10 Novel alkyne 2 was prepared in four chromatography-free steps (see ESI, page S3‡), with inclusion of a glycine unit as part of 2 to guarantee sufficient bulk for the rotaxane stopper. Novel amino macrocycle 3 was synthesized in four steps from commercially available starting materials (see ESI, pages S6 & S9‡).¶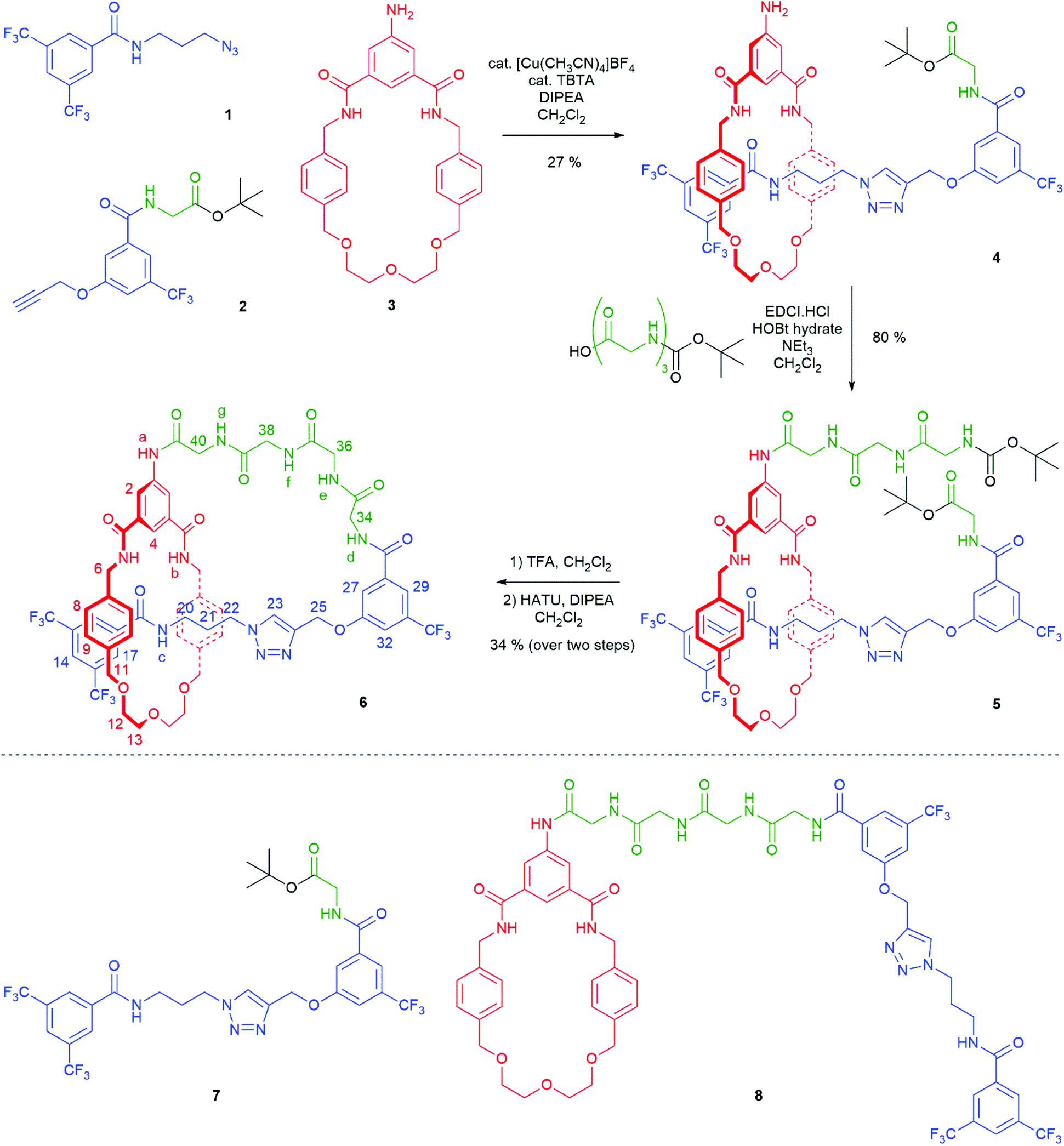 | ||
| Scheme 1 Synthesis of tetra-glycine [1]rotaxane 6via “scaffold” [2]rotaxane 4, and structures of axle 7 and unthreaded “[1]rotaxane” isomer 8. | ||
[2]Rotaxane 4 was synthesized using our recently reported hydrogen bond templated strategy.10–12 Macrocycle 3 was dissolved in CH2Cl2 in the presence of 1.1 equivalents of azide 1 and 1.1 equivalents of alkyne 2. Catalytic [Cu(CH3CN)4]BF4 and TBTA and 1.2 equivalents of DIPEA were added and the reaction stirred overnight at room temperature under an inert atmosphere. After aqueous work-up and careful silica chromatography, [2]rotaxane 4 was isolated in 27% yield.15
The 1H NMR spectra of [2]rotaxane 4, along with that of non-interlocked macrocycle 3 and axle 7 (for synthesis of axle 7 see ESI, page S30‡) are shown in Fig. 2. The upfield shift and splitting of aromatic macrocycle protons 8 and 9 and the upfield shifts of axle protons 17, 20, 21, 22, 23 and 25 in [2]rotaxane 4 compared to macrocycle 3 and axle 7 are consistent with the threading of the axle component through the macrocycle. The downfield shift of internal isophthalamide proton 4 is indicative of hydrogen bonding to the carbonyl oxygen of the axle amide. Evidence to support the co-conformation of [2]rotaxane 4 depicted in Scheme 1 is provided by the appearance of through-space correlations in the 1H ROESY NMR spectrum between protons 8 and 9 of the macrocycle and axle proton environments 17, 20 and 21 (see ESI, page S16‡). For [2]rotaxane 4, the resonances of macrocyclic protons 6 and 11 are split due to the directionality of the threaded axle that causes the protons attached to the same carbon to become inequivalent. In addition, the molecular ion peak [M + Na]+ for [2]rotaxane 4 was observed in the positive-ion electrospray mass spectrum at m/z = 1209 Da (see ESI, page S17‡).
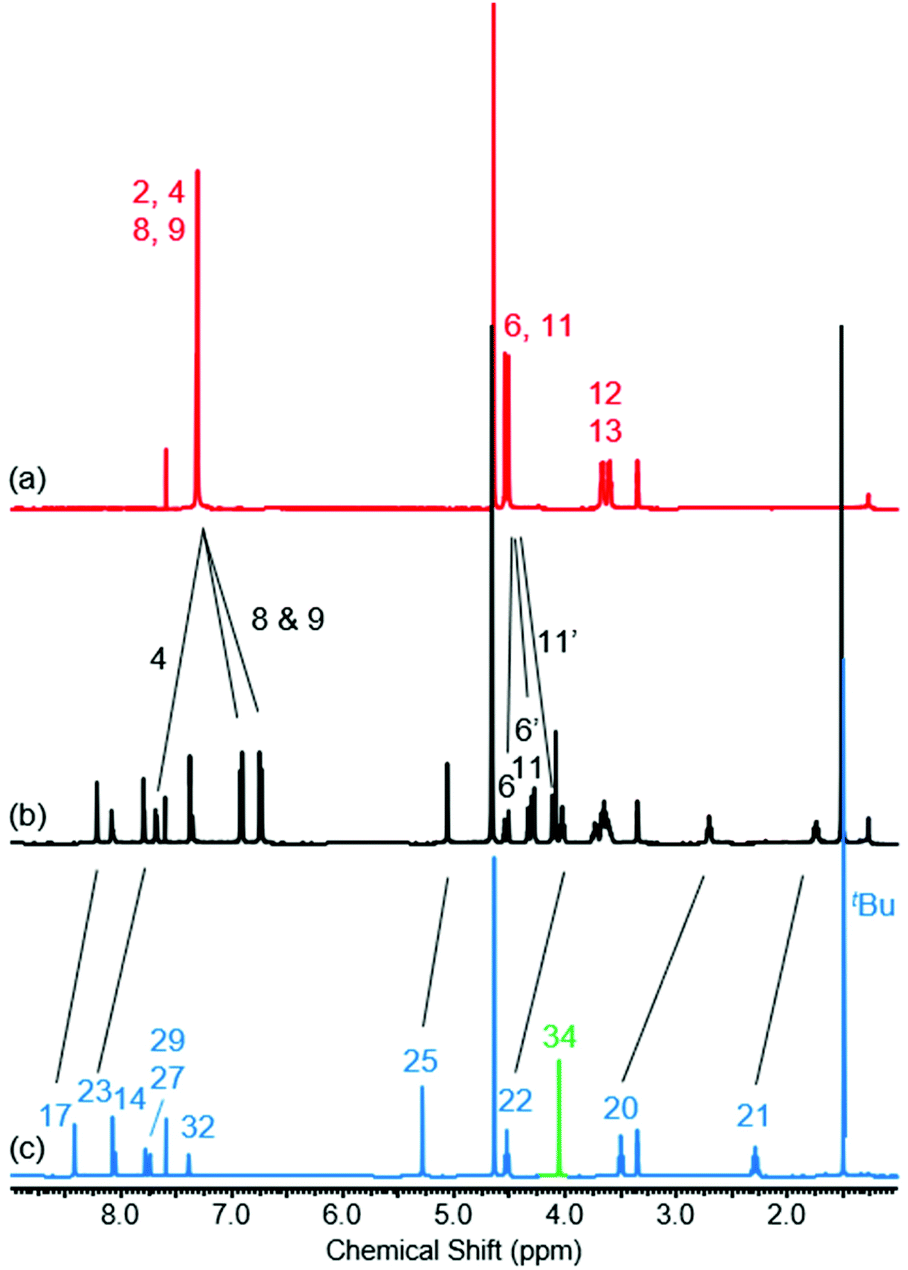 | ||
Fig. 2
1H NMR spectra of (a) macrocycle 3, (b) [2]rotaxane 4 and (c) axle 7 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CDCl3/CD3OD, 400 MHz, 298 K). For atom labels see Scheme 1. :1 CDCl3/CD3OD, 400 MHz, 298 K). For atom labels see Scheme 1. | ||
With [2]rotaxane 4 in-hand, preparation of the target [1]rotaxane was executed. Boc–Gly–Gly–Gly was appended to the macrocyclic amine group of [2]rotaxane 4 by use of EDC coupling in 80% yield (using three equivalents of the protected tripeptide). Treatment of Boc-peptide appended [2]rotaxane 5 with TFA in CH2Cl2 simultaneously removed both the Boc and tert-butyl ester. Cyclization was carried out using HATU16 at a dilute concentration of 2 mmol L−1 of deprotected 5 in CH2Cl2 to minimize the effect of competing oligomerization reactions, with [1]rotaxane 6 being isolated in 34% yield (over two steps from 5) after aqueous work-up and silica gel column chromatography. Confirmation of the successful isolation of target [1]rotaxane 6 was ascertained by NMR spectroscopy and mass spectrometry – a molecular ion peak [M + Na]+ for 6 being observed in the positive-ion electrospray mass spectrum at m/z = 1306 Da (see ESI, page S24‡).
To study the structural effects of [1]rotaxane 6 being interlocked, the unthreaded analogue 8 was also prepared (for details of synthesis see ESI, page S33‡). Inspection of the 1H NMR spectra (recorded in D6-DMSO due to the very limited solubility of 8, Fig. 3) reveals the anticipated shielding of protons 8, 9, 17, 20, 21, 22 and 25 (but interestingly not 23) and deshielding of proton 4 for [1]rotaxane 6 compared to unthreaded 8. In addition, the axle amide proton c is notably upfield in 6 compared to 8, consistent to being present within the shielding macrocyclic cavity of [1]rotaxane 6. There is also the expected marked splitting of macrocyclic resonances 6 and 11 for [1]rotaxane 6 arising from the directionality of the threaded axle. Other notable differences in chemical shift between the two molecules are for amide proton a (significantly greater than for any of the glycine amide protons d–g) and aromatic axle proton 27, tentatively attributed to differences in hydrogen bonding involving these protons (either intramolecularly and/or with the strongly hydrogen bond accepting DMSO solvent).
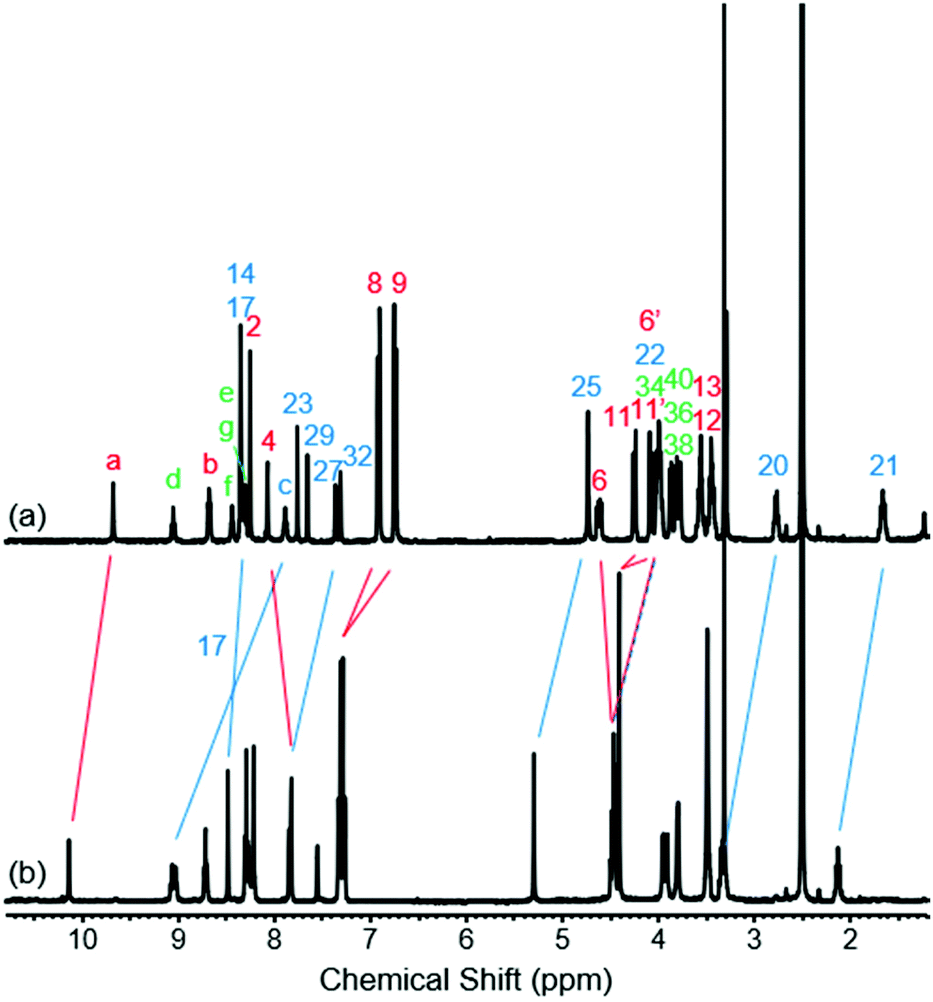 | ||
| Fig. 3 1H NMR spectra of (a) [1]rotaxane 6 and (b) unthreaded isomer 8 (D6-DMSO, 400 MHz, 298 K). For atom labels see Scheme 1. | ||
NMR experiments at elevated temperatures provided no evidence of dethreading of 6 or threading of 8. However, they were used to study variation in the chemical shift of NH resonances as a function of temperature, as has been used in the structural characterization of proteins in aqueous solution (see ESI, pages S45–S47‡).17 In aqueous solution, temperature coefficients Δδ/ΔT > −4.5 ppb K−1 are attributed to intramolecular hydrogen bonds, while those that are more negative (<−4.5 ppb K−1) are deemed to be freely interacting with solvent. The limited literature data for rigid peptides in D6-DMSO is consistent with all coefficients being slightly less negative on average.18
For unthreaded 8, all amide temperature coefficients are in the range −3.3 to −4.5 ppb K−1, which is consistent with relatively unrestricted solvent accessibility. For [1]rotaxane 6, the bulk of the NH environments have similar coefficients (in the range of −3.0 to −4.3 ppb K−1) but there are two key exceptions. Axle amide c has a coefficient of −1.1 ppb K−1 implying it is inaccessible to solvent and macrocycle protons b have an unusually negative coefficient of −6.7 ppb K−1. We attribute these deviations to the various non-covalent interactions arising from the axle component threading through the macrocycle cavity.
Further structural comparison between 6 and 8 was possible from 1H diffusion NMR experiments (see ESI, pages S48 & S49‡).19 For [1]rotaxane 6 the diffusion coefficient D = 1.28 ± 0.03 × 10−10 m2 s−1, whereas for unthreaded analogue 8D = 1.10 ± 0.03 × 10−10 m2 s−1. In other words, the threaded [1]rotaxane diffuses faster which is consistent with a more compact 3D structure in solution. The observed D for both 6 and 8 is consistent with monomeric forms in solution,20 and is also in agreement with a comparison of the DFT-simulated (B3LYP/6-31G*) structures of [1]rotaxane 6 and its unthreaded isomer 8 (Fig. 4).21
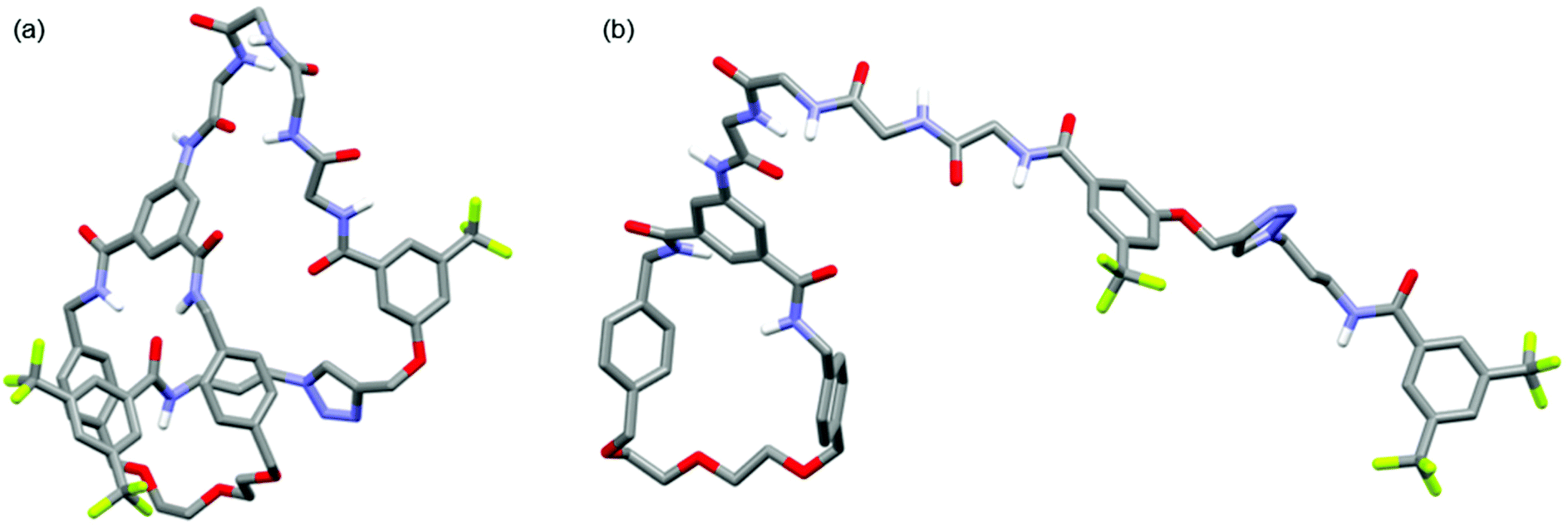 | ||
| Fig. 4 Minimum energy DFT calculated structures of (a) [1]rotaxane 6 and (b) unthreaded isomer 8 (C–H hydrogen atoms omitted for clarity). | ||
Conclusions
An expedient route to a peptide containing amide hydrogen bond templated [1]rotaxane has been demonstrated by the synthesis and structural characterization of a tetra-glycine containing [1]rotaxane. Looking forward, substitution of Boc–Gly–Gly–Gly by other aliphatic side-chained peptide sequences should be straightforward, and in principle with modifications to protecting group strategy more exotic amino acid side-chains (or non-peptide motifs) could be incorporated. Hence delicate bioactive functionality could be displayed in the loop region, with protection against enzymatic degradation9 and/or conformational control being provided by the entangled [1]rotaxane structure. Work on some of these avenues of research, as well as developing alternative synthetic routes to novel [1]rotaxanes, are ongoing in our laboratories.Experimental
General information
Commercially available solvents and chemicals were used without further purification unless stated. Dry solvents, NEt3 and DIPEA were purchased dry and stored under an inert atmosphere. Cu(CH3CN)4BF4 was stored in desiccator over P4O10. Deionised water was used in all cases.Silica gel with a 60 Å particle size was used as the stationary phase for column chromatography. Analytical TLC was used to monitor the progress of column chromatography and analytical TLC plates were examined under short wavelength (λ = 254 nm) UV light. Preparatory TLC was carried out on silica gel possessing a fluorescent indicator to allow for examination with short wavelength UV light.
IR spectra of neat samples were recorded on an Agilent Technologies Cary 630 FTIR spectrometer. NMR spectra of samples dissolved in deuterated solvent were recorded on a Bruker AVANCE III 400 spectrometer at 298 K. 1D NMR spectra were referenced using residual solvent peaks, and assigned using standard 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC NMR experiments, with data being reported according to the atom labels as defined in the structures below. Electrospray mass spectra of dissolved samples, diluted in CH3CN or CH3OH, were recorded on a Shimadzu LCMS IT ToF instrument. Melting points were recorded on a Gallenkamp capillary melting point apparatus and are uncorrected.
Experimental procedures
:1 to 96:4) to yield pure product 4 as a colourless film (52 mg). In addition, axle 7 (102 mg) was also isolated. Further pure product 4 was isolated after prep TLC of mixed fractions (17 mg). Total yield of [2]rotaxane 4: 69 mg, 27%.
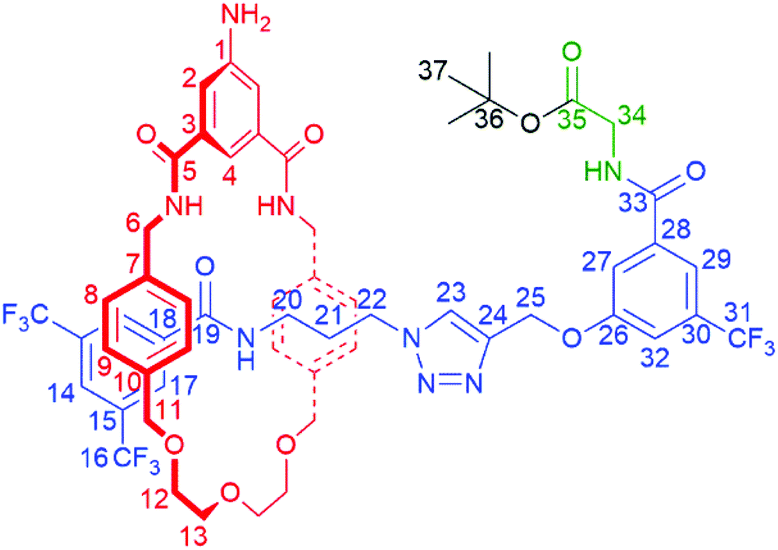 :2).
:2).
ν
max
/cm
−1
(neat): 3350 (N–H), 2920 (C–H), 1740 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester), 1650 (CO amide), 1600, 1520, 1450, 1360, 1280, 1230, 1130, 1030.
O ester), 1650 (CO amide), 1600, 1520, 1450, 1360, 1280, 1230, 1130, 1030.
δ
H
(400 MHz; CDCl
3
/CD
3
OD 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1): 8.18 (2H, s, C17H), 8.05 (1H, s, C14H), 7.75–7.76 (2H, m, C23H & C29H), 7.67 (1H, t, 4J = 1.5 Hz, C4H), 7.64 (1H, s, C27H), 7.43 (1H, t, 3J = 5.4 Hz, C20NH), 7.36 (2H, d, 4J = 1.5 Hz, C2H), 7.32 (1H, s, C32H), 6.88 (4H, d, 3J = 7.9 Hz, C8H), 6.71 (4H, d, 3J = 7.9 Hz, C9H), 5.02 (2H, s, C25H), 4.49 (2H, d, 2J = 14 Hz, C6H – diastereotopic) 4.25–4.30 (4H, m, C6H – diastereotopic & C11H – diastereotopic) 4.05–4.08 (4H, m, C11H – diastereotopic & C34H), 3.99 (2H, t, 3J = 7.0 Hz, C22H), 3.54–3.72 (8H, m, C13H & C12H), 2.67 (2H, br s, C20H), 1.66–1.73 (2H, m, C21H), 1.47 (9H, s, C37H).
:1): 8.18 (2H, s, C17H), 8.05 (1H, s, C14H), 7.75–7.76 (2H, m, C23H & C29H), 7.67 (1H, t, 4J = 1.5 Hz, C4H), 7.64 (1H, s, C27H), 7.43 (1H, t, 3J = 5.4 Hz, C20NH), 7.36 (2H, d, 4J = 1.5 Hz, C2H), 7.32 (1H, s, C32H), 6.88 (4H, d, 3J = 7.9 Hz, C8H), 6.71 (4H, d, 3J = 7.9 Hz, C9H), 5.02 (2H, s, C25H), 4.49 (2H, d, 2J = 14 Hz, C6H – diastereotopic) 4.25–4.30 (4H, m, C6H – diastereotopic & C11H – diastereotopic) 4.05–4.08 (4H, m, C11H – diastereotopic & C34H), 3.99 (2H, t, 3J = 7.0 Hz, C22H), 3.54–3.72 (8H, m, C13H & C12H), 2.67 (2H, br s, C20H), 1.66–1.73 (2H, m, C21H), 1.47 (9H, s, C37H).
δ
C
(100 MHz; CDCl
3
/CD
3
OD 1:1): 170.0 (C35), 168.7 (C5), 167.9 (C33), 165.0 (C19), 159.4 (C26), 149.1 (C1), 143.1 (C24), 138.1 (C7), 137.1 (C10), 137.1 (sic, C28), 136.7 (C18), 136.0 (C3), 132.8 (q, 2J = 32 Hz, C30), 132.3 (q, 2J = 34 Hz, C15), 129.6 (C9), 129.1 (C17), 129.0 (C8), 125.7 (C23), 125.3 (br, C14), 124.0 (q, 1J = 271 Hz, C16), 118.0 (C2), 117.6 (br, C29), 117.3 (C27) 115.9 (br, C32), 114.2 (C4), 82.9 (C36), 74.3 (C11), 71.6 (C13), 70.4 (C12), 62.4 (C25), 48.7 (C22), 44.8 (C6), 43.1 (C34), 38.1 (C20), 29.6 (C21), 28.4 (C37). Quartet for C31 lost in baseline.
δ
F
(377 MHz; CDCl
3
/CD
3
OD 1:1): −63.1 (C16F3), −63.4 (C31F3).
m/z (ES): 1209.4047 ([M + Na]+ C57H59F9N8NaO10 requires 1209.4103).
:2 to 92:8) to yield pure product 5 as a white film (68 mg, 80%).
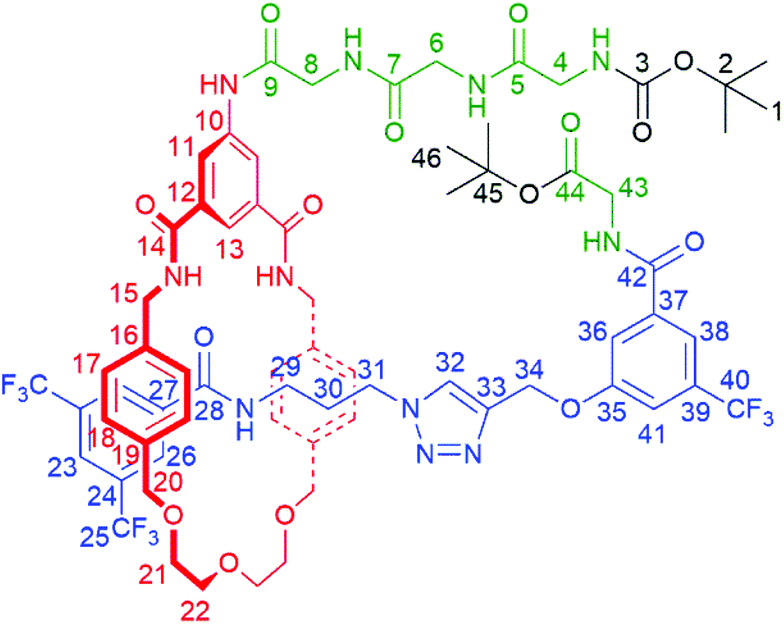 :4).
:4).
ν
max
/cm
−1
(neat): 3320 (N–H), 3070 (C–H), 2920 (C–H), 1650 (CO), 1600, 1520, 1450, 1370, 1280, 1130, 1030.
δ H (400 MHz; CDCl 3 ): 9.45 (1H, s, C9NH), 8.19 (4H, s, C11H & C26H), 8.07–8.10 (2H, br s, C5HNH or C7HNH & C42HNH), 8.04 (1H, s, C13H), 8.00 (1H, s, C23H), 7.90 (1H, br s, C5HNH or C7HNH), 7.84 (2H, br s, C14NH), 7.81 (1H, s, C32H), 7.70 (1H, s, C38H), 7.58 (1H, s, C36H), 7.24 (2H, s, C28NH & C41H), 6.94 (4H, d, 3J = 7.9 Hz, C17H), 6.74 (4H, d, 3J = 7.9 Hz, C18H), 6.28 (1H, br s, C3HNH), 4.99 (2H, s, C34H), 4.60–4.65 (2H, m, C15H – diastereotopic), 4.29 (2H, d, 2J = 10 Hz, C20H – diastereotopic), 4.20–4.25 (2H, m, C15H – diastereotopic), 4.15 (2H, d, 3J = 5.5 Hz, C43H), 4.09 (2H, d, 2J = 10 Hz, C20H – diastereotopic), 4.01 (6H, m, C6H, C8H & C31H), 3.87 (2H, d, 3J = 5.5 Hz, C4H), 3.56–3.75 (8H, m, C21H & C22H), 2.65–2.69 (2H, app q, C29H), 1.70–1.73 (2H, m, C30H), 1.51 (9H, s, C46H), 1.35 (9H, s, C1H).
δ C (100 MHz; CDCl 3 ): 171.9 (C5), 170.7 (C7 or C9), 169.9 (C44), 168.0 (C7 or C9), 166.8 (C14), 166.2 (C42), 164.0 (C28), 158.5 (C35), 156.7 (C3), 142.1 (C33), 138.8 (*), 137.3 (C16), 136.2 (C19), 136.0 (*), 135.8 (*), 135.1 (*), 131.4 (q, 2J = 32 Hz, C24 and/or C39), 128.8 (C18), 128.3 (C17 & C26), 124.8 (C32), 124.5 (C23), 123.1 (q, 1J = 271 Hz, C25 and/or C40), 121.6 (C11), 120.3 (br, C13), 117.0 (br, C38), 116.4 (C36), 115.0 (br, C41), 82.7 (C45), 80.1 (C2), 73.6 (C20), 70.8 (C22), 69.5 (C21), 62.0 (C34), 47.7 (C31), 44.5 (C15), 44.3 (C4), 43.7 (C6 or C8), 43.4 (C6 or C8), 42.8 (C43), 37.3 (C29), 28.7 (C30), 28.2 (C1), 28.1 (C46). * = C10 or C12 or C27 or C37.
δ F (377 MHz; CDCl 3 ): −62.5 (C25F3), −62.7 (C40F3).
m/z (ES): 1480.5282 ([M + Na]+ C68H76F9N11NaO15 requires 1480.5271).
:4 to 90:10) to yield pure product 6 as a white film (6.0 mg, 34%).
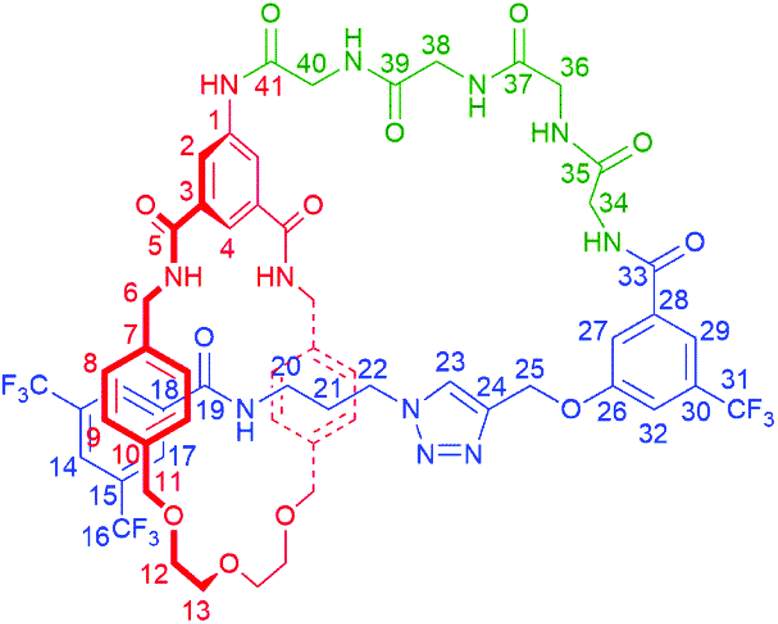 :6).
:6).
ν
max
/cm
−1
(neat): 3300 (N–H), 3080 (C–H), 2920 (C–H), 2870 (C–H), 1650 (CO), 1600, 1530, 1450, 1360, 1330, 1280, 1170, 1130, 1030.
δ
H
(400 MHz; CDCl
3
/CD
3
OD 1:1): 8.23 (2H, d, 4J = 1.5 Hz, C2H), 8.14 (1H, s, C23H), 8.11 (2H, br s, C17H), 8.02 (2H, app t, C4H & C14H), 7.65 (1H, s, C29H), 7.38 (1H, s, C27H), 7.15 (1H, s, C32H), 6.91 (4H, d, 3J = 8.0 Hz, C8H), 6.76 (4H, d, 3J = 8.0 Hz, C9H), 4.83 (2H, s, C25H), 4.60–4.62 (2H, obscured doublet, C6H – diastereotopic), 4.30 (2H, d, 2J = 10 Hz, C11H – diastereotopic), 4.19 (2H, s, C34H), 4.12–4.17 (4H, m, C22H & C6H – diastereotopic), 4.09 (2H, d, 2J = 10 Hz, C11H – diastereotopic), 4.03 (2H, s, C38H or C40H), 3.99 (2H, s, C36H), 3.92 (2H, s, C38H or C40H), 3.71–3.76 (2H, m, C13H – diastereotopic), 3.55–3.66 (6H, C12H & C13H – diastereotopic), 2.61 (2H, t, 3J = 7.4 Hz, C20H), 1.76–1.83 (2H, app quin, C21H).
δ
C
(100 MHz; CDCl
3
/CD
3
OD 1:1): 172.3 (C35), 172.1 (C37/C39/C41), 171.6 (C37/C39/C41), 169.4 (C37/C39/C41), 168.3 (C5 & C33), 164.7 (C19), 159.2 (C26), 142.9 (C24), 139.3 (C1 or C3), 138.1 (C7), 137.1 (C10), 136.8 (C18 or C28), 136.5 (C18 or C28), 136.1 (C1 or C3), 132.7 (q, 2J = 33 Hz, C30), 132.2 (q, 2J = 34 Hz, C15), 129.4 (C9), 128.9 (C8 & C17), 126.5 (C23), 125.3 (br, C14), 122.7 (C2), 121.5 (C4), 117.9 (br, C29), 117.0 (C27), 115.2 (br, C32), 74.2 (C11), 71.6 (C13), 70.3 (C12), 62.4 (C25), 48.6 (C22), 45.0 (C6), 44.3 (C34), 44.3 (sic, C38 or C40), 43.8 (C38 or C40), 43.6 (C36), 37.8 (C20), 29.5 (C21). Quartets for C16 & C31 not fully resolved in 120–130 ppm region.
δ
F
(377 MHz; CDCl
3
/CD
3
OD 1:1): −63.1 (C16F3), −63.5 (C31F3).
m/z (ES): 1306.3971 ([M + Na]+ C59H58F9N11NaO12 requires 1306.4015).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. H. E. expresses his appreciation for funding from Lancaster University (Faculty of Science & Technology Research Grant) and the Joy Welch Educational Charitable Trust. We also wish to thank the following colleagues at Lancaster University: Dr Mike Coogan & Dr Nathan Halcovitch for the collecting and processing of X-ray crystallographic data, Dr David Rochester for the recording of mass spectra and Dr Michael Peach for computational advice.Notes and references
- For reviews about lasso peptides see: (a) M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC; (b) J. D. Hegemann, H. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS; (c) H. Martin-Gómez and J. Tulla-Puche, Org. Biomol. Chem., 2018, 16, 5065–5080 RSC.
- (a) R. A. Salomón and R. N. Farías, J. Bacteriol., 1992, 174, 7428–7435 CrossRef PubMed; (b) M. Iwatsuki, H. Tomoda, R. Uchida, H. Gouda, S. Hirono and S. Ōmura, J. Am. Chem. Soc., 2006, 128, 7486–7491 CrossRef CAS; (c) T. A. Knappe, U. Linne, S. Zirah, S. Rebuffat, X. Xie and M. A. Marahiel, J. Am. Chem. Soc., 2008, 130, 11446–11454 CrossRef CAS; (d) W. L. Cheung-Lee, M. E. Parry, A. J. Cartagena, S. A. Darst and A. J. Link, J. Biol. Chem., 2019, 294, 6822–6830 CrossRef CAS PubMed.
- (a) R. Katahira, K. Shibata, M. Yamasaki, Y. Matsuda and M. Yoshida, Bioorg. Med. Chem., 1995, 3, 1273–1280 CrossRef CAS PubMed; (b) T. A. Knappe, U. Linne, X. Xie and M. A. Marahiel, FEBS Lett., 2010, 584, 785–789 CrossRef CAS PubMed.
- (a) G. Helynck, C. Dubertret, J. F. Mayaux and J. Leboul, J. Antibiot., 1993, 46, 1756–1757 CrossRef CAS PubMed; (b) N. R. Braddman, F. J. Piscotta, J. Hauver, E. A. Campbell, A. J. Link and S. A. Darst, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 1273–1278 CrossRef PubMed.
- M. Chen, S. Wang and X. Yu, Chem. Commun., 2019, 55, 3323–3326 RSC.
- T. A. Knappe, F. Manzenrieder, C. Mas-Moruno, U. Linne, F. Sasse, H. Kessler, X. Xie and M. A. Marahiel, Angew. Chem., Int. Ed., 2011, 50, 8714–8717 CrossRef CAS PubMed.
- Selected examples of synthetic [1]rotaxanes: (a) K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568–13569 CrossRef CAS PubMed; (b) Z. Xue and M. F. Mayer, J. Am. Chem. Soc., 2010, 132, 3274–3276 CrossRef CAS PubMed; (c) H. Li, H. Zhang, Q. Zhang, Q.-W. Zhang and D.-H. Qu, Org. Lett., 2012, 14, 5900–5903 CrossRef CAS PubMed; (d) H. Li, J.-N. Zhang, W. Zhou, H. Zhang, Q. Zhang, D.-H. Qu and H. Tian, Org. Lett., 2013, 15, 3070–3073 CrossRef CAS PubMed; (e) B. Xia and M. Xue, Chem. Commun., 2014, 50, 1021–1023 RSC; (f) P. Waelès, C. Clavel, K. Fournel-Marotte and F. Coutrot, Chem. Sci., 2015, 6, 4828–4836 RSC; (g) Y. Wang, J. Sun, Z. Liu, M. S. Nassar, Y. Y. Botros and J. F. Stoddart, Chem. Sci., 2017, 8, 2562–2568 RSC; (h) A. Saura-Sanmartin, A. Martinez-Cuezva, A. Pastor, D. Bautista and J. Berna, Org. Biomol. Chem., 2018, 16, 6980–6987 RSC; (i) J.-J. Yu, L.-Y. Zhao, Z.-T. Shi, Q. Zhang, G. London, W.-J. Liang, C. Gao, M.-M. Li, X.-M. Cao, H. Tian, B. L. Feringa and D.-H. Qu, J. Org. Chem., 2019, 84, 5790–5802 CrossRef CAS PubMed.
- C. Clavel, K. Fournel-Marotte and F. Coutrot, Molecules, 2013, 18, 11553–11575 CrossRef CAS PubMed.
- F. Saito and J. W. Bode, Chem. Sci., 2017, 8, 2878–2884 RSC.
- B. E. Fletcher, M. J. G. Peach and N. H. Evans, Org. Biomol. Chem., 2017, 15, 2797–2803 RSC.
- N. H. Evans and G. R. Akien, Supramol. Chem., 2018, 30, 758–764 CrossRef.
- C. E. Gell, T. A. McArdle-Ismaguilov and N. H. Evans, Chem. Commun., 2019, 55, 1576–1579 RSC.
- For a recent review on the hydrogen bond templated synthesis of rotaxanes (and catenanes) see: N. H. Evans, Eur. J. Org. Chem., 2019, 3320–3343 CrossRef CAS.
- Selected examples of other, non-CuAAC click, stoppering of simple amide half-axles threaded through mono-isophthalamide macrocycles to generate [2]rotaxanes: (a) D. A. Leigh and A. R. Thomson, Org. Lett., 2006, 8, 5377–5379 CrossRef CAS; (b) A. Vidonne and D. Philp, Tetrahedron, 2008, 64, 8464–8475 CrossRef CAS; (c) T. Kosikova, N. I. Hassan, D. B. Cordes, A. M. Z. Slawin and D. Philp, J. Am. Chem. Soc., 2015, 137, 16074–16083 CrossRef CAS PubMed; (d) A. Vidonne, T. Kosikova and D. Philp, Chem. Sci., 2016, 7, 2592–2603 RSC.
- The modest isolated yield of [2]rotaxane 4 is attributed to oxidation of the macrocyclic amino functionality, in particular during chromatographic purification.
- HATU – (1-[bis(dimethylaminomethylene]-1H-1,2,3-triazolo[4,5-b]pyridinium oxide hexafluorophosphate) – is widely used for the macrocyclization of peptides. For reviews of this topic see: (a) C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed; (b) Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC; (c) D. G. Rivera, G. M. Ojeda-Carralero, L. Reguera and E. V. Van der Eycken, Chem. Soc. Rev., 2020, 49, 2039–2059 RSC.
- (a) N. J. Baxter and M. P. Williamson, J. Biomol. NMR, 1997, 9, 359–369 CrossRef CAS PubMed; (b) T. Cierpicki and J. Otlewiski, J. Biomol. NMR, 2001, 21, 249–261 CrossRef CAS PubMed.
- M. Llinas and M. P. Klein, J. Am. Chem. Soc., 1975, 97, 4731–4737 CrossRef CAS.
- For reviews on the use of diffusion NMR experiments in supramolecular chemistry see: (a) S. V. Kharlamov and S. K. Latypov, Russ. Chem. Rev., 2010, 79, 635–653 CrossRef CAS; (b) L. Avram and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199–3202 CrossRef CAS PubMed.
- The DFT structure of [1]rotaxane 6 reveals that all amides in the loop (a & d–g) can be in a trans conformation. Depiction of amide a as cis in Scheme 1 arises from attempting to achieve a 2D depiction of [1]rotaxane 6 without unusual bond lengths or angles.
Footnotes |
| † Underlying data for this paper are provided in the Experimental section and ESI,‡ along with electronic copies of NMR spectra (including fid files) and computational output files being available from: DOI: 10.17635/lancaster/researchdata/372. |
| ‡ Electronic supplementary information (ESI) available: Further experimental procedures; copies of spectral characterization data; crystallographic data for macrocycle 3; details of computational modelling. CCDC 1995143. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ob01190h |
| § Current address: Institute of Organic Chemistry, Albert-Ludwigs-University of Freiburg, Albertstrasse 21, 79104 Freiburg, Germany. |
| ¶ Single crystals of macrocycle 3 suitable for X-ray structure determination were obtained by slow evaporation of a chloroform/methanol solution. The solved structure (CCDC 1995143‡) and further details are to be found in ESI, page S50.‡ |
| This journal is © The Royal Society of Chemistry 2020 |