
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Methyl-β-carboline alkaloids: structure-dependent photosensitizing properties and localization in subcellular domains†
M. Paula
Denofrio‡
a,
Federico A. O.
Rasse-Suriani‡
ab,
Jose M.
Paredes
c,
Federico
Fassetta
a,
Luis
Crovetto
 *c,
Maria D.
Giron
d,
Rafael
Salto
d,
Bernd
Epe
e and
Franco M.
Cabrerizo
*a
*c,
Maria D.
Giron
d,
Rafael
Salto
d,
Bernd
Epe
e and
Franco M.
Cabrerizo
*a
aInstituto Tecnológico de Chascomús (INTECH), Universidad Nacional de San Martín (UNSAM) - Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Av. Intendente Marino Km 8.2, CC 164 (B7130IWA), Chascomús, Argentina. E-mail: fcabrerizo@intech.gov.ar
bInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas (INIFTA), CCT-La Plata, Universidad Nacional de La Plata, Diag. 113 y 64 (1900), La Plata, Argentina
cDepartment of Physical Chemistry, Faculty of Pharmacy, Unidad de Excelencia en Química Aplicada a Biomedicina y Medioambiente (UEQ), University of Granada, Cartuja Campus, 18071 Granada, Spain. E-mail: luiscrovetto@ugr.es
dDepartment of Biochemistry and Molecular Biology II, Faculty of Pharmacy, Unidad de Excelencia en Quimica Aplicada a Biomedicina y Medioambiente (UEQ), University of Granada, Cartuja Campus, 18071 Granada, Spain
eInstitute of Pharmacy and Biochemistry, University of Mainz, Staudingerweg 5, Mainz, Germany
First published on 1st July 2020
Abstract
N-Methyl-β-carboline (βC) alkaloids, including normelinonine F (1b) and melinonine F (2b), have been found in a vast range of living species playing different biological, biomedical and/or pharmacological roles. Despite this, molecular bases of the mechanisms through which these alkaloids would exert their effect still remain unknown. Fundamental aspects including the photosensitizing properties and intracellular internalization of a selected group of N-methyl-βC alkaloids were investigated herein. Data reveal that methylation of the βC main ring enhances its photosensitizing properties either by increasing its binding affinity with DNA as a biomolecular target and/or by increasing its oxidation potential, in a structure-dependent manner. As a general rule, N(9)-substituted βCs showed the highest photosensitizing efficiency. With the exception of 2-methyl-harminium, all the N-methyl-βCs investigated herein induce a similar DNA photodamage profile, dominated largely by oxidized purines. This fact represents a distinctive behavior when comparing with N-unsubstituted-βCs. On the other hand, although all the investigated compounds might accumulate mainly into the mitochondria of HeLa cells, methylation provides a distinctive dynamic pattern for mitochondrial uptake. While rapid (passive) diffusion is most probably reponsible for the prompt uptake/release of neutral βCs, an active transport appears to mediate the (reatively slow) uptake of the quaternary cationic βCs. This might be a consequence of a distinctive subcellular localization (mitochondrial membrane and/or matrix) or interaction with intracellular components. Biomedical and biotechnological implications are also discussed herein.
Introduction
β-Carbolines (βCs) are a set of endogenous alkaloids present in a vast range of living species.1–8 The βC skeleton construction occurs through the catalysed Pictet–Spengler reaction.9–11 In particular, N-methyl-β-carboline alkaloids, including normelinonine F (1b) and melinonine F (2b), were isolated from different mammals’ body tissues and fluids such as brain, cerebrospinal and plasma.1–7 A broad spectrum of biological, biomedical and pharmacological activities has been reported for these alkaloids:(i) unsubstituted full aromatic norharmane (1a) crosses the blood–brain barrier penetrating into the brain,12,13 where it is enzymatically converted into the quaternary βCs 1b and 2,9-dimethyl-norharmanium (1d).14
(ii) 1b, 1d and other related quaternary βCs were described as potential pathogenic agents in several neurological disorders, including Parkinson's disease.15–17 The main mechanism through which these alkaloids would exert their effect seems to be the mitochondrial respiratory inhibition.18
(iii) 1b was also found to be a potent inhibitor of acetylcholinesterase (AchE), a target for the clinical treatment of Alzheimer and other memory impairments of vascular origin.19
(iv) on the contrary, 9-methyl-norharmane (1c) would exert neuroprotective and neuron-differentiating effects.20
(v) βC derivatives having a short alkyl or benzyl substituent at position N(9) were found to be promising antitumor and antiproliferative agents. Compounds substituted at both N(9) and C(3) positions showed a remarkable enhancement of the antitumor activity with a concomitant decrease in the acute toxicity and neurotoxicity. The magnitude and extent of the latter effects strongly depend on the chemical nature of substituents.21
(vi) N-Methyl-βCs are also quite promising antimicrobial agents with remarkable activity against a great variety of microorganisms.22–36 For example, 1b and 2b showed anti-Plasmodium activity, even against the chloroquine- and pyrimethamine-resistant Plasmodium falciparum K1 type.32,33 In addition, 1b and its chloro-derivatives displayed potent cyanobactericidal and algicidal activity against a group of photosynthetic aquatic organisms including Microcystis aeruginosa, Synechococcus sp. and Kirchneriella contorta.34 In a very recent study, several βC derivatives were identified as novel and potent antiviral agents against Herpes simplex-1 and -2 virus. In particular, 9-methyl-derivatives 1c and 2c showed the highest inhibitory effect on virus replication.35 The mechanism of action appears to be at the transcriptional level where these alkaloids could bind to the viral genome and block transcription, giving rise to a delay (or even suppression) in the early and late protein expression.35,36 Interestingly, 1c and 2c could also play a role at the post-translational level, interfering with the late location of HHV infected cell polypeptide 0 (ICP0) at the cytoplasm where this protein normally inhibits antiviral signalling and promotes viral replication.
(vii) Due to their planar chemical structure, most of the βCs37–40 including 1b and 2b interact with DNA.41 Partial intercalation of the indolic ring into the stacked base-pairs is the dominant mode of interaction, placing the βC-pyridine ring in a polar-protic environment.37–40
(viii) The photosensitizing properties of a selected group of βC alkaloids have been demonstrated on both cell-free and intracellular DNA and its components37–40,42–46 Interestingly, harmine (3a)38 as well as 3c40 and chloro-harmine derivatives39 were shown to be the most efficient photosensitizers among all the βCs investigated to date. Thus, the methoxy group placed at position C(7) would play a key role enhancing the photosensitizing properties of the βC ring.
On the other hand, the broad variety of tuneable physicochemical and photochemical properties of βCs make these alkaloids quite a promising set of molecules for a great variety of biotechnological applications. Recently, βCs were used as photosensitizer agents and fluorescence markers carried by selective vehicles, based on albumin–folate conjugates, for tumour cell targeted PDT.45 Although encouraging results were obtained, the overall photodynamic efficiency depicted was relatively low. Thus, the search for novel and more efficient photosensitizers based on small and biocompatible molecules is still a challenge.
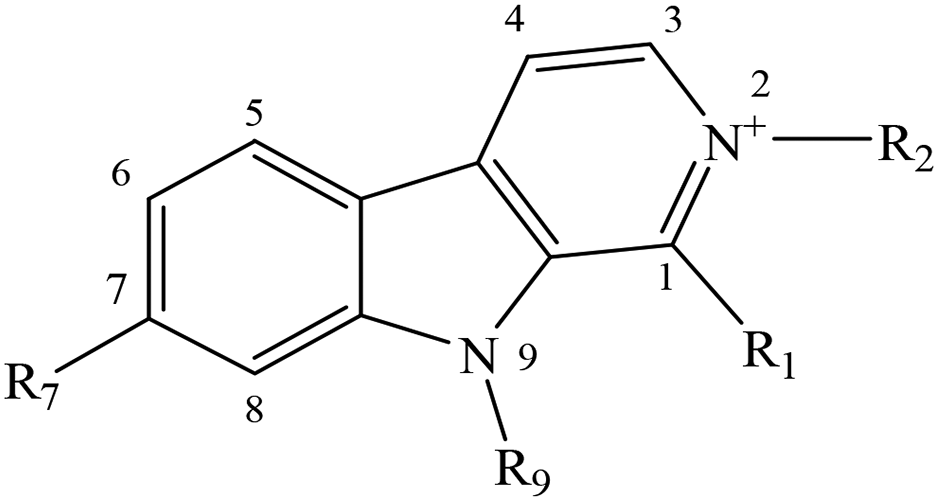
Despite the well-established biological and biomedical relevance of N-methyl-βCs, the molecular bases of the mechanisms through which these alkaloids would exert their effect still remain unclear. To gain further insight with these regards, fundamental aspects including the photosensitizing properties as well as the intracellular internalization of a selected group of N-methyl-βC alkaloids (Table 1) were investigated herein.
|
|
||||
|---|---|---|---|---|
| Short name (abbreviation) | R1 | R2 | R7 | R9 |
| Norharmane (1a) | –H | –H | –H | –H |
| 2-Methyl-norharmanium or normelinonine F (1b) | –H | –CH3 | –H | –H |
| 9-Methyl-norharmane (1c) | –H | –H | –H | –CH3 |
| 2,9-diMethyl-norharmanium (1d) | –H | –CH3 | –H | –CH3 |
| Harmane (2a) | –CH3 | –H | –H | –H |
| 2-Methyl-harmanium or melinonine F (2b) | –CH3 | –CH3 | –H | –H |
| 9-Methyl-harmane (2c) | –CH3 | –H | –H | –CH3 |
| 2,9-diMethyl-harmanium (2d) | –CH3 | –CH3 | –H | –CH3 |
| Harmine (3a) | –CH3 | –H | –OCH3 | –H |
| 2-Methyl-harminium (3b) | –CH3 | –CH3 | –OCH3 | –H |
| 9-Methyl-harmine (3c) | –CH3 | –H | –OCH3 | –CH3 |
| 2,9-diMethyl-harminium (3d) | –CH3 | –CH3 | –OCH3 | –CH3 |
Experimental
General
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480 carrying the plasmid ptac-denV) provided by L. Mullenders, Leiden. Endonuclease III (Endo III) from E. coli was kindly provided by S. Boiteux, Fontenay aux Roses, France. All repair endonucleases were tested for their incision at reference modifications according to the procedure described elsewhere.49
000 cells per cm2 on 25 mm coverslips to reach a 70–80% confluence. (i) Cells preparation for FLIM microscopy experiments: HeLa and HEK239 cells were washed (2 times) with phosphate buffered saline medium (PBS) and incubated during 5, 10, 15, 20 or 35 min with PBS and the corresponding βC (10 μM) at 37 °C. (ii) Cells preparation for colocalization studies: HeLa cells in DMEM free of FBS were first incubated with 16 nM of Mitotracker® and Lysotracker® in two different sets of experiments. After 20 min of incubation, 1d (10 μM) was added and co-incubated during 2 h. Cells were then washed (3 times) with PBS and fixed using filtered paraformaldehyde (2%). Fixed samples were again washed (3 times) with PBS and prepared for confocal microscope analysis.
480 carrying the plasmid ptac-denV) provided by L. Mullenders, Leiden. Endonuclease III (Endo III) from E. coli was kindly provided by S. Boiteux, Fontenay aux Roses, France. All repair endonucleases were tested for their incision at reference modifications according to the procedure described elsewhere.49
000 cells per cm2 on 25 mm coverslips to reach a 70–80% confluence. (i) Cells preparation for FLIM microscopy experiments: HeLa and HEK239 cells were washed (2 times) with phosphate buffered saline medium (PBS) and incubated during 5, 10, 15, 20 or 35 min with PBS and the corresponding βC (10 μM) at 37 °C. (ii) Cells preparation for colocalization studies: HeLa cells in DMEM free of FBS were first incubated with 16 nM of Mitotracker® and Lysotracker® in two different sets of experiments. After 20 min of incubation, 1d (10 μM) was added and co-incubated during 2 h. Cells were then washed (3 times) with PBS and fixed using filtered paraformaldehyde (2%). Fixed samples were again washed (3 times) with PBS and prepared for confocal microscope analysis.
Calf thymus DNA (ctDNA) absorption coefficient determination
241) was used as dialysis membrane.
The method was validated by determining the molar absorption coefficient at 260 nm (ε260 nm) of 2′-deoxyadenosine 5′-monophosphate (dAMP) in aqueous solution (ESI Fig. 2c and d†). When comparing data obtained herein (15.824 ± 30 M−1 cm−1) with the gravimetrically obtained value (15.060 M−1 cm−1)52 a relative error of ∼4% was observed.
Binding studies
The interaction of βC derivatives with ctDNA, in KH2PO4−NaOH (pH 7.4, 10 mM) buffer solutions, was studied by both steady-state and time-resolved fluorescence spectroscopy. Equipment and data analysis used were described elsewhere.40 The use of ctDNA as a model of genetic material allows a direct comparison with the information reported in the literature for related βCs.38–40,44DNA photoproduct measurements
The number of cyclobutane pyrimidine dimers (CPDscalc) were calculated as the difference between the number of sites recognized during incubation with both Endo IV and T4 endo V and the number of AP sites recognized by Endo IV alone. Likewise, the number of oxidatively generated damage on pyrimidine nucleobases (Ox-Pycalc) were calculated by combining the use of both Endo III and Endo IV enzymes. To further identify the photosensitized DNA damage, two different control experiments were carried out: light-controls (PM2 DNA irradiated in the absence of N-methyl-βCs) and dark-controls (mixtures of PM2 and N-methyl-βCs, at the highest concentrations, kept in the dark).
Fluorescence imaging microscopy
FLIM experiments were carried out on a MicroTime 200 instrument (PicoQuant), using a LDH-375 (PicoQuant) pulsed laser as the excitation source with a repetition rate of 20 MHz. The laser was directed into the sample using a dichroic mirror (F33-375RDC, AHF/Chroma) and an oil immersion objective (1.4 NA, 100×) of an inverted microscope (IX-71, Olympus). The emitted fluorescence was filtered by a cutoff filter (F76-405LP, AHF/Chroma) and focused onto a 75 μm pinhole. The detection filter was a FB450-40 bandpass filter (Thorlabs), and the fluorescence was detected using a single-photon avalanche diode (SPCM-AQR 14, PerkinElmer). Photon counting, imaging reconstruction and data acquisition were performed with a TimeHarp 200 TCSPC module (PicoQuant). Raster scanned images were recorded with a 512 × 512 pixels resolution. The fluorescence images were analyzed using the image processing package Fiji.56Colocalization studies were performed in a Leica SPS II confocal microscope equipped with a 405 nm diode laser and 633 nm HeNe laser. Excitations were sequentially switched between 405 and 633 nm, to pump 1d and the biomarkers, respectively. Emission signals were recorded in two different emission ranges: 410–490 and 640–750 nm, to collect 1d (cyan channel) and the red the biomarkers (red channel) fluorescent emission, respectively. The objective used was a PL APO 63x/1.2 CS water immersion. Fluorescence was acquired using a hybrid detector (HyD, Leica).
Results and discussion
DNA damage photoinduced by N-methyl-βCs
The photosensitizing properties of a set of N-methyl-βCs (Table 1) were explored herein using supercoiled extracellular DNA of bacteriophage PM2. DNA shows several sites of attack leading place to a broad spectrum of chemical reactions. The use of the latter DNA model, in combination with a set of incision repair endonucleases, allows the evaluation of all types of putative photosensitizing mechanisms.39 PM2 DNA was subject to UVA (365 nm) irradiation, in the presence of different concentrations of N-methyl-βCs. The number of single-strand breaks (SSBs) as well as DNA modifications recognized by the DNA repair glycosylase Fpg, which are mainly oxidatively modified purines (8-oxo-7,8-dihydroguanine and formamidopyrimidines) and sites of base loss (AP sites), were subsequently quantified (Fig. 1, left column). Similar dose-dependencies have already been reported for other related βCs.37–40,44,45,57 Briefly, the number of DNA modifications increases linearly with the compounds’ concentration. For all the investigated alkaloids Fpg-sensitive sites were found to be produced with the highest efficiency, whereas SSBs were produced with a very low or even null efficiency (<0.07 modifications/104 bp). Note that all DNA damage is mediated by the photoexcited βCs, since no damage was observed in both light-control (see data points at the y-intercept in Fig. 1a, left column) and dark-controls (see the empty stars at the maximum βC concentration depicted in Fig. 1a, left column).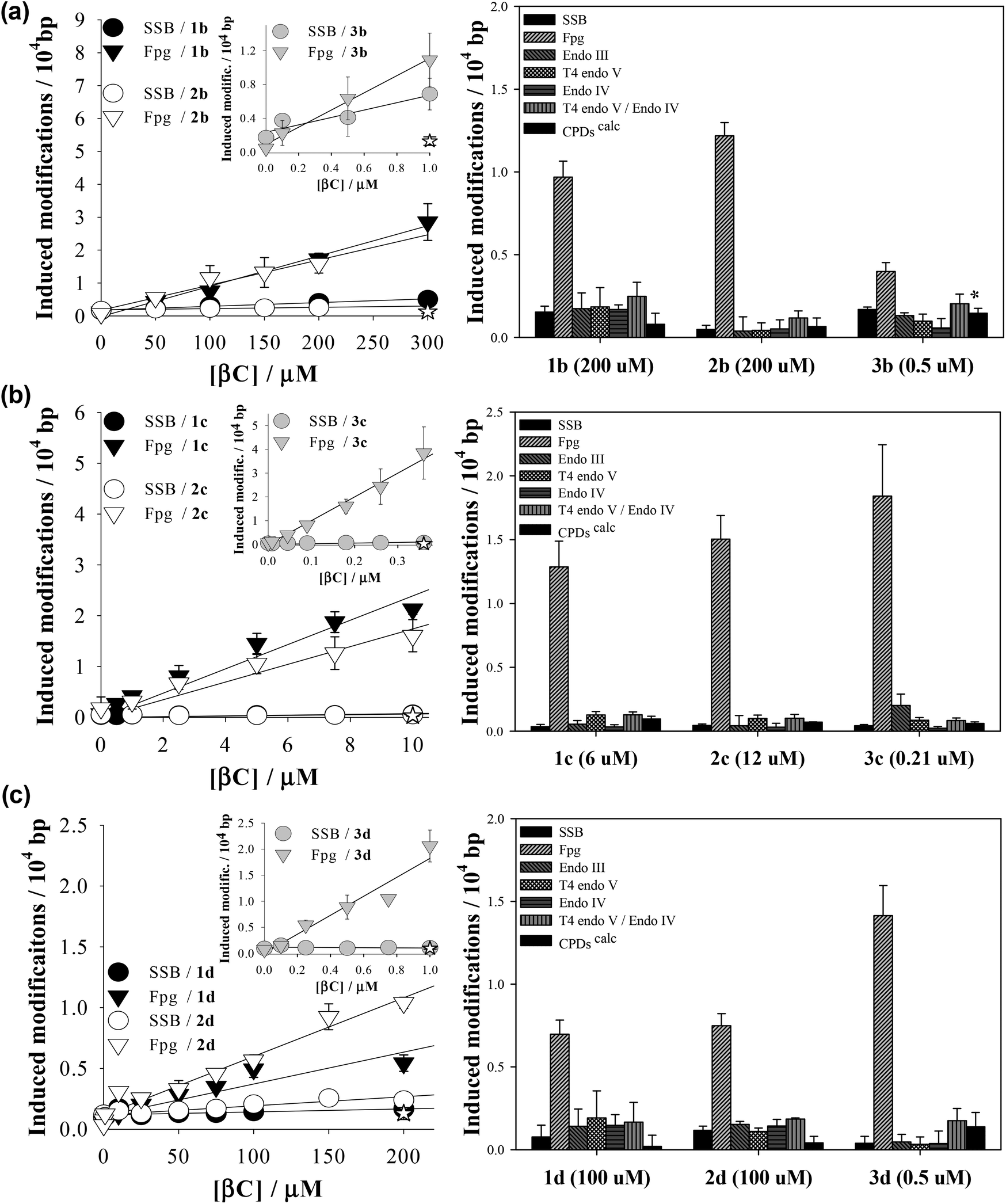 | ||
| Fig. 1 Left column: SSBs (circles) and Fpg-sensitive modifications (down triangles) induced in PM2 DNA by exposure to UVA light (366 ± 20 nm; 20 minutes) in the presence of different concentrations of (a) 2-methyl-βCs, (b) 9-methyl-βCs and (c) 2,9-dimethyl-βCs in phosphate buffer solutions (pH 7.4). Right column: DNA damage profiles showing the numbers of SSBs and several types of endonuclease sensitive modifications induced in PM2 DNA by photoexcited N-methyl-βCs. The concentrations are indicated in parenthesis. Data are the means of 5 independent experiments (±S.D.). | ||
Since βCs can lead to the formation of a wide range of DNA photoproducts,38,39 DNA damage profiles (i.e., the number of various other types of repair enzymes-sensitive modifications) photoinduced by N-methyl-βCs were also determined (Fig. 1, right column). In particular, four different repair endonucleases were used to selectively quantify different type of DNA modifications: • Fpg to detect oxidatively damaged purines and AP-sites (see above), • Endo III to recognize oxidized pyrimidines as well as AP sites, • T4 endo V to recognize cyclobutane pyrimidine dimers (CPDs) and a number of specific types of AP-sites, and • Endo IV to sense all types of AP-sites.
Data depict qualitative and quantitative structure–activity relationships:
(i) with the exception of 3b, all the N-methyl-derivatives investigated induce specific DNA base damage. I. e., Fpg-sensitive modifications represent the main types of photoproducts, while oxidized pyrimidines (Ox-Pycalc) as well as CPDs and SSBs are absent or generated in low relative yields. This contrasts with the DNA damage spectra previously determined for unsubstituted βC rings (1a, 2a and 3a), which induce a broader spectrum of DNA modifications (ESI Fig. 1†).38 It is generally accepted that Fpg-sensitive sites are produced either by type I or type II photosensitization mechanisms. It was demonstrated that the methylation of βCs’ ring has no significant effect on the intrinsic capability to photosensitize reactive oxygen species (ROS) formation7 but shifts the oxidation potential of the βC main ring towards more positive values.58 In addition, electronic excited βCs show a characteristic separation of charge. Thus, as it was demonstrated for N(9)-methyl βCs (1c, 2c and 3c),40 electron/proton exchanges (type I oxidative processes) would be enhanced and, hence, the formation of Fpg-sensitive sites. Among the nucleobases, guanine is the most susceptible target of an oxidative attack, both by type I and type II mechanism.
(ii) On the contrary, 3b showed a broader damage profile. A close look at the data reveals that the yield of oxidized purines (8-oxoG) is reduced relative to that of AP-sites, oxidized pyrimidines as well as CPDs. It has been demonstrated that photosensitized formation of CPDs mainly occurs through triplet–triplet energy transfer (TTET) from the triplet electronic excited state of the photosensitizer to the triplet electronic state of the pyrimidine (mostly, thymine). This process is thermodynamically favoured for those photosensitizers having energy triplet values (ET) larger than 267 kJ mol−1.59 Although the ET value of 3b was not determined, it was demonstrated that the unsubstituted derivative 3a has the required energy (274.4 kJ mol−1).60
Considering that methylation at position N(2) induces an only small change in the relative energy of the electronic states (i.e., bathochromic shift ∼5 nm), it is expected that 3b also has the required energy for triplet–triplet sensitization. This hypothesis should be further investigated.
(iii) The 9-methyl-derivatives (1c, 2c, 3c) show a higher photosensitizing efficiency than the corresponding cationic species (i.e., ten-times lower concentrations of 9-methyl-βCs give rise to a similar amount of DNA damage as 2-methyl-βCs and 2,9-dimethyl-βCs). This fact can be accounted for by the lower polarity of the 9-methyl-βC molecules that can give rise to a higher hydrophobic interaction with the DNA double helix (partial intercalation). On the other hand, the positive net charge placed on the 2-methyl- and 2,9-dimethyl-βC cationic species would enhance their electrostatic interaction with the negative charges placed on the phosphate groups of the DNA skeleton reducing their intercalation capability (see below).
(iv) Harmine derivatives (3a–3d) showed the highest photosensitizing efficiency. I. e., the photosensitizerś concentration (absorbance) needed to induce the same amount of DNA-damage was one or two orders of magnitude lower than the required concentration for the corresponding N-methyl-norharmane (1a–1d) and N-methyl-harmane (2a–2d). Therefore, the methoxy group at C(7) enhances the overall photosensitizing activity of the βC-ring. This could be a consequence of an increased capability to generate ROS7 and/or of an increased binding affinity to DNA (Fig. 1).
Interaction between N-methyl-βCs and calf thymus DNA
Due to the varying content of water and counter ions, ε of nucleic acids cannot reliably be based on weight. Many years ago, Chargaff et al.62 introduced a highly accurate method based on the fact that nucleic acids contain one phosphorus atom per nitrogenous base. As such, the absorbance of a given solution of nucleic acid can directly be related to the nucleobase concentration by measuring the total phosphorus content. Based on this method, Felsenfeld et al.63 and Hirschman et al.64 reported (more than 60 years ago) an average value of 6600 M−1 cm−1 (or 13200 Mbp−1 cm−1, when expressed in terms of base-pairs) for the molar absorption coefficient of ctDNA at 260 nm (ε260 nm). Although the latter value seems to be considerably underestimated and it varies with the experimental conditions (see ESI†), it is frequently adopted by the scientific community in quantitative studies using almost all types of dsDNA material under different experimental conditions.65
In this context, the ε260 nm value for ctDNA reconstituted according to the general recommendation of the fabricant were re-examined and accurately determined herein by using a more sensitive technique (i.e., microwave-plasma-emission atomic spectroscopy). Total phosphorus concentration ([P]) and UV-visible absorption spectra were measured from both dialyzed and non-dialyzed ctDNA samples. To further evaluate potential interferences due to pH or ionic strength changes, stock solutions were diluted in different solvents. All the cases showed a linear trend when depicting the corrected absorbance A260 nmcorvs. [P] (ESI Fig. 2e†). The corresponding ε260 nm obtained from the slopes are listed in ESI Table 1.†
With the exception of ctDNA dialyzed in milliQ water, ε260 nm observed under all the experimental conditions were the same, within the experimental error (〈ε260 nm〉 = 7.9 (±0.5) × 103 M−1 cm−1 or 15.8 × 103 Mbp−1 cm−1). This is reflected in the lack of any substantial change in the normalized spectra (ESI Fig. 2f†), suggesting the absence of significant changes of the ctDNA structure when subject to variation of the local environment (ionic strength and/or type of cations). On the contrary, changes in the UV-visible spectrum observed for dialyzed (in milliQ water) ctDNA material due to denaturation correlates with the highest ε260 nm value observed (8.9 (±0.2) × 103 M−1 cm−1).
Fluorescence quenching of βC alkaloids by calf-thymus DNA: steady-state and time-resolved spectroscopy
The extent of DNA photosensitized damage shown above might be modulated by the magnitude and mode of interaction between DNA and the photosensitizers in the electronic ground and/or excited states. It has been demonstrated that small changes of the molecular structure of βCs, as well as of the environment (solvent and pH) strongly impact the physicochemical and photochemical properties of these alkaloids.53,60,66–69 To further investigate this influence, titration of N-methyl-βC solutions with calf thymus DNA (ctDNA) were monitored by fluorescence spectroscopy (Fig. 2 and ESI Fig. 3 to 9†).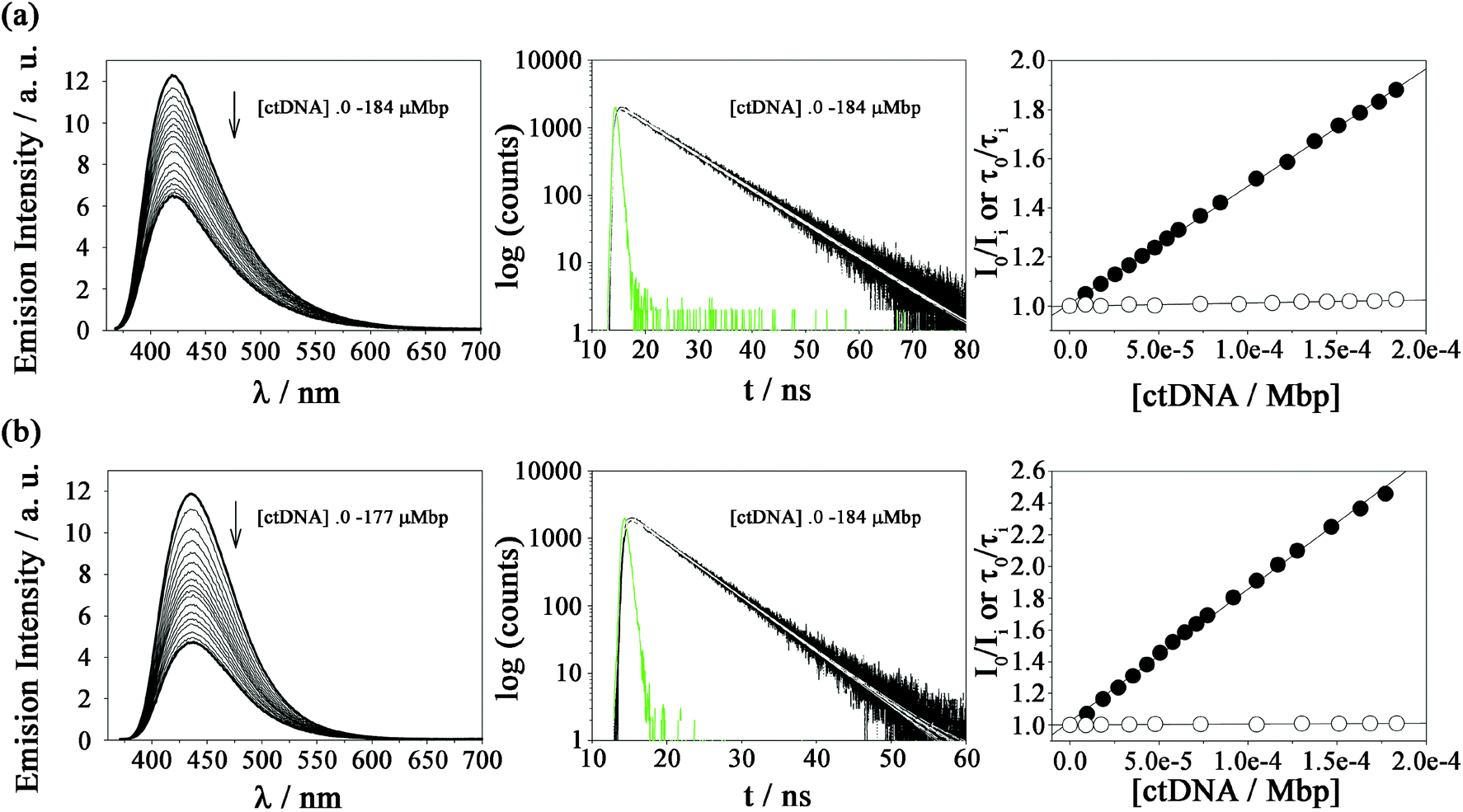 | ||
| Fig. 2 Steady-state and time-resolved emission of (a) 3b (15 μM) and (b) 3d (25 μM) in air-equilibrated buffered solution (pH 7.4), recorded in the presence of increasing amounts of ctDNA (μMbp). Left: corrected emission spectra (λexc = 360 nm and 363 nm for 3b and 3d, respectively). Arrows indicate the variation in the emission spectra upon increasing [ctDNA] (initial and final concentrations are highlighted in black). Middle: βC fluorescence decays recorded at wavelengths of the corresponding emission maximum (black lines, λexc = 341 nm), prompt signal (green line) and mono-exponential fitting curves (white lines). Right: Stern–Volmer plots of the emission intensities (I) (black circles) and lifetimes (τ) (white circles). | ||
Steady-state fluorescence experiments, performed under three different pH conditions, show the same qualitative trend for all the investigated compounds. Briefly, the emission bands (centred at ∼450 nm) exhibit significant changes in terms of intensity when [ctDNA] is increased. The corresponding Stern–Volmer plots (i.e., I0/I vs. [ctDNA]) showed a linear dependence (Fig. 2). The extent of the interaction, quantified by the slope (KSS) of the Stern–Volmer plot depends on the chemical structure of the βC derivative (Table 2). When comparing these data, the following conclusions can be drawn:
(i) The larger the number of substituent (methyl and methoxy groups) in the βC ring, the larger the overall binding affinity values observed: KSS(3a–3d) > KSS(2a–2d) > KSS(1a–1d). DNA can quench a given fluorophore through several deactivation mechanisms. However, in the case of βCs, it has been demonstrated that these alkaloids mainly interact with ctDNA giving rise to the formation of a ground-state static complex (βCs/ctDNA). In such complexes, the combination of both electrostatic and non-polar forces contributes to the overall interaction. The same mechanism is also expected for quaternary βC derivatives, 1b, 2b, 3b, 1d, 2d and 3d, (see time-resolved fluorescence data presented below). In consequence, if KSS is interpreted as a binding constant, the dependence described above can be ascribed to non-polar contributions: the higher the number of alkyl-substituents, the lower the polarity of the βC-ring enhancing their hydrophobic interaction (partial intercalation).
(ii) N(9)-Substituted βCs show larger KSS values than those observed for N(9)-unsubstituted derivatives: K(9-methyl-βCs)SS > K(2,9-di methyl-βCs)SS ≫ K(βCs)SS > K(2-methyl−-βCs)SS. This might well be because of the decrease in the polarity of the βC ring due to the substitution of the hydrogen atom by the methyl group and, therefore, is more likely to enhance the hydrophobic interaction mode of the βC ring.
(iii) N(2)-Substituted βCs show KSS values slightly lower than those observed for the corresponding N(2)-unsubstituted derivatives: K(2,9-di methyl-βCs)SS < K(9-methyl-βCs)SS and K(2-methyl-βCs)SS < K(βCs)SS. This seems counterintuitive since the net positive charge present in quaternary βCs should enhance the coulombic attractions with the negatively charged DNA backbone. However, the methyl groups placed at N(2) might either introduce steric hindrance or block the chance to establish hydrogen bonding between βCs and nucleobases. This can explain the decrease in the overall strength of the interaction observed.
(iv) The dependence of KSS with the pH has already been demonstrated for N(2)-unsubstituted-βCs (i.e., βCs and 9-methyl-βCs): the lower the pH, the higher the binding affinity. The latter fact was accounted for by the fact that under acidic conditions, protonated species of N(2)-unsubstituted-βCs are present in the solution. Thus, the contribution of electrostatic forces (attraction between the cationic alkaloids and the negatively charged phosphate groups of the DNA) to the overall interaction would be enhanced. On the contrary, the pH-dependence was not observed for quaternary βCs (1b, 2b, 3b, 1d, 2d and 3d). This is accounted for by the fact that, in the whole pH-range investigated (4.8–9.5), only one acid–base species of quaternary βCs is present in the bulk solution.
(v) The presence of ctDNA does not induce changes in the shape or the position of the emission maxima. Thus, the polarity of the environment/surroundings of the fluorophores (at least, with respect to the pyridine ring of the βCs) is not significantly modified. This confirms, once more, that βCs are not fully intercalated into the DNA helix.
| Compound | K SS M−1 in bp−1 103 | K D M−1 in bp−1 103 |
|---|---|---|
| a Data obtained from ref. 38. b Data obtained from ref. 40. c Data obtained from ref. 39. | ||
| 1a | 1.82 (±0.03) | 0.52 (±0.02)a |
| 1b | 0.6 (±0.1) | 0 |
| 0.7 (±0.1) (pH 4.8) | ||
| 0.6 (±0.1) (pH 9.5) | ||
| 1c | 2.7 (±0.1)b | 0.25 (±0.05)a |
| 1d | 1.8 (±0.1) | 0 |
| 1.8 (±0.1) (pH 4.8) | ||
| 2.5 (±0.1) (pH 9.5) | ||
| 2a | 4.65 (±0.03) | 0.54 (±0.02)a |
| 2b | 2.48 (±0.02) | 0 |
| 2c | 6.4 (±0.7)b | 0.90 (±0.04)a |
| 2d | 3.7 (±0.1) | 0 |
| 3a | 7.7 (±0.2)c | 0.33 (±0.03)a |
| 3b | 5.9 (±0.3) | 0 |
| 5 (±1) (pH 4.8) | ||
| 6 (±1) (pH 9.5) | ||
| 3c | 16.8 (±0.5)b | — |
| 3d | 9.9 (±0.1) | 0 |
The almost null contribution of the dynamic quenching to the overall interaction between N(2)-unsubstituted-βCs and ctDNA has already been demonstrated for related βCs. To further evaluate potential dynamic contributions to the overall emission quenching described above, fluorescence lifetimes of quaternary βCs (2-Me-βCs and 2,9-diMe-βCs) were measured in the presence of increasing ctDNA concentrations. Decays shown in Fig. 2, ESI Fig. 7 and 8† were the same in the whole ctDNA concentration range investigated, indicating the lack of dynamic contribution to the overall fluorescence quenching induced by ctDNA. This is also represented in the corresponding Stern–Volmer plot (τ0/τ vs. [ctDNA]) showing slopes equal to cero (see the dynamic quenching constants, KD, listed in Table 2). The data confirm that static interactions dominate the overall interactions between the investigated βCs and ctDNA.
Cellular uptake and release of norharmane derivatives
Changes in the βC's chemical structure, including N-methylation, can modulate both the dynamics of the uptake into cells and/or the specific intracellular localization of the βC. Although cellular uptake of βCs has been suggested, the fact that these alkaloids exhibit similar spectral patterns to endogenous chromophores makes quite a challenge to univocally establish their precise intracellular localization by using conventional fluorescence microscopy. To further address this, in vitro cultures of HeLa and HEK293 cell lines were co-incubated with four different βC derivatives and monitored by FLIM. Compounds 1a–1d derivatives were used herein as representative examples.When co-incubated with βCs in PBS (extracellular [βC] = 10–15 μM), cells were able to take up all four investigated compounds within 20 min. All βC derivatives show a similar image pattern, staining the same intracellular structures (Fig. 3). Interestingly, the compounds present a specific intracellular accumulation pattern (Fig. 3, left column) in some internal structures. Although the accumulation is not specific, it is sufficient to isolate these regions using home-coded macros in Fiji. The second and third columns in Fig. 3 show the lifetime images of the complete cells and from the internal structures where the compounds are accumulated, respectively. Fig. 3 also shows arrows traced where we reconstruct histograms of lifetime and intensity. These histograms show the changes in intensity and lifetime along the arrow traced. For a better clarity, we indicate under grey bars, the increase in fluorescence due to a structure where the compounds are accumulated. As can be observed, the increases in intensity are coincident with lower fluorescence lifetimes. Fig. 3 suggests that all βC derivatives show a similar intensity pattern, due, probably, to a staining of the same intracellular structures. Also, this is in agreement with our previous studies which indicated that fluorescence lifetimes of the compounds are dependent on the environment.7 This fact, together with the long fluorescence lifetimes of all the compounds, open the possibility to use this parameter to monitor changes in the microenvironment inside these structures and to introduce a time-gate in the decay traces registered in FLIM to discard completely the autofluorescence of cells.
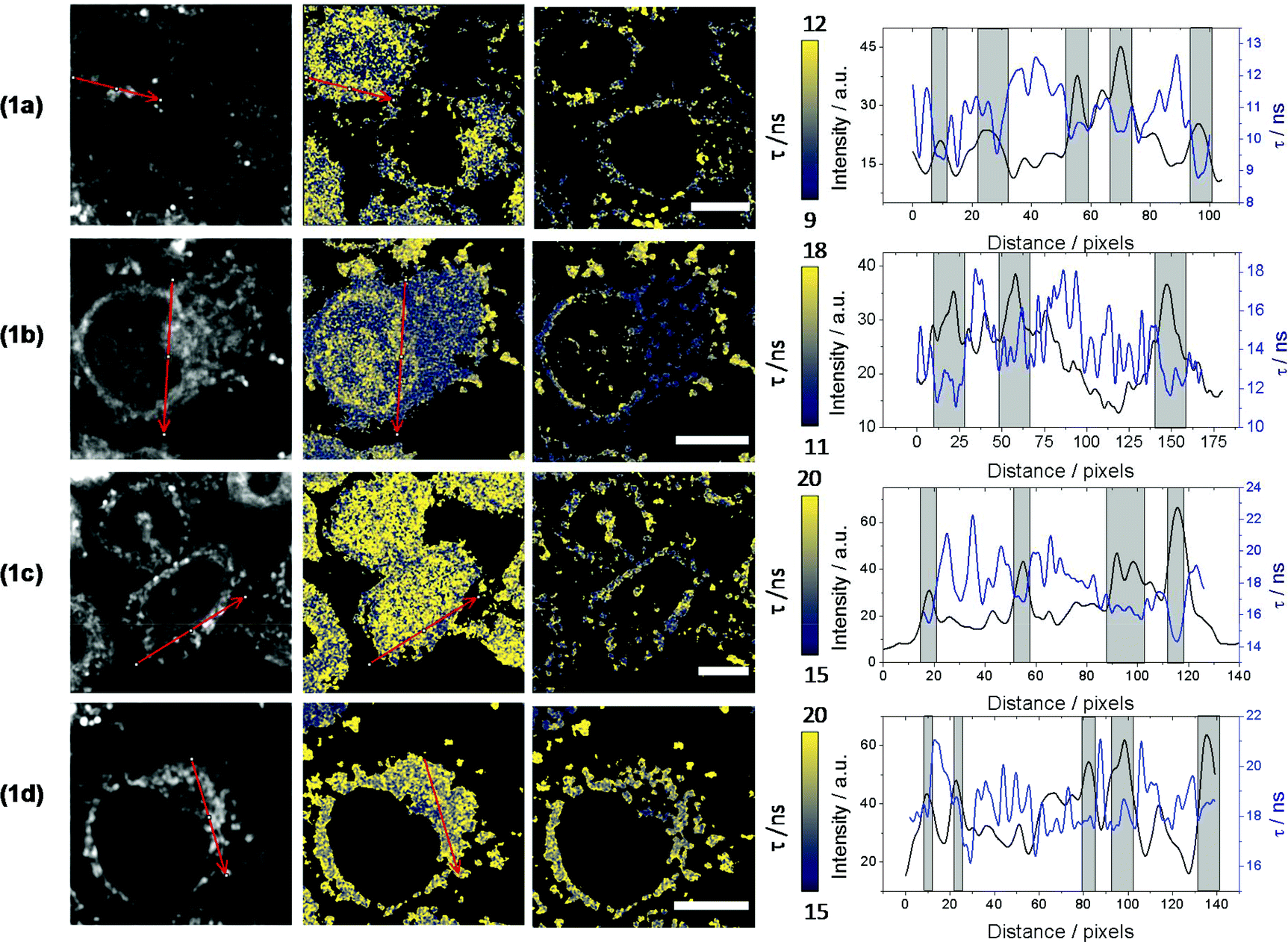 | ||
| Fig. 3 Fluorescence images of HeLa cells incubated with compounds 1a, 1b, 1c and 1d. Column 1: fluorescence intensity images of the dye. Column 2: fluorescence lifetime images of dye in the whole cell. Column 3: fluorescence lifetime images in the accumulate intracellular organelles. Scale bar = 10 μm. Column 4: plot of intensities and lifetimes along the selected arrows. | ||
Surprisingly, data also showed that the dynamic of both uptake and release processes strongly depends on the chemical structure of the alkaloid. Briefly, neutral βCs (1a and 1c) showed a faster uptake than quaternary βCs (1b and 1d). This is evidenced by the fact that the maximum emission is reached before 5–10 min of incubation with 1c or 1a (Fig. 4a and ESI Fig. 10a,† respectively), whereas up to 20 min of incubation were needed for both cationic derivatives (Fig. 4b and ESI Fig. 10b†). Upon washing (two times) the cell cultures with PBS free of βC, a prompt and drastic decrease of the fluorescence intensities of cells preincubated with both neutral derivatives (1a and 1c) was observed, reaching the cell auto-fluorescence background levels. On the contrary, fluorescence intensity of washed cells pre-incubated with 1b and 1d remains nearly constant within the time window of analysis (30 min).
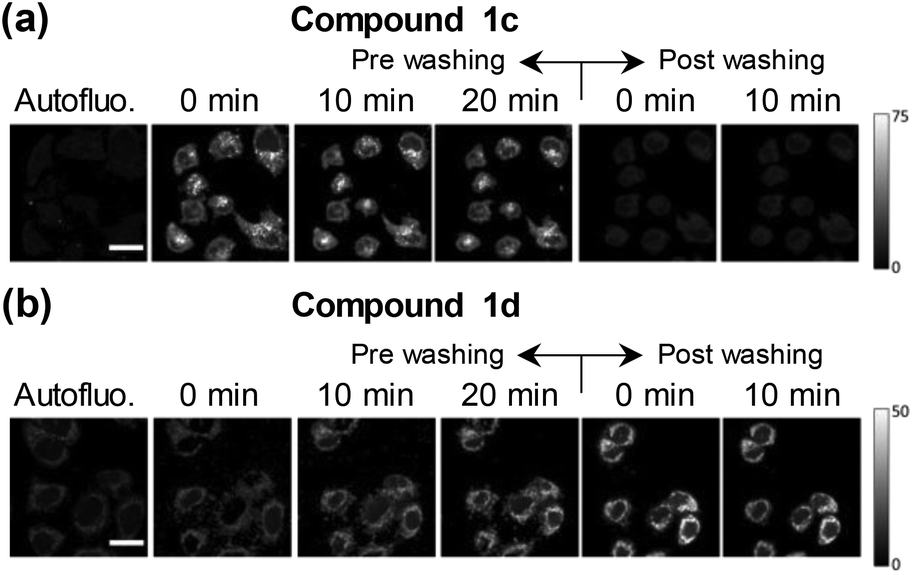 | ||
| Fig. 4 Examples of fluorescence images obtained from HEK293 cells co-incubated with compounds (a) 1c and (b) 1d. Column 1: HEK293 autofluorescence. Columns 2 to 4: fluorescence images obtained after 0, 10 and 20 minutes of addition of the corresponding βC, respectively. Columns 5 and 6: images obtained 0 and 10 minutes after washing HEK293 cells with PBS. Scale bar = 20 μm. | ||
The facts described in the paragraph above are consistent with a free diffusion of 1a and 1c between intracellular compartments and the extracellular space. Thus, a passive uptake/release would be the operative mechanism for neutral βCs. On the contrary, the permeability of the cellular membrane for the polar quaternary βC cations is probably slow, and both processes (uptake and release) may even be mediated by an active transport. These aspects need to be further investigated.
Intracellular localization was further characterized by using two different red fluorescent biomarkers, Mitotracker® and Lysotracker®, that selectively stain mitochondria and lysosome, respectively. To this aim, 1d was chosen as a representative example, due to its intrinsic photophysical properties setting it aside from all the other investigated compounds including high fluorescent emission quantum yield (ΦF ∼ 0.75), highest fluorescent lifetime (τF = 25.9 ns) and lack of fluorescence pH-dependence.7 Results show that 1d is mostly localized in mitochondria (Fig. 5a and ESI Fig. 11†). This is better represented by the white pixels in the colocalized image. Moreover, a Pearson's coefficient value, r, of 0.65 ± 0.03 was obtained (see ESI†), indicating the dye is mainly localized in mitochondria. In contrast, the relatively low Pearson's coefficient (0.25 ± 0.02) calculated from Lysotracker® images (Fig. 5b and ESI Fig. 12†) suggest a low or null internalization into the lysosomes.
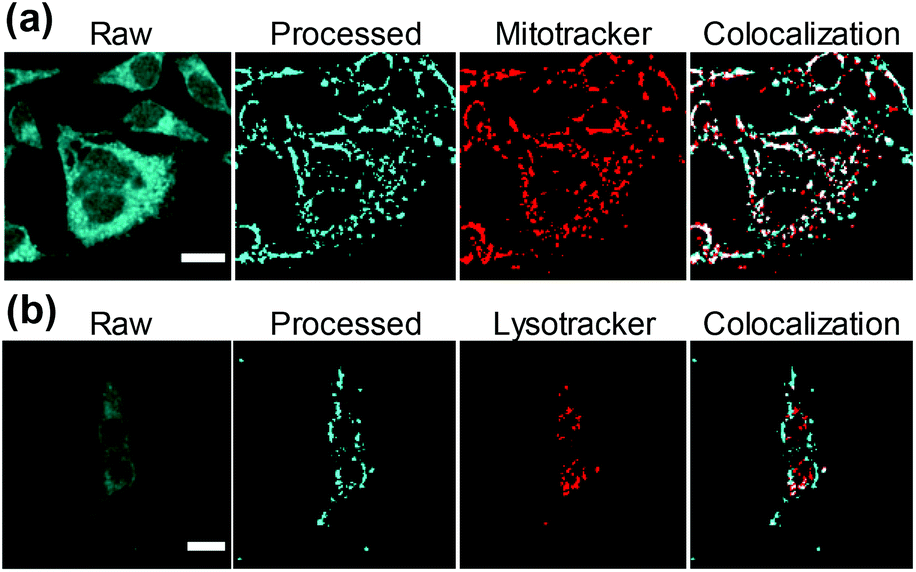 | ||
| Fig. 5 Fluorescence images of HeLa cells co-incubated with compound 1d (cyan). The figure shows the raw fluorescence images (first column) and the processed images isolating the regions where the compound accumulates. The two different biomarkers (red): (a) Mitotracker® and (b) Lysotracker® are in the third column. The merge from processed and biomarker images and the colocalization pixels (white) are represented in the fourth column. Scale bar = 20 μm. | ||
Conclusions
The study describes the structural and mechanistic basis underlying the photosensitizing properties and the cellular distribution of a selected group of N-methyl-βC alkaloids (Table 1). Data clearly show that small changes in the chemical structure of the βC ring allow a fine-tuning of its intrinsic physicochemical properties, enabling specific responses and/or applications. Briefly,(a) methylation enhances its photosensitizing properties either by increasing its binding affinity with macromolecules such as DNA and/or by increasing its oxidation potential. The latter fact is (indirectly) evidenced by the DNA photodamage spectra induced by these derivatives. With the exception of 3b, all the N-methyl derivatives investigated showed a quite selective damage generation, where oxidized purines represent the major type of photoproducts formed.
(b) The quite interesting photochemical and photosensitizing properties of N-methyl-βCs (i. e, the bathochromic shift of the absorption bands as well as the enhancement on the photosensitizing properties induced by methylation) make these derivatives suitable for different biotechnological applications. N(9)-Methyl-βCs could represent excellent candidates for the development of novel and more efficient albumin-folate-βC conjugates as selective vehicles for tumour cell targeted PDT.45
(c) Our data demonstrate that all the investigated compounds are taken up by HeLa and HEK293 cells. Data reveal that:
(i) lifetime changes depend on the microenvironment. This can have particular ramifications providing an open future for the development of biotechnological applications based on small molecules derived from βC chemical structures.70
(ii) These compounds mainly accumulate into mitochondria. Therefore, although other intracellular components could be involved, mitochondrial DNA could be an important target for the βC-mediated photoinactivation.
(iii) Neutral βCs show a faster uptake and release kinetic than the quaternary derivatives, suggesting that different cellular uptake pathways (or mechanisms) might operate in each case. This distinctive dynamic pattern observed provides valuable information to further understand some of the biological effects described in the introductory section. In particular, they can help to understand/assess the neurotoxic effect of quaternary βCs played at the mitochondrial domain. Also, these results shed light into the mechanism of anti-parasitic22 and antiviral35 activity of 1c, 2c and 3c.
(d) As a secondary outcome of this study, accurate absorption coefficients of ctDNA under different conditions were established. Values were determined by using a more sensitive technique (i.e., microwave-plasma-emission atomic spectroscopy) than previously applied. The greater accuracy of the new values is expected to have a strong impact on binding constants measurements, quantitative real-time PCR,71 among others. In this regard, values reported herein might represent an excellent alternative to standardize the preparation and quantification of ctDNA solutions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Grants PICT-2015-0374, 2016-0370 and 2018-3193 (ANPCyT, Argentina), and CTQ2017-85658-R and CTQ2014-55474-C2-2-R (Spanish Ministry of Economy and Competitiveness). MPD and FMC are research members of CONICET. Authors thanks to Carlos G. Alberici for his technical support, and Fernando Villarruel for the artwork.Notes and references
- J. Torreilles, M. C. Guerin and A. Previero, Biochimie, 1985, 67, 929–947 CrossRef CAS PubMed.
- J. Adachi, Y. Mizoi, T. Naito, Y. Ogawa, Y. Uetani and I. Ninomiya, J. Nutr., 1991, 121, 646–652 CrossRef CAS PubMed.
- S. Manabe, J. Yuan, T. Takahashi and R. C. Urban Jr., Exp. Eye Res., 1996, 63, 179–186 CrossRef CAS PubMed.
- K. Pari, C. S. Sundari, S. Chandani and D. Balasubramanian, J. Biol. Chem., 2000, 275, 2455–2462 CrossRef CAS PubMed.
- R. Spijkerman, R. van den Eijnden, D. van de Mheen, I. Bongers and D. Fekkes, Eur. Neuropsychopharmacol., 2002, 12, 61–71 CrossRef CAS PubMed.
- U. Breyer-Pfaff, G. Wiatr, I. Stevens, H. Jörg Gaertner, G. Mundle and K. Mann, Life Sci., 1996, 58, 1425–1432 CrossRef CAS PubMed.
- F. A. O. Rasse-Suriani, F. S. García-Einschlag, M. Rafti, T. Schmidt De León, P. M. David Gara, R. Erra-Balsells and F. M. Cabrerizo, Photochem. Photobiol., 2018, 94, 36–51 CrossRef CAS PubMed.
- C. Liu, M. N. Masuno, J. B. MacMillan and T. F. Molinski, Angew. Chem., Int. Ed., 2004, 43, 5951–5954 CrossRef CAS PubMed.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef PubMed.
- Q. Chen, C. Ji, Y. Song, H. Huang, J. Ma, X. Tian and J. Ju, Angew. Chem., Int. Ed., 2013, 52, 9980–9984 CrossRef CAS PubMed.
- D. Pressnitz, E.-M. Fischereder, J. Pletz, C. Kofler, L. Hammerer, K. Hiebler, H. Lechner, N. Richter, E. Eger and W. Kroutil, Angew. Chem., Int. Ed., 2018, 57, 10683–10687 CrossRef CAS PubMed.
- B. Greiner, C. Fahndrich, S. Strauss and H. Rommelspacher, Naunyn-Schmiedeberg's Arch. Pharmacol., 1983, 322, 140–146 CrossRef CAS PubMed.
- D. Fekkes and W. T. Bode, Life Sci., 1993, 52, 2045–2054 CrossRef CAS PubMed.
- M. G. Thomas, D. Sartini, M. Emanuelli, M. J. van Haren, N. I. Martin, D. M. Mountford, D. J. Barlow, F. Klamt, D. B. Ramsden, M. Reza and R. B. Parsons, Biochem. J., 2016, 473, 3253–3267 CrossRef CAS PubMed.
- M. A. Collins and E. J. Neafsey, in Neurotoxic Factors in Parkinson's Disease and Related Disorders, ed. A. Storch and M. A. Collins, Kluwer Academic/Plenum Publishers, New York, 2000, pp. 115–129 Search PubMed.
- D. A. Gearhart, M. A. Collins, J. M. Lee and E. J. Neafsey, Neurobiol. Dis., 2000, 7, 201–211 CrossRef CAS PubMed.
- D. A. Gearhart, E. J. Neafsey and M. A. Collins, Neurochem. Int., 2002, 40, 611–620 CrossRef CAS PubMed.
- R. Albores, E. J. Neafsey, G. Drucker, J. Z. Fields and M. A. Collins, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 9368–9372 CrossRef CAS PubMed.
- Y. Tadokoro, T. Nishikawa, T. Ichimori, S. Matsunaga, M. J. Fujita and R. Sakai, ACS Omega, 2017, 2, 1074–1080 CrossRef CAS PubMed.
- J. Hamann, C. Wernicke, J. Lehmann, H. Reichmann, H. Rommelspacher and G. Gille, Neurochem. Int., 2008, 52, 688–700 CrossRef CAS PubMed.
- R. Cao, Q. Chen, X. Hou, H. Chen, H. Guan, Y. Ma, W. Peng and A. Xu, Bioorg. Med. Chem., 2004, 12, 4613–4623 CrossRef CAS PubMed.
- M. L. Alomar, F. A. Rasse-Suriani, A. Ganuza, V. M. Coceres, F. M. Cabrerizo and S. O. Angel, BMC Res. Notes, 2013, 6, 193 CrossRef CAS PubMed.
- C. Di Giorgio, F. Delmas, E. Ollivier, R. Elias, G. Balansard and P. Timon-David, Exp. Parasitol., 2004, 106, 67–74 CrossRef CAS PubMed.
- B. B. Mishra, R. K. Singh, A. Srivastava, V. J. Tripathi and V. K. Tiwari, Mini-Rev. Med. Chem., 2009, 9, 107–123 CrossRef CAS PubMed.
- S. Lala, S. Pramanick, S. Mukhopadhyay, S. Bandyopadhyay and M. K. Basu, J. Drug Targeting, 2004, 12, 165–175 CrossRef CAS PubMed.
- A. Gellis, A. Dumètre, G. Lanzada, S. Hutter, E. Ollivier, P. Vanelle and N. Azas, Biomed. Pharmacother., 2012, 66, 339–347 CrossRef CAS PubMed.
- D. J. McKenna, Pharmacol. Ther., 2004, 102, 111–129 CrossRef CAS PubMed.
- L. T. D. Tonin, M. R. Panice, C. V. Nakamura, K. J. P. Rocha, A. O. D. Santos, T. Ueda-Nakamura, W. F. D. Costa and M. H. Sarragiotto, Biomed. Pharmacother., 2010, 64, 386–389 CrossRef CAS PubMed.
- T. Satou, A. Horiuchi, N. Akao, K. Koike, K. Fujita and T. Nikaido, Exp. Parasitol., 2005, 110, 134–139 CrossRef CAS PubMed.
- G. M. Olmedo, L. Cerioni, M. M. González, F. M. Cabrerizo, V. A. Rapisarda and S. I. Volentini, Food Microbiol., 2017, 62, 9–14 CrossRef CAS PubMed.
- G. M. Olmedo, L. Cerioni, M. M. González, F. M. Cabrerizo, S. I. Volentini and V. A. Rapisarda, Front. Microbiol., 2017, 8, 347 Search PubMed.
- G. Van Baelen, S. Hostyn, L. Dhooghe, P. Tapolcsányi, P. Mátyus, G. Lemière, R. Dommisse, M. Kaiser, R. Brun, P. Cos, L. Maes, G. Hajós, Z. Riedl, I. Nagy, B. U. W. Maes and L. Pieters, Bioorg. Med. Chem., 2009, 17, 7209–7217 CrossRef CAS PubMed.
- C. W. Wright, J. D. Phillipson, S. O. Awe, G. C. Kirby, D. C. Warhurst, J. Quetin-Leclercq and L. Angenot, Phytother. Res., 1996, 10, 361–363 CrossRef CAS.
- J. F. Blom, T. Brütsch, D. Barbaras, Y. Bethuel, H. H. Locher, C. Hubschwerlen and K. Gademann, Org. Lett., 2006, 8, 737–740 CrossRef CAS PubMed.
- M. M. Gonzalez, F. M. Cabrerizo, A. Baiker, R. Erra-Balsells, A. Osterman, H. Nitschko and M. G. Vizoso-Pinto, Int. J. Antimicrob. Agents, 2018, 52, 459–468 CrossRef CAS PubMed.
- P. Bag, D. Ojha, H. Mukherjee, U. C. Halder, S. Mondal, A. Biswas, A. Sharon, L. Van Kaer, S. Chakrabarty, G. Das, D. Mitra and D. Chattopadhyay, Antiviral Res., 2014, 105, 126–134 CrossRef CAS PubMed.
- M. M. Gonzalez, M. Pellon-Maison, M. A. Ales-Gandolfo, M. R. Gonzalez-Baró, R. Erra-Balsells and F. M. Cabrerizo, Org. Biomol. Chem., 2010, 8, 2543–2552 RSC.
- M. M. Gonzalez, M. Vignoni, M. Pellon-Maison, M. A. Ales-Gandolfo, M. R. Gonzalez-Baro, R. Erra-Balsells, B. Epe and F. M. Cabrerizo, Org. Biomol. Chem., 2012, 10, 1807–1819 RSC.
- J. G. Yañuk, M. P. Denofrio, F. A. O. Rasse-Suriani, F. D. Villarruel, F. Fassetta, F. S. García Einschlag, R. Erra-Balsells, B. Epe and F. M. Cabrerizo, Org. Biomol. Chem., 2018, 16, 2170–2184 RSC.
- M. Vignoni, F. A. O. Rasse-Suriani, K. Butzbach, R. Erra-Balsells, B. Epe and F. M. Cabrerizo, Org. Biomol. Chem., 2013, 11, 5300–5309 RSC.
- M. Caprasse and C. Houssier, Biochimie, 1983, 65, 157–167 CrossRef CAS PubMed.
- M. M. Gonzalez, F. A. O. Rasse-Suriani, C. A. Franca, R. Pis Diez, Y. Gholipour, H. Nonami, R. Erra-Balsells and F. M. Cabrerizo, Org. Biomol. Chem., 2012, 10, 9359–9372 RSC.
- M. M. Gonzalez, M. P. Denofrio, F. S. Garcia Einschlag, C. A. Franca, R. Pis Diez, R. Erra-Balsells and F. M. Cabrerizo, Phys. Chem. Chem. Phys., 2014, 16, 16547–16562 RSC.
- M. Vignoni, R. Erra-Balsells, B. Epe and F. M. Cabrerizo, J. Photochem. Photobiol., B, 2014, 132, 66–71 CrossRef CAS PubMed.
- K. Butzbach, F. A. O. Rasse-Suriani, M. M. Gonzalez, F. M. Cabrerizo and B. Epe, Photochem. Photobiol., 2016, 92, 611–619 CrossRef CAS PubMed.
- J. G. Yañuk, M. L. Alomar, M. M. Gonzalez, F. Simon, R. Erra-Balsells, M. Rafti and F. M. Cabrerizo, Phys. Chem. Chem. Phys., 2015, 17, 12462–12465 RSC.
- M. Salditt, S. N. Braunstein, R. D. Camerini-Otero and R. M. Franklin, Virology, 1972, 48, 259–262 CrossRef CAS PubMed.
- S. Boiteux, T. R. O'Connor, F. Lederer, A. Gouyette and J. Laval, J. Biol. Chem., 1990, 265, 3916–3922 CAS.
- E. Müller, S. Boiteux, R. P. Cunningham and B. Epe, Nucleic Acids Res., 1990, 18, 5969–5973 CrossRef PubMed.
- Milli-Q water was used as dialysis solvent in order to ensure a complete removal of phosphate ions that might initially be provided by the starting ctDNA material.
- Dialysis at fix pH and ionic strength was performed using ammonium chloride buffer (10 mM, pH 7.4) due to its null interference with the analytical methods. Note that the detector of the MP-EAS equipment is not sensitive to chloride ions and, on the other hand, ammonium ions are evaporated in the torch prior to reaching the detector.
- M. J. Cavaluzzi and P. N. Borer, Nucleic Acids Res., 2004, 32, e13–e13 CrossRef PubMed.
- M. M. Gonzalez, J. Arnbjerg, M. Paula Denofrio, R. Erra-Balsells, P. R. Ogilby and F. M. Cabrerizo, J. Phys. Chem. A, 2009, 113, 6648–6656 CrossRef CAS PubMed.
- B. Epe, J. Hegler and P. Lester, in Methods Enzymol., Academic Press, 1994, vol. 234, pp. 122–131 Search PubMed.
- B. Epe, Photochem. Photobiol. Sci., 2012, 11, 98–106 RSC.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- I. Maisuls, F. M. Cabrerizo, P. M. David-Gara, B. Epe and G. T. Ruiz, Chem. – Eur. J., 2018, 24, 12902–12911 CrossRef CAS PubMed.
- J. G. Yañuk, F. M. Cabrerizo, F. G. Dellatorre and M. F. Cerdá, Energy Rep., 2020, 6, 25–36 CrossRef.
- V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell, F. Bosca and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 7409–7414 CrossRef CAS PubMed.
- F. A. O. Rasse-Suriani, M. Paula Denofrio, J. G. Yañuk, M. Micaela Gonzalez, E. Wolcan, M. Seifermann, R. Erra-Balsells and F. M. Cabrerizo, Phys. Chem. Chem. Phys., 2016, 18, 886–900 RSC.
- A. Prisecaru, Z. Molphy, R. G. Kipping, E. J. Peterson, Y. Qu, A. Kellett and N. P. Farrell, Nucleic Acids Res., 2014, 42, 13474–13487 CrossRef CAS PubMed.
- S. Zamenhof and E. Chargaff, J. Biol. Chem., 1949, 178, 531–532 CAS.
- G. Felsenfeld and S. Z. Hirschman, J. Mol. Biol., 1965, 13, 407–427 CrossRef CAS PubMed.
- S. Z. Hirschman and G. Felsenfeld, J. Mol. Biol., 1966, 16, 347–358 CrossRef CAS PubMed.
- Note that Fensenfeld's (1965) and Hirschman's (1966) manuscripts have been cited ∼120 and ∼80 times, respectively. In addition, according to one of the world's most popular internet search engines, more than 2,390,000 and 215,000 results match with the searches of “DNA + 6600” and “DNA + 13200”, respectively.
- M. M. Gonzalez, M. L. Salum, Y. Gholipour, F. M. Cabrerizo and R. Erra-Balsells, Photochem. Photobiol. Sci., 2009, 8, 1139–1149 RSC.
- F. M. Cabrerizo, J. Arnbjerg, M. P. Denofrio, R. Erra-Balsells and P. R. Ogilby, ChemPhysChem, 2010, 11, 796–798 CrossRef CAS PubMed.
- D. Hrsak, L. Holmegaard, A. S. Poulsen, N. H. List, J. Kongsted, M. P. Denofrio, R. Erra-Balsells, F. M. Cabrerizo, O. Christiansen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2015, 17, 12090–12099 RSC.
- O. I. Tarzi, M. A. Ponce, F. M. Cabrerizo, S. M. Bonesi and R. Erra-Balsells, ARKIVOC, 2005, vii, 295–310 Search PubMed.
- S. Hotha, J. C. Yarrow, J. G. Yang, S. Garrett, K. V. Renduchintala, T. U. Mayer and T. M. Kapoor, Angew. Chem., Int. Ed., 2003, 42, 2379–2382 CrossRef CAS PubMed.
- J. L. Love, P. Scholes, B. Gilpin, M. Savill, S. Lin and L. Samuel, J. Microbiol. Methods, 2006, 67, 349–356 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01122c |
| ‡ Equal contribution of both authors |
| This journal is © The Royal Society of Chemistry 2020 |