
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral 1,5-disubstituted 1,2,3-triazoles – versatile tools for foldamers and peptidomimetic applications†
Anna
Said Stålsmeden
 a,
Andrew J.
Paterson
a,
Imola Cs.
Szigyártó
b,
Linda
Thunberg
c,
Johan R.
Johansson
*d,
Tamás
Beke-Somfai
*b and
Nina
Kann
*a
a,
Andrew J.
Paterson
a,
Imola Cs.
Szigyártó
b,
Linda
Thunberg
c,
Johan R.
Johansson
*d,
Tamás
Beke-Somfai
*b and
Nina
Kann
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: kann@chalmers.se
bResearch Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar tudósok krt. 2, H-1117 Budapest, Hungary. E-mail: beke-somfai.tamas@ttk.mta.hu
cEarly Chemical Development, Pharmaceutical Sciences, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden
dMedicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden. E-mail: johan.x.johansson@astrazeneca.com
First published on 19th February 2020
Abstract
1,4- and 1,5-Disubstituted triazole amino acid monomers have gained increasing interest among peptidic foldamers, as they are easily prepared via Cu- and Ru-catalyzed click reactions, with the potential for side chain variation. While the latter is key to their applicability, the synthesis and structural properties of the chiral mono- or disubstituted triazole amino acids have only been partially addressed. We here present the synthesis of all eight possible chiral derivatives of a triazole monomer prepared via a ruthenium-catalyzed azide alkyne cycloaddition (RuAAC). To evaluate the conformational properties of the individual building units, a systematic quantum chemical study was performed on all monomers, indicating their capacity to form several low energy conformers. This feature may be used to effect structural diversity when the monomers are inserted into various peptide sequences. We envisage that these results will facilitate new applications for these artificial oligomeric compounds in diverse areas, ranging from pharmaceutics to biotechnology.
Introduction
The structural and functional properties of natural biomolecules can be mimicked by synthetic compounds. Such man-made molecules can retain the beneficial properties of their natural counterparts while also benefiting from additional useful attributes. They can provide increased stability against enzymatic degradation,1 better designability, and diverse side-chain chemistry,2 characteristics that are often desired for novel bioactive molecules. Consequently, peptidomimetics and protein engineering often employ synthetic, non-natural, amino acid-derived insertions to improve characteristics of natural compounds.3In the last two decades, it has become apparent that peptidic oligomers containing, or composed almost entirely of, non-natural amino acids can fold into well-defined secondary structures, leading to the emerging field of peptidic foldamers, or foldamers.3c,4 Since the initial studies demonstrating helical oligomers containing homologated derivatives of natural amino acids,2,5 a wide variety of compounds have been investigated in this direction.3c Numerous reports have also pointed out that these compounds will have a propensity to fold into secondary structures. Furthermore they can provide antibacterial,6 antiviral,7 and cell-penetrating activity,8 as well as having self-assembling properties.9 This may lead to applications in pharmaceutics or bionanotechnology.
One advantage of constructing foldamers from non-natural amino acids is that standard techniques for peptide coupling can be applied for assembling these monomers into a sequence.3c,10 Such monomers may include synthetic amino acids based on a cycloalkyl, aromatic or heteroaromatic scaffold.11 1,4-Disubstituted 1,2,3-triazoles, easily accessible via the copper-catalyzed azide alkyne cycloaddition (CuAAC) reaction,12 have also been applied in this context. Examples include oligo(phenyl-amide-triazole)-type foldamers that function as halide ion receptors,13 oxazolidinone-triazole units incorporated into dipeptide mimics for use as foldamer building blocks.14 as well as other examples of peptide backbone modifications using 1,4-disubstituted 1,2,3-triazoles.15 In contrast to CuAAC, the ruthenium-catalyzed cycloaddition reaction (RuAAC) instead provides access to the 1,5-disubstituted 1,2,3-triazole with a different spatial arrangement of the carboxylic acid and amino functionality used for linking the monomeric units.16 Homo- and heterochiral peptidomimetics have been developed from enantiomerically pure propargylamines and α-azido acids using the RuAAC reaction.17 NMR and MD simulations have shown the presence of a β-turn-like structure in homochiral peptido-triazolamers, while a heterochiral triazolamer shows a polyproline-like helix. Moreover, a well defined protocol for the synthesis of chiral propargylamines, with a side chain in the α-position, is feasible using Ellman's tert-butylsulfinamide as a chiral auxiliary.18 The RuAAC reaction has been frequently utilized in medicinal chemistry applications,19 including the synthesis of peptidomimetics,20 but its use in the area of foldamers is as yet very limited. In earlier reports from our group, we have developed microwave-assisted methodology for the synthesis of 1,5-disubstituted 1,2,3-triazole monomers via RuAAC and also investigated the structures and conformations of δ-peptides prepared from these monomers.21 In addition, we have compared the conformational properties of triazole monomers prepared via CuAAC and RuAAC.22 In this study, we instead focus on the conformational effects of introducing chiral substituents, on either side or both sides of the triazole. During the preparation of this manuscript, a related report, concerning the synthesis of some 1,5-disubstituted peptidotriazolomers, was published by Sewald and co-workers.17 While there are some touching points between the two studies, the present study describes the complete set of eight possible combinations for mono- and disubstituted chiral derivatives. Thus, these studies are different both in terms of the triazoles prepared, as well as with respect to the computational studies performed. Our results from these investigations are described herein.
Results and discussion
Monomer synthesis
The chiral alkyne 1 and azide 2 (Fig. 1) used in this study originate from the amino acid alanine. Employing both enantiomers, in combination with the corresponding non-chiral non-methylated N-Boc propargylamine 3 and methyl 2-azidoacetate 4, allowed the synthesis of eight different stereoisomers of the triazole derivatives (Fig. 2). The chiral alkynes (R)-1 and (S)-1 were initially prepared via a Bestmann–Ohira reaction23 from Boc-protected (R)- and (S)-alaninal, where the (R)-enantiomer was obtained via Dess–Martin oxidation24 of Boc-(R)-alaninol, and the (S)-form was commercially available. While both reactions are documented to proceed without causing racemisation,23,25 we found that erosion of the stereochemistry had taken place for both enantiomers of 1 in our case. Raines has reported the preparation of (S)-1via reduction of the corresponding Weinreb amide of alanine, followed by a Bestmann–Ohira reaction of the formed aldehyde in situ, and this may be a better option to avoid prolonged handling of the sensitive aldehyde.20b We instead opted for using commercially available (R)-1 and (S)-1 for the continued studies. For compound 2, both enantiomers were prepared via diazo transfer from the corresponding amine by using 2-azido-1,3-dimethylimidazolinium hexafluoro-phosphate (ADMP), following a report by Kitamura et al.26 The non-chiral derivatives 3 and 4 are both commercially available. With all starting materials in place, reaction conditions for the RuAAC coupling were then investigated. This involved screening various catalysts, including the two most commonly used for RuAAC, i.e. Cp*RuCl(PPh3)2 and Cp*RuCl(COD), as well as [Cp*RuCl2]n and [Cp*RuCl]4. A solution of (S)-1 and azido ester 4 in THF was heated to 60 or 80 °C for 20 minutes under microwave conditions, and products were purified by HPLC or flash chromatography. The results are shown in Table 1. All the catalysts screened, with the exception of [Cp*RuCl]4, provided triazole (S)-5 in moderate to good yields, and showed no significant differences in enantiomeric excess. The preparation of the full set of chiral triazoles was then pursued using either Cp*RuCl(COD) or [Cp*RuCl2]n as the catalyst. | ||
| Fig. 1 Chiral and non-chiral alkynes and azides used in this study. | ||
 | ||
| Fig. 2 Chiral 1,5-subsituted triazoles prepared via RuAAC. See Experimental section for method used. | ||

Alkyne (R)-1 was reacted with 4 using 4 mol% [Cp*RuCl2]n as the catalyst, affording (R)-5, i.e. the enantiomer of the earlier prepared triazole (S)-5, in a somewhat lower yield (52%). Conventional heating at 100 °C, using Cp*RuCl(COD) as the catalyst, was found to be more successful in this case, affording (R)-5 in 79% yield (Fig. 2) and high enantiomeric excess (>99%). Subsequently, the (R)- and (S)-enantiomers of chiral azide 2 were applied and combined with N-Boc-propargylamine 3, yielding the two enantiomers of triazole 6, with the methyl substituent in the α-position relative to the ester, in 84 and 87% yield respectively. 2D NOESY NMR confirmed the 1,5-regioselectivity of the 1,2,3-triazoles (see ESI†). However, some loss of stereochemistry was found for compounds (R)-6 and (S)-6, with approximately 10% of the other enantiomer formed alongside the desired product. As the chiral centre next to the alkyne 1 remains intact during the reaction, as seen in Table 1, the cause of this partial racemisation was pinpointed to the methyl-substituted carbon of azides 2. To determine if this racemisation occurs during the synthesis of the chiral precursor azides 2, or during the RuAAC reaction itself, azides 2 were subjected to a CuAAC reaction at room temperature, using CuSO4 as the catalyst (Scheme 1). Both 1,4-triazoles (R,R)-8 and (S,R)-8 were formed in good yields (92% and 87% respectively) and good to excellent stereoselectivity, indicating that the minor loss of stereochemistry is most likely occuring during the triazole formation itself, rather than in the preparation of 2. With these results in hand, all four stereoisomers of the dimethylated triazole 7 (Fig. 2) were prepared in the same fashion. Azide (R)-2 was first combined with both enantiomers of alkyne 1 to produce the diastereomers (R,R)- and (R,S)-7 in similar yields as for compounds 6. The same reaction starting instead from (S)-2 afforded the second set of diastereomers (S,R)- and (S,S)-7 in 95% and 77% yield, respectively. Interestingly, all these compounds were obtained with excellent enantioselectivity and good to excellent diastereoselectivity. The loss of stereochemical integrity for compounds 6 may thus be a result of the lower degree of sterical hindrance in these compounds, allowing racemisation via enolisation to take place, while the extra methyl group in compounds 7 may in some manner block this process. More detailed studies are needed, however, to elucidate the mechanism for this racemisation.
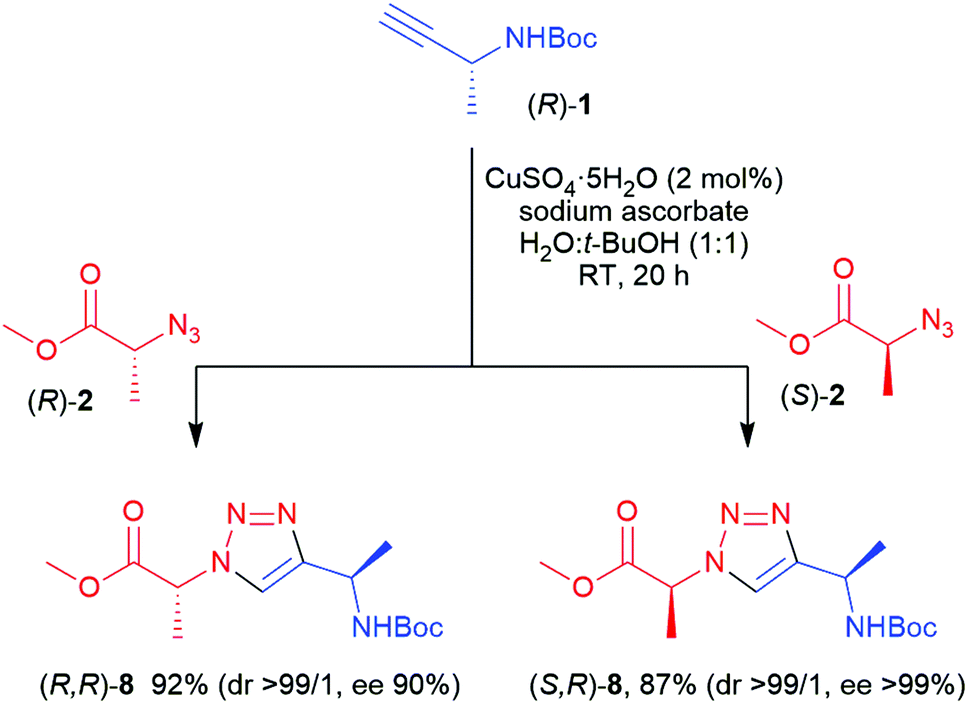 | ||
| Scheme 1 Chiral 1,4-substituted triazoles prepared via CuAAC. | ||
Trimer synthesis
To verify that the chiral triazoles prepared could be used for foldamer synthesis, a trimer was prepared from triazole (S)-5 (Scheme 2). N-Methyl amide (S)-9 was first synthesized via hydrolysis of (S)-5 with LiOH to the corresponding carboxylic acid, followed by HATU-mediated amide formation, affording (S)-9 in 65% yield. A dimer of (S)-5 was then prepared. In one experiment, (S)-5 was Boc-deprotected with trifluoroacetic acid, and in a second experiment, (S)-5 was again hydrolyzed with LiOH. The two parts were then coupled directly without further purification, using HATU as the coupling agent and DIPEA as the base, affording dimer (S,S)-10 in 78% yield. (S)-9 was then deprotected and subsequently coupled with hydrolysed (S,S)-10, affording trimer (S,S,S)-11 in 46% yield as a white solid. 2D NOESY NMR was performed on (S,S,S)-11 to study the conformational properties of the trimer. The 1H–1H NOESY was recorded at 24 mM concentration in DMSO-d6, using a mixing time of 500 ms. In comparison to a corresponding non-chiral trimer reported by us earlier,21 with the N-methyl amide replaced by a methyl ester, the results are much less conclusive. Where the non-chiral structure displays a long range coupling between the Boc tert-butyl group and the terminal methyl group, i.e. between the two ends of the oligomer, indicating a turn-like conformer, no such correlation can be seen for (S,S,S)-11. This is perhaps not surprising, as a greater backbone flexibility would be expected for the corresponding non-chiral trimer. | ||
| Scheme 2 Synthesis of trimer (S,S,S)-11. | ||
Of more interest is a weak short-range coupling between the triazole CH signals and the methyl substituents on the chiral carbons in (S,S,S)-11, indicating a twisted conformer that allows these groups to come into proximity, rather than a structure with a straighter elongated backbone chain. Excluding expected correlations, no other interactions of interest could be discerned.
Molecular modelling
With the full set of chiral triazoles at hand, our attention was then directed towards the conformational properties of these compounds. To explore the conformational properties of the eight chiral derivatives prepared, quantum chemical (QM) calculations were performed employing three different levels of theory. QM methods are regularly employed in understanding structural and energetic properties of non-natural amino acid compounds. In contrast to natural amino acids and proteins, proper parametrization of these exotic residues is often missing, making extensive use of molecular dynamics (MD) simulations less reliable.27 Previously, the potential energy hypersurface of an achiral 1,5-disubstituted 1,2,3-triazole amino acid was explored in an exhaustive scan.22 In the present study, 20 of these previously found achiral conformers were employed to construct the eight chiral models, which resulted the general structure of Ac-5Tzl-NHMe (for more details see Computational methods, and ESI, Tables S1–S8†). Since the R and S conformers could be considered enantiomer pairs, these were tested against the same initial torsional angles from results of the achiral triazole calculations, and conformational enantiomers were not considered. This way we obtained the relative energies for only one side of the asymmetric potential energy hypersurface (PEHS) for each model, where those of the enantiomer pairs can be converted into each other by their inversion center. Therefore the obtained results are discussed for the enantiomer pairs together. Considering the (R)-5 and (S)-5 models, respectively, 9 and 7 out of the located 18 and 17 conformers are within 3 kcal mol−1 in relative energy, counted from the most stable conformer 2′ and 2 (see ESI†). This indicates that in principle both derivatives have over ten conformers which have low relative energies and thus could be the structural building units of several low energy secondary structures. For (R)-6 and (S)-6, there are 6 and 11, respectively, low energy conformers, i.e. below 3 kcal mol−1 relative energy, with the most stable conformers being 2 and 1 (see ESI†), respectively. These results indicate that the single substituted models closely resemble to energetic properties of the achiral triazole derivative where numerous low-energy secondary structures can be found. In contrast, for the (R,R)-7 and (S,S)-7 enantiomer pair, there are only 3 and 5 low energy conformers, with 6 and 1 (see ESI†), respectively, being the most stable. The (R,S)-7 and (S,R)-7 have 3 and 7 low energy conformers, respectively, with conformers 2 and 2′ as most stable ones (see ESI†). The lower number of stable conformers for the disubstituted models most likely originate from steric interactions.Overall, all chiral compounds have several low energy conformers, which may result in conformational diversity when homo-oligomers are synthesized from these monomers.
Structure and energetics of heptamers
The eight sets of secondary structures were investigated in order to predict the potentially most preferred structure for each substitution pattern. To allow formation of defined secondary structures, longer peptide sequences, heptamers, were employed. All these sets were investigated in an aqueous environment, as detailed in the Experimental section under Computational methods. These conformations have intramolecular hydrogen bonds formed between relevant parts of the amide groups. Our nomenclature for the conformations is based on the pseudo-cycles formed by hydrogen bonds, using the number of atoms in the peptide backbone involved in forming the H-bond. Thus, conformers H8, H10, H14, and H16 were investigated for both right (P) and left handed (M) structures.When using water as an environment, for (R)-6, (R)-5, and (R,R)-7, the most preferred secondary structure is the right-handed H16 conformation, H16P (Fig. 3 and Table S9†) while the second most stable conformation is H14P. Similarly, due to being conformational enantiomers, for (S)-6, (S)-5, and (S,S)-7, the most preferred structure is also H16, but its left handed form, H16M. For these compounds, the above H16 conformations have a high relative stability compared to the other investigated secondary structures, as their energy is nearly 9 kcal mol−1 lower than that of the second most stable conformer, H14. Note, that for (R)-5 and (R,R)-7 the H10P is quite close in relative free energy to H14P. Thus, for these compounds it is very likely that an oligomer with the above chiral substitution patterns could fold into a helical structure, such as H16, H14 or H10. In general, when investigating handedness there is a strong preference towards either P or M, as for (R)-6, and (S)-6, respectively, where the least stable conformer is H14 for both of them, but with the inverse handedness. For the (R)-5 and (S)-5 substitution patterns we see a very similar relative energy distribution, and preference of handedness is also pronounced because all conformers belonging to the preferred P, or M subsets have higher or lower relative energies than its inverse conformer, respectively. For (R,R)-7 and (S,S)-7, H16 is again the most stable conformer with H14 being the second most stable one, however, here these conformers have a somewhat higher stability relative to the other conformers investigated (Fig. 3).
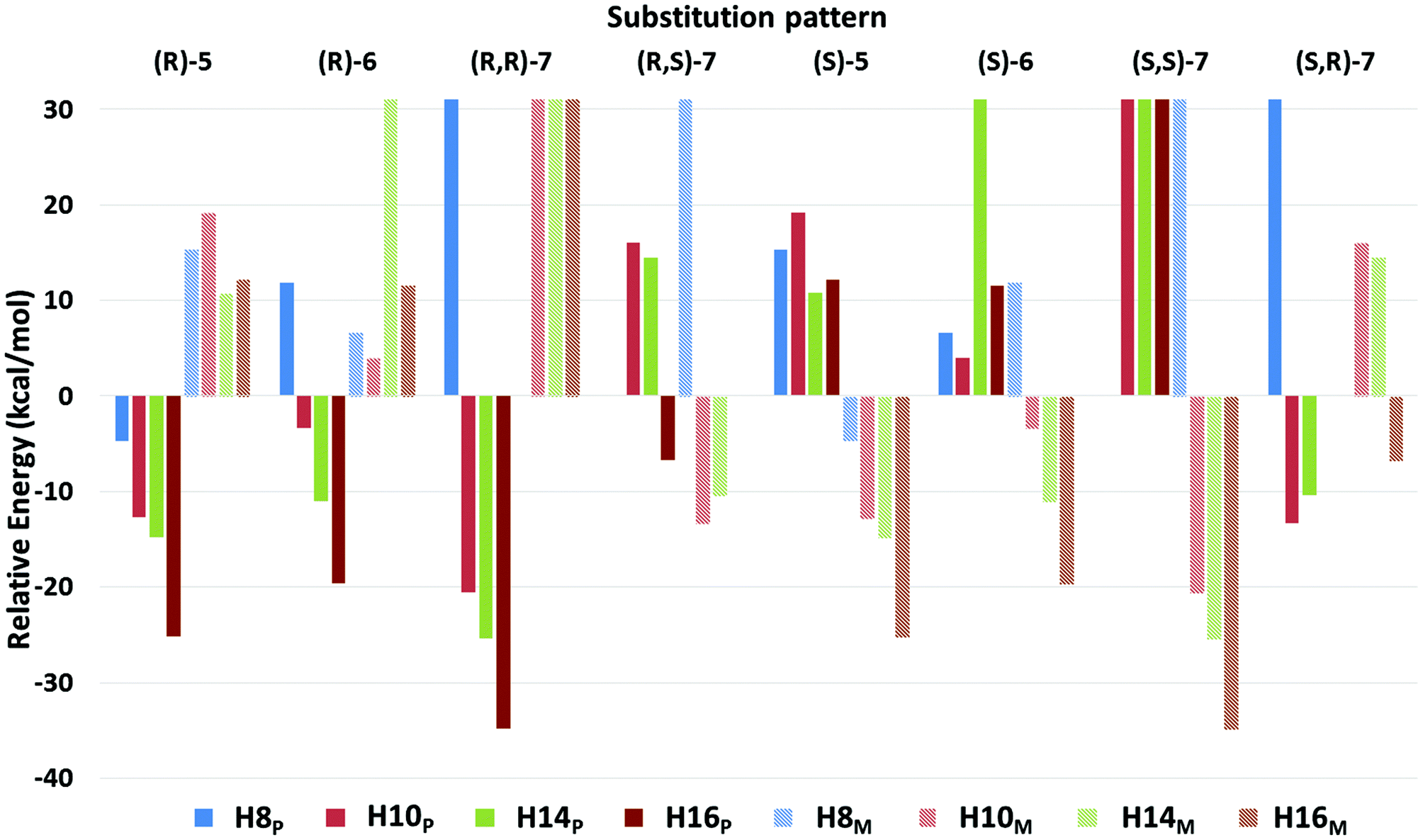 | ||
| Fig. 3 The relative free energies of the investigated secondary structures for hepta-homooligomers using all 8 possible substitution patterns. Values were obtained at the ωB97X-D/6-31+G(d,p)//ωB97X-D/6-31G(d) level of theory using water as solvent and involving entropy contributions as well as zero point and thermal energy corrections. Relative values were determined for each peptide using the average energy value of all secondary structures as reference point. For more details, see section Computational methods. | ||
In contrast to the above six substitution patterns, from a structural point of view the oligomers built from (S,R)-7 and (R,S)-7 monomers seem to be the most interesting. In the case of these two oligomers, the relative energy of the investigated conformers have values much closer to each other. For (R,S)-7, the most stable conformer is H10M, however its relative energy is only ∼3 kcal mol−1 from the second most stable conformer, H14M (Fig. 4). Furthermore, the third most stable conformer is H16P, which has the opposite handedness as the above two conformers, yet its energy is still within 7 kcal mol−1 to that of H10M. Similarly, in case of (S,R)-7, the same energy distribution can be observed. Based on these results for (S,R)-7, (R,S)-7 systems, there is probably a steric factor introduced which renders various secondary structures to comparable stability. This will probably give rise to a conformational diversity that is comparable to that observed for natural peptidic compounds in living systems. Note, that for homooligomers, the above results are in contrast to the conformational diversity observed for monomeric compounds. Once the monosubstituted Tzl amino acids are oligomerized, there is a clear preference for one secondary structure among the ones investigated. This observation is also valid for disubstituted compounds with both substitutions having the same chirality, (S,S)-7 and (R,R)-7. In contrast to the above, the mixed substitution patterns, (S,R)-7 and (R,S)-7, may result in conformational diversity for homooligomeric foldamers as several secondary structure elements have close relative energies, irrespective of handedness.
 | ||
| Fig. 4 The three most stable conformation of (S,R)-7 heptahomooligomer. Calculations were performed at the ωB97X-D/6-31+G(d,p)//ωB97X-D/6-31G(d) level of theory. | ||
Conclusions
The complete set of possible chiral combinations for 1,5-disubstituted 1,2,3-triazole units have been prepared via a ruthenium-catalyzed azide alkyne cycloaddition (RuAAC), in moderate to good yields. These chiral monomeric triazole building blocks are intended for applications in the area of foldamer synthesis. The precursor alkynes and azides used for the RuAAC reaction all originate from the amino acids glycine and alanine. To demonstrate the applicability of the 1,5-triazole monomer building blocks in the synthesis of foldamers, a trimer structure was also prepared. A conformational and structural investigation of these monomers was then carried out to predict the effect of introducing these units into a foldamer. Computational studies on the monomers indicate that several low energy conformers are present for all eight compounds, where several secondary structures may be built with low relative energies for peptidic molecules containing these compounds as single insertions. This is especially indicated for the monosubstituted compounds, where the number of lower energy conformers exceeds ten. However, when considering homooligomer foldamers built from these compounds, relative energies show a single most stable conformer for 6 out of the 8 substitution patterns. The only two exceptions are (R,S) and (S,R), where several secondary structures result in relative energies very close to each other, indicating that structural diversity of the backbone, as a key feature for fine-tuning structural properties with various side chain patterns, is present. Overall, we anticipate that the outcome of these studies can be of use in the design and synthesis of new classes of foldamers from non-natural amino acids based on a 1,5-disubstituted 1,2,3-triazole scaffold.Experimental
General experimental methods
Starting materials were purchased from commercial sources unless otherwise stated. All reactions were performed under an inert atmosphere (argon or nitrogen). Chiral alkynes 1 were initially synthesized from commercial Boc-(R)-alaninol and Boc-(S)-alaninal in one or two steps, but were subsequently purchased from Ark Pharm Inc. (tert-butyl (S)-but-3-yn-2-yl-carbamate, 99.5% ee) and J&W PharmLab LLC (tert-butyl (R)-but-3-yn-2-ylcarbamate, 98.8% ee). THF was purchased as anhydrous grade. Automated flash chromatography was performed on a Biotage Isolera One. 1H, 13C and 2D NOESY NMR were acquired on a Varian MR 400 MHz instrument or on a Bruker Avance III 500 MHz spectrometer fitted with a cryoprobe in CDCl3 or DMSO, using residual solvent peaks for reference. FTIR was performed on a PerkinElmer Frontier FTIR Spectrometer with an ATR device using Spectrum software. The enantiomeric excess was determined by chiral SFC, using a Chiralpak IC column (25% EtOH in CO2) or a Lux Cellulose-5 column (25% EtOH in CO2) or a Lux Amylose-1 column (10% EtOH in CO2) at 120 bar, 40 °C, flow rate 3.5 mL min−1, 220 nm detection (see ESI† for chromatographic data for each compound).Experimental procedures
Synthesis of chiral propargylic amines
Synthesis of chiral azides
General procedure for the RuAAC reaction
RuAAC reactions were performed using microwave heating (Method A) or conventional oil bath heating (Method B).Method A: The corresponding alkyne (0.44 mmol) and azide (0.44 mmol) were charged in a Biotage microwave vial with a stirrer bar and dissolved in anhydrous THF (3 mL). Cp*RuCl(COD) (8.4 mg, 0.02 mmol) was added and the vial was sealed, evacuated and refilled with nitrogen three times. The reaction mixture was then heated to 60 °C for 20 min in a microwave reactor or heated in an oil bath at 100 °C for 15 h. The resulting solution was filtered through a syringe filter and concentrated. The crude product was purified by preparative HPLC on a Kromasil C8 column (10 μm 250 × 20 ID mm), using a gradient of 5–45% acetonitrile in a H2O/acetonitrile/formic acid (95/5/0.2) buffer, over 20 minutes with a flow rate of 19 mL min−1. The compounds were detected by UV at 225 nm. Pure fractions were combined and lyophilized to obtain the product.
Method B, exemplified by the formation of (S)-6: To a microwave vial equipped with a stirrer bar was added methyl (S)-2-azidopropanoate (26 mg, 0.2 mmol) and N-Boc-propargylamine (34 mg, 0.2 mmol) along with Cp*RuCl(COD) (3.5 mg, 0.009 mmol) and THF (1 mL). The vessel was sealed in an argon atmosphere and stirred at 100 °C for 15 h or until crude TLC analysis indicated no more starting materials were being consumed (visualized by staining with ninhydrin solution and heating). The crude product was purified by automated flash chromatography on a Biotage Isolera One system, using a gradient of 30–60% EtOAc in petroleum ether. Pure fractions were combined and concentrated by rotary evaporation to afford the product.
General procedure for the CuAAC reaction
To a Biotage microwave vial equipped with a stirrer bar was added the corresponding azide (26 mg, 0.2 mmol) and alkyne (34 mg, 0.2 mmol) along with CuSO4·5H2O (1 mg, 0.004 mmol), sodium ascorbate (2.5 mg, 0.012 mmol) and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2O:tBuOH (1 mL). The vessel was sealed in an air atmosphere and stirred at room temperature for 20 h at which point TLC analysis indicated the reaction was complete. The crude reaction mixture was then diluted in CH2Cl2 (10 mL) and water (10 mL) and the aqueous layer was extracted a further two times with CH2Cl2 (2 × 10 mL). The combined organic fractions were then washed with brine (10 mL) and dried using MgSO4. The MgSO4 was then removed from the organic fractions by filtration, concentrated, and purified by flash column chromatography (40–60% EtOAC/hexane) to afford the pure products.
:1 mixture of H2O:tBuOH (1 mL). The vessel was sealed in an air atmosphere and stirred at room temperature for 20 h at which point TLC analysis indicated the reaction was complete. The crude reaction mixture was then diluted in CH2Cl2 (10 mL) and water (10 mL) and the aqueous layer was extracted a further two times with CH2Cl2 (2 × 10 mL). The combined organic fractions were then washed with brine (10 mL) and dried using MgSO4. The MgSO4 was then removed from the organic fractions by filtration, concentrated, and purified by flash column chromatography (40–60% EtOAC/hexane) to afford the pure products.
Synthesis of dimer (S,S)-10
Synthesis of trimer (S,S,S)-11
Computational methods
All computations were carried out using the Gaussian 09 software package.33 In contrast to experiments, for the calculations the monomer amino acids had N-methyl and acetamide protecting groups at their C- and N-terminals, respectively, abbreviated as Ac-5Tzl-NHMe. In case of the longer models the C-terminal protecting group was NH2, resulting the general Ac-(5Tzl)7-NH2 form. Nevertheless, the same nomenclature was applied for the substitution patterns as for the experiments (Fig. 2). For the monomers, the 27 minima obtained previously in an exhaustive conformational search for the achiral models at the RHF/3-21G level of theory,22 were employed in the current study as initial conformations. Out of these 7 starting conformers were completely planar, with relative energies of their achiral version higher than 20 kcal mol−1. Consequently, these were omitted for the current chiral models, leaving 20 starting test conformers for all eight models. The total of 160 optimisations were primarily performed at the RHF/3-21G level of theory. Further on, the optimised structures were submitted to calculations using Becke's three parameter functional with the Lee–Yang–Parr exchange functional (B3LYP),34 the ωB97X-D functional by Chai and Head-Gordon,35 which included dispersion correction and long-range electron correlation corrections, as well as M06-2X functional developed by Zhao and Truhlar.36 For all functionals, the 6-311++G(2d,2p) basis set was employed. The obtained results for all the conformers are found in the ESI.†To estimate secondary structure preference of longer oligomers composed of the investigated 8 monomers, previously defined secondary structures were employed.21 Initially, five secondary structures, four helical, and one extended, were modified with the 8 chiral substitution patterns and optimised at the ωB97x-D/6-31G(d) level of theory. To predict preference of helicity handedness, both P and M helices were optimised resulting a total of 9 secondary structures. Considering the large size and the unfavorable nature of the extended conformation, for many substitution patterns this conformer experienced convergence problems during the calculations, therefore this conformer was excluded from further investigation. To validate the critical points, i.e. to confirm that they are local minima, as well as to obtain zero point and thermal corrections to the energy, and entropy contributions to provide Gibbs free energy values, normal mode analysis was performed on the obtained conformers at the ωB97x-D/6-31G(d) level of theory. These optimised conformers were also submitted to point energy calculations employing the same functional and the 6-31+G(d,p) basis set. Further on, environmental effects of water were considered also by single point energy calculations at the samel level of theory, using the implicit solvent model integral equation formalism for polarizable continuum model (IEFPCM) (Tables S1–S9†).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The following sources of funding are gratefully acknowledged: the Swedish Research Council (N. K., 2015-05360), the Swedish Research Council Formas (N. K., 2015-1106), the Carl Trygger Foundation (A. P., CTS 16:235), the Hungarian Scientific Research Fund (T. B.-S., OTKA-109588), the Momentum program of the Hungarian Academy of Sciences (T. B.-S., LP2016-2), the National Competitiveness and Excellence Program (NVKP_16-1-2016-0007), the Excellence program BIONANO_GINOP-2.3.2-15-2016-00017, the Nemzeti Versenyképességi és Kiválósági Program, Hungary (NVKP_16-1-2016-0007), the Gazdaságfejlesztési és Innovációs Operatív Program, Hungary (GINOP-2.3.2-15-2016-00017), and a Marie Curie fellowship (T. B.-S., MSCA-IF BARREL 660030).References
- (a) J. V. Schreiber, J. Frackenpohl, F. Moser, T. Fleischmann, H. P. E. Kohler and D. Seebach, ChemBioChem, 2002, 3, 424–432 CrossRef CAS PubMed; (b) D. F. Hook, F. Gessier, C. Noti, P. Kast and D. Seebach, ChemBioChem, 2004, 5, 691–706 CrossRef CAS.
- D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodivers., 2004, 1, 1111–1239 CrossRef CAS.
- (a) T. Kimmerlin, D. Seebach and D. Hilvert, Helv. Chim. Acta, 2002, 85, 1812–1826 CrossRef CAS; (b) G. V. M. Sharma, B. S. Babu, K. V. S. Ramakrishna, P. Nagendar, A. C. Kunwar, P. Schramm, C. Baldauf and H. J. Hofmann, Chem. – Eur. J., 2009, 15, 5552–5566 CrossRef CAS PubMed; (c) T. A. Martinek and F. Fulop, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- K. Gademann, T. Hintermann and J. V. Schreiber, Curr. Med. Chem., 1999, 6, 905–925 CAS.
- W. S. Horne and S. H. Gellman, Acc. Chem. Res., 2008, 41, 1399–1408 CrossRef CAS PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. Degrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756 CrossRef CAS PubMed.
- N. Umezawa, M. A. Gelman, M. C. Haigis, R. T. Raines and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 368–369 CrossRef CAS PubMed.
- J. Kim, S. Kwon, S. H. Kim, C. K. Lee, J. H. Lee, S. J. Cho, H. S. Lee and H. Ihee, J. Am. Chem. Soc., 2012, 134, 20573–20576 CrossRef CAS PubMed.
- (a) R. V. Nair, K. N. Vijayadas, A. Roy and G. J. Sanjayan, Eur. J. Org. Chem., 2014, 7763–7780 CrossRef CAS; (b) J. S. Laursen, J. Engel-Andreasen and C. A. Olsen, Acc. Chem. Res., 2015, 48, 2696–2704 CrossRef CAS PubMed.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed.
- (a) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) E. Haldon, M. C. Nicasio and P. J. Perez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC; (d) C. L. Wang, D. Ikhlef, S. Kahlal, J. Y. Saillard and D. Astruc, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- Y. Wang, J. F. Xiang and H. Jiang, Chem. – Eur. J., 2011, 17, 613–619 CrossRef CAS PubMed.
- N. Castellucci and C. Tomasini, Eur. J. Org. Chem., 2013, 3567–3573 CrossRef CAS.
- (a) W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed; (b) T. Boddaert, J. Sola, M. Helliwell and J. Clayden, Chem. Commun., 2012, 48, 3397–3399 RSC; (c) K. B. Salah, S. Das, N. Ruiz, V. Andreu, J. Martinez, E. Wenger, M. Amblard, C. Didierjean, B. Legrand and N. Inguimbert, Org. Biomol. Chem., 2018, 16, 3576–3583 RSC.
- (a) L. Zhang, X. G. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. C. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. T. Zhao, Z. Y. Lin, G. C. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS.
- O. Kracker, J. Gora, J. Krzciuk-Gula, A. Marion, B. Neumann, H. G. Stammler, A. Niess, I. Antes, R. Latajka and N. Sewald, Chem. – Eur. J., 2018, 24, 953–961 CrossRef CAS.
- M. Wunsch, D. Schroder, T. Frohr, L. Teichmann, S. Hedwig, N. Janson, C. Belu, J. Simon, S. Heidemeyer, P. Holtkamp, J. Rudlof, L. Klemme, A. Hinzmann, B. Neumann, H. G. Stammler and N. Sewald, Beilstein J. Org. Chem., 2017, 13, 2428–2441 CrossRef.
- J. R. Johansson, T. Beke-Somfai, A. S. Stålsmeden and N. Kann, Chem. Rev., 2016, 116, 14726–14768 CrossRef CAS PubMed.
- (a) D. S. Pedersen and A. Abell, Eur. J. Org. Chem., 2011, 2399–2411 CrossRef CAS; (b) A. Tam, U. Arnold, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 12670–12671 CrossRef CAS PubMed; (c) J. K. Pokorski, L. M. M. Jenkins, H. Q. Feng, S. R. Durell, Y. W. Bai and D. H. Appella, Org. Lett., 2007, 9, 2381–2383 CrossRef CAS PubMed; (d) D. Tietze, M. Tischler, S. Voigt, D. Imhof, O. Ohlenschlager, M. Gorlach and G. Buntkowsky, Chem. – Eur. J., 2010, 16, 7572–7578 CrossRef CAS PubMed; (e) M. Empting, O. Avrutina, R. Meusinger, S. Fabritz, M. Reinwarth, M. Biesalski, S. Voigt, G. Buntkowsky and H. Kolmar, Angew. Chem., Int. Ed., 2011, 50, 5207–5211 CrossRef CAS PubMed; (f) J. Q. Zhang, J. Kemmink, D. T. S. Rijkers and R. M. J. Liskamp, Chem. Commun., 2013, 49, 4498–4500 RSC; (g) S. Ferrini, J. Z. Chandanshive, S. Lena, M. C. Franchini, G. Giannini, A. Tafi and M. Taddei, J. Org. Chem., 2015, 80, 2562–2572 CrossRef CAS; (h) X. Yang, L. P. Beroske, J. Kemmink, D. T. S. Rijkers and R. M. J. Liskamp, Tetrahedron Lett., 2017, 58, 4542–4546 CrossRef CAS; (i) S. Pacifico, A. Kerckhoffs, A. J. Fallow, R. E. Foreman, R. Guerrini, J. McDonald, D. G. Lambert and A. G. Jamieson, Org. Biomol. Chem., 2017, 15, 4704–4710 RSC; (j) E. Engholm, N. Stuhr-Hansen and O. Blixt, Tetrahedron Lett., 2017, 58, 2272–2275 CrossRef CAS.
- J. R. Johansson, E. Hermansson, B. Norden, N. Kann and T. Beke-Somfai, Eur. J. Org. Chem., 2014, 2703–2713 CrossRef CAS.
- N. Kann, J. R. Johansson and T. Beke-Somfai, Org. Biomol. Chem., 2015, 13, 2776–2785 RSC.
- D. Habrant, V. Rauhala and A. M. P. Koskinen, Chem. Soc. Rev., 2010, 39, 2007–2017 RSC.
- (a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef CAS.
- M. Kitamura, S. Kato, M. Yano, N. Tashiro, Y. Shiratake, M. Sando and T. Okauchi, Org. Biomol. Chem., 2014, 12, 4397–4406 RSC.
- C. Baldauf and H. J. Hofmann, Helv. Chim. Acta, 2012, 95, 2348–2383 CrossRef CAS.
- J. Ryan, M. Siauciulis, A. Gomm, B. Macia, E. O'Reilly and V. Caprio, J. Am. Chem. Soc., 2016, 138, 15798–15800 CrossRef CAS.
- B. Rajagopal, Y. Y. Chen, C. C. Chen, X. Y. Liu, H. R. Wang and P. C. Lin, J. Org. Chem., 2014, 79, 1254–1264 CrossRef CAS PubMed.
- J. A. Fehrentz and B. Castro, Synthesis, 1983, 676–678 CrossRef CAS.
- R. V. Hoffman and H. O. Kim, Tetrahedron, 1992, 48, 3007–3020 CrossRef CAS.
- A. Paul, H. Bittermann and P. Gmeiner, Tetrahedron, 2006, 62, 8919–8927 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2006, 120, 215–241 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational data, determination of enantiomeric excess for monomers, 1H and 13C NMR spectra. See DOI: 10.1039/d0ob00168f |
| This journal is © The Royal Society of Chemistry 2020 |