
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated access to well-defined ionic oligosaccharides†
Yuntao
Zhu
 a,
Theodore
Tyrikos-Ergas
ab,
Kevin
Schiefelbein
a,
Andrea
Grafmüller
c,
Peter H.
Seeberger
ab and
Martina
Delbianco
*a
a,
Theodore
Tyrikos-Ergas
ab,
Kevin
Schiefelbein
a,
Andrea
Grafmüller
c,
Peter H.
Seeberger
ab and
Martina
Delbianco
*a
aDepartment of Biomolecular Systems, Max-Planck-Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: martina.delbianco@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
cDepartment of Theory, Max-Planck-Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany
First published on 4th February 2020
Abstract
Ionic polysaccharides are part of many biological events, but lack structural characterisation due to challenging purifications and complex synthesis. Four monosaccharides bearing modifications not found in nature are used for the automated synthesis of a collection of ionic oligosaccharides. Structural analysis reveals how the charge pattern affects glycan conformation.
Polysaccharides bearing charged functional groups are ubiquitous in Nature, including chitosan, pectin, and glycosaminoglycans (GAGs).1 These polysaccharides play important roles in plant and bacteria cell–wall formation,2,3 cellular interactions,4 and signaling.5 Due to their unique gelation properties and tendency to bind metals6 and/or proteins,5 they are useful biomaterials for drug delivery and tissue engineering.2,7–10 Little is known on how a particular charge pattern can affect the material properties,11–13 partially due to the lack of pure samples with well-defined structures. Polysaccharides, generally extracted from natural sources as heterogeneous mixtures, limit definitive structure–property correlations. A better understanding of how the charge pattern affects carbohydrate conformation and aggregation is necessary for the design of functional carbohydrate materials, as already demonstrated for soft materials based on peptides and proteins.14,15
Automated Glycan Assembly (AGA) is a powerful method for the quick production of well-defined natural and unnatural oligosaccharides.16–18 Many natural ionic oligosaccharides have been prepared by AGA,19–23 however several complications make the synthesis of these glycans challenging.24 Uronic acid building blocks (BBs) suffer from low reactivity during glycosylation.19,20,23 Glycosylation of natural 2-amino-2-deoxysugars often require tedious protecting group manipulations.20,25 Regioselective sulfation is challenging and the poor stability of sulfated moieties can be troublesome due to migration and/or cleavage during further synthetic manipulations.20,26 An alternative approach to access glycans with a well-defined charge pattern relies on the insertion of unnatural charged sugar units, that are much easier to handle during the glycan synthesis.
Here, we describe four unnatural BBs based on a carboxylic acid masked as methyl ester and an amine masked as azide. All BBs are prepared in gram scale from commercially available precursors and show good reactivity and stability upon storage. The AGA of a collection of oligosaccharides with well-defined charge patterns illustrates the utility of these BBs. Cellulose-like structures were prepared, due to the importance of cellulose and its ionic derivatives, such as carboxymethyl cellulose.27,28 Deprotection methods on solid support, such as methanolysis and Staudinger reduction, were optimised to provide the desired compounds in good yields.29,30 Molecular dynamics (MD) simulations indicate the importance of the charge pattern for the oligosaccharide conformation.
The insertion of unnatural functionalities in defined position of the cellulose backbone is a powerful approach to manipulate the hydrogen-bonding and modulate the aggregation of cellulose.29 To gain a better understanding of the roles played by the charged moieties for oligosaccharide conformation, four glucose BBs were synthesised (BB-1–4, Scheme 1). Unnatural BBs bearing a methyl carboxylate or an azide were designed to reveal negative or a positive charge, upon global deprotection. The different position of the substituent on the glucose ring (position 3 vs. 6) permits to further manipulate the H-bond network.
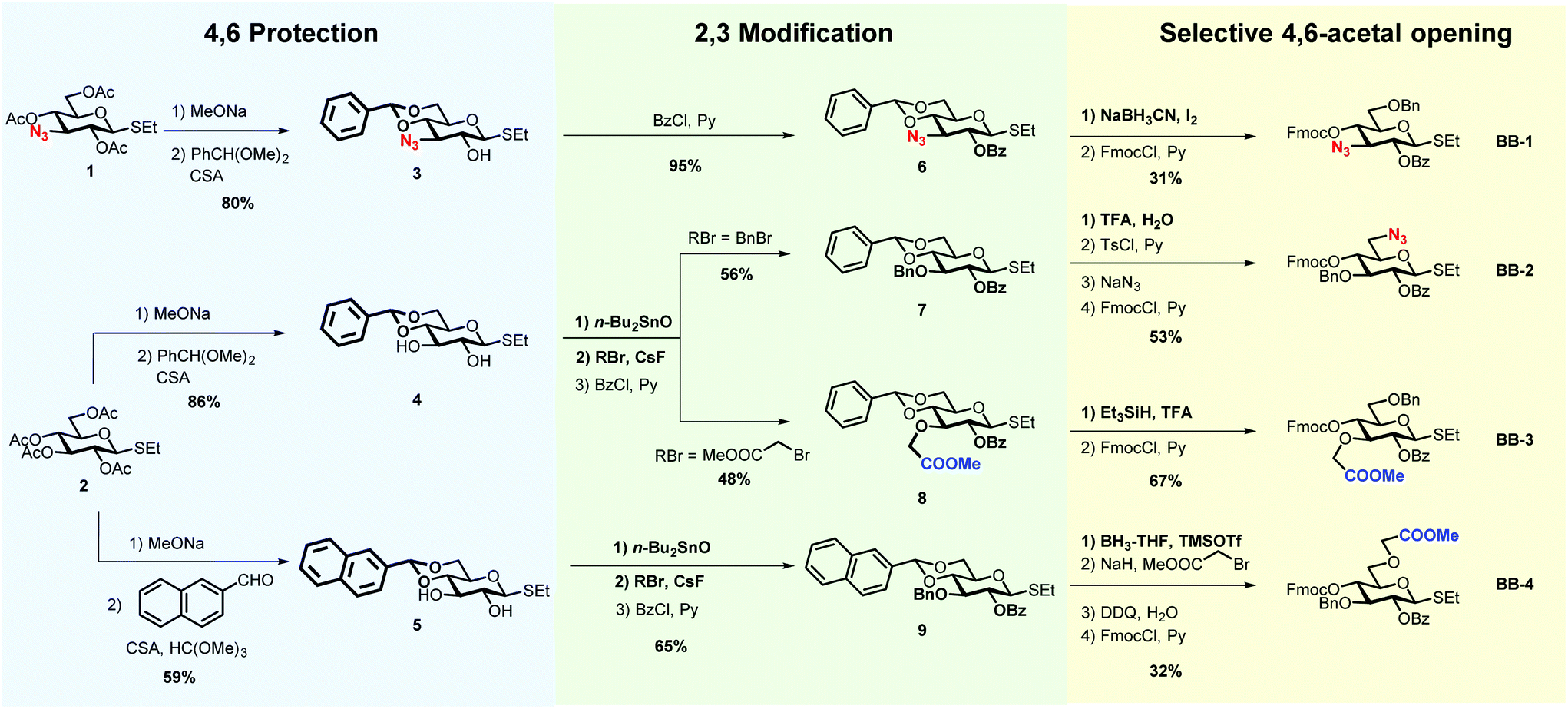 | ||
| Scheme 1 Strategies for the synthesis of four unnatural BBs bearing masked charged functionalities. | ||
The BB design follows AGA considerations.18 Thioglycosides are stable to prolonged storage and show good reactivity during glycosylations.31 Benzoyl groups (Bz) were used as participant protecting groups to direct the formation of the β-(1–4) linkage. The fluorenylmethoxycarbonyl (Fmoc) was selected as a temporary protecting group that can be easily removed in the deprotection cycle, before chain elongation. The remaining positions were selectively functionalised with either methyl carboxylate, azides or benzyl ethers (Bn).
All BBs can be prepared following a similar strategy based on the selective protection of position 4 and 6 with cyclic acetals, regioselective functionalisation of positions 2 and 3, and selective acetal opening (Scheme 1). The azido-functionalised compound 1, obtained in seven steps starting from glucose diacetone (see ESI†), served for the synthesis of BB-1. Peracetylated ethyl β-D-thioglucopyranoside (2) allowed for the synthesis of BB2–4. Methanolysis of the acetyl groups, followed by 4,6-O-benzylidene protection using benzaldehyde dimethyl acetal catalysed by camphorsulfonic acid, afforded compounds 3–4.32 Alternatively, the 4,6-O-naphthylmethylene acetal was introduced using 2-naphthaldehyde catalysed by camphorsulfonic acid and trimethyl orthoformate, yielding compound 5.33
2-O-Benzoyl protected 6 was obtained readily upon treatment with benzoyl chloride and pyridine. Treatment with dibutyltin oxide, followed by addition of the desired alkyl bromide in the presence of cesium fluoride (CsF), permitted the regioselective functionalisation of compounds 4–5.34,35 The addition of CsF to the stanylidene acetal facilitated the nucleophilic substitution with the relatively unreactive, but sterically not very demanding, bromomethyl acetate. Benzoylation afforded compounds 7–9.
Selective opening of the 4,6-O-acetal liberated the desired hydroxyl groups for further chemical modification.36 Compound 8 was subjected to reductive ring opening conditions, in the presence of triethylsilane and trifluoroacetic acid (TFA), to reveal the 4-hydroxyl group. Similar conditions were not compatible with compound 6, resulting in an additional azide reduction. Treatment with sodium cyanoborohydride and iodine afforded the target product (BB-1 precursor). Cleavage of the 4,6-O-benzylidene using TFA–water, followed by regioselective functionalisation of the primary alcohol with 4-toluenesulfonyl chloride (TsCl) permitted the introduction of the azide group in position 6 (BB-2 precursor). Compound 9 was reacted with borane–tetrahydrofuran (BH3–THF) complex and trimethylsilyl trifluoromethanesulfonate (TMSOTf) to liberate the 6-hydroxyl group, that was subsequently subjected to etherification, using bromomethyl acetate and NaH (BB-4 precursor). The introduction of Fmoc completed all BBs syntheses.
BB1–4 were used to prepare a collection of charged oligocellulose analogues by AGA (Scheme 2). Merrifield resin bearing the photolabile linker (S0) was selected as solid support.37 Unmodified glucose units were introduced using the commercially available BB-0. A Staudinger reduction on solid phase was developed, adapting the conditions from a previously reported method.30 The resin was suspended in THF–water and ammonia and triethylphosphine were added, liberating the desired amino group in 24 h. Methanolysis on solid phase29 to hydrolyse the benzoyl groups was performed with trace amounts of water, permitting the simultaneous hydrolysis of the methyl esters. UV cleavage released the partially protected oligosaccharides that, upon hydrogenation catalysed by Pd/C, afforded the target compounds.
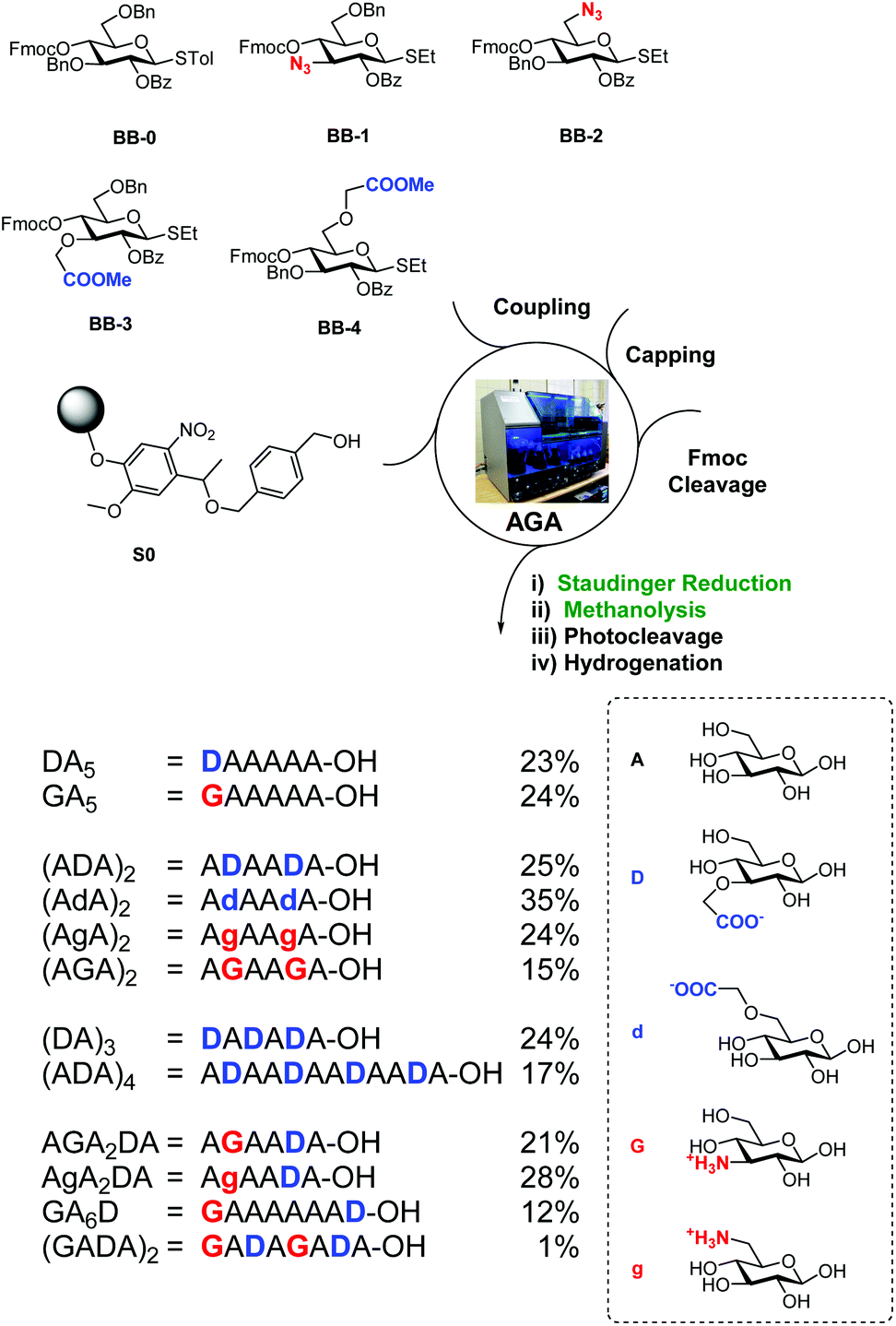 | ||
| Scheme 2 AGA of tailor-made charged cellulose-like oligosaccharides. Yields of isolated products after AGA, deprotection, and purification are reported. | ||
Twelve analogues with different charge patterns were prepared (Scheme 2). All four unnatural BBs showed good reactivity during glycosylation and stability during post AGA manipulation. The modified BBs were successfully positioned at the non-reducing end of the glycan (e.g. DA5 and GA5) or internally (e.g. (AGA)2). Multiple substitutions were also tolerated without any significant loss in yield (e.g. (ADA)4). Zwitterionic compounds (e.g. AGA2DA) were prepared, suggesting the possibility of using such BBs for the preparation of GAG analogues.
These compounds are important substrates to evaluate the effect of the charge pattern on the oligosaccharide structure. MD simulations were performed to systematically examine the effect of each modification on glycan conformation (Fig. 1). Four analogues with the same sequence were compared (i.e. (AXA)2). All the modifications resulted in more flexible structures when compared to the unsubstituted analogue A6 (see ESI†). Specific intramolecular hydrogen bonds stabilise particular conformations that can trigger the formation of different materials. The substitution at position 6 (g and d) produces a drastic effect on the ω torsion angles, much less affected by the modification at position 3 (G and D). The interaction between the NH3+(6) and the OH(3) of the adjacent sugar (R + 1) stabilises the gt rotamers (red and yellow plots, Fig. 1).
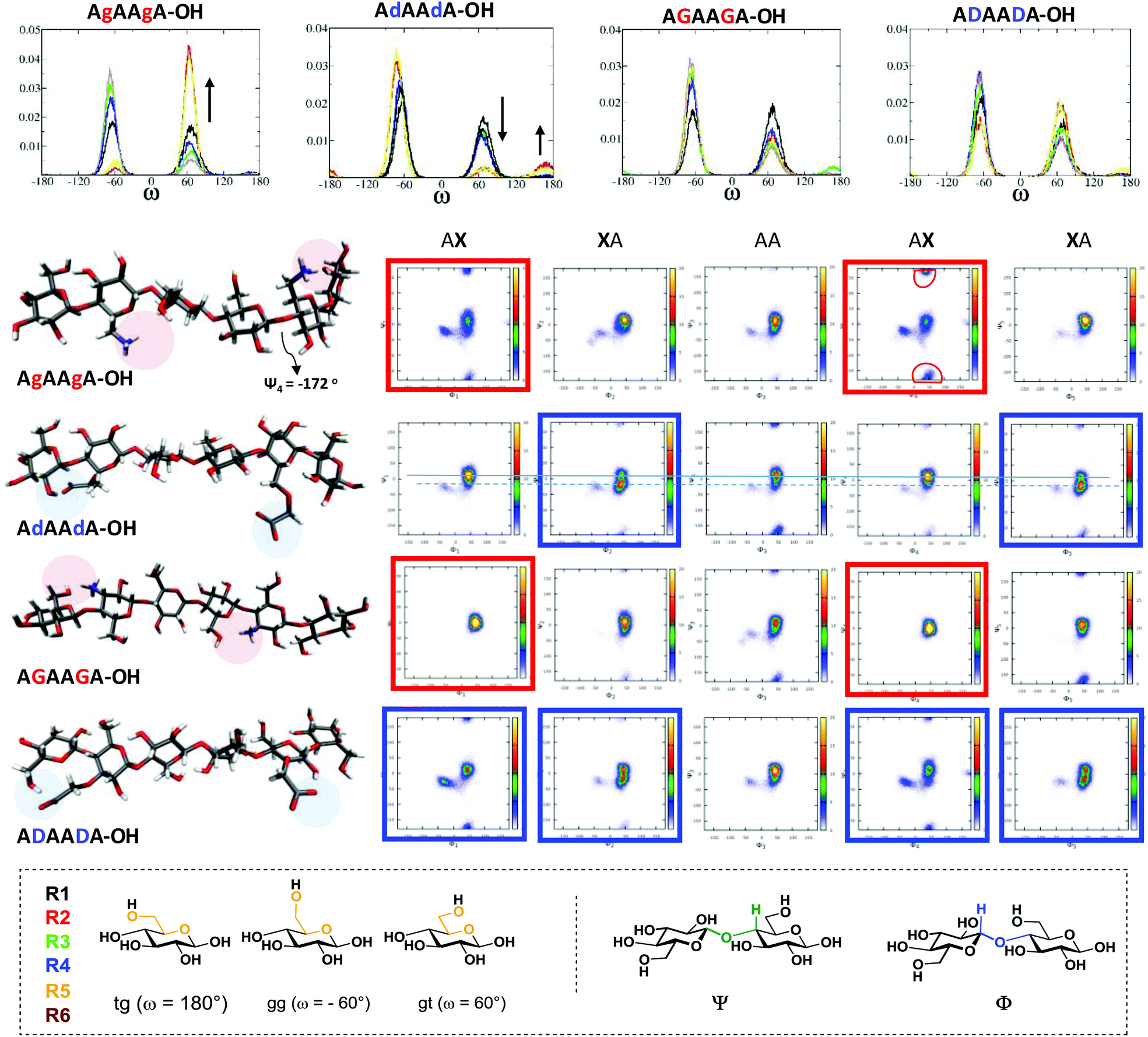 | ||
| Fig. 1 Dihedral analysis (ω, Ψ and Φ) obtained by MD simulations and representative snapshots of the oligomers showing specific intramolecular interactions (highlighted with circles) due to the modifications. The effect of the modification on the ω torsion angles is highlighted with arrows (top graphs). The most affected Ψ and Φ dihedrals are marked by red or blue boxes. For (AdA)2 the two blue lines serve as guide to the eye for comparing different minima. The residues are numbered from the non-reducing end (R1) to the reducing end (R6). | ||
This also affects the glycosidic bond geometry with a high population at negative degrees (−172°) for Ψ1, Ψ4 ((AgA)2 red boxes). The opposite trend is observed when position 6 is substituted with COO− (d). Due to sterics, the gg conformation is preferred, with a small percentage of tg stabilised by a COO−⋯OH(2) interaction (Fig. 1). This interaction also affects the dihedrals of the adjacent glycosidic bond ((AdA)2 blue boxes). The substitutions in position 3 influence predominantly the glycosidic bond geometry. The interaction between the NH3+(3) and the O(5) of the previous residue (R-1) preserved a cellulose-like character ((AGA)2 red boxes). In contrast, the carboxylate at position 3 can engage in additional H-bonds, as observed between COO− and OH(2) of the same residue as well as the OH(6) of the previous sugar (R-1), resulting in remarkable changes of the Φ and Ψ dihedrals ((ADA)2 blue boxes).
Conclusions
Four monosaccharide BBs bearing masked carboxylic acid or amino groups are synthesised. Their good reactivity and stability make them useful BBs for creation of ionic oligosaccharides. Twelve well-defined ionic cellulose analogues were prepared by AGA. Multiply charged oligosaccharides as well as zwitterionic compounds are accessible. MD simulations demonstrate how the nature and the position of the modification (3 vs. 6) plays a major role for the flexibility and conformation of such oligosaccharides. These differences could play a major role in the formation of supramolecular assembly based on charged polysaccharides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Max-Planck Society, the Minerva Fast Track Program, and the MPG-FhG Cooperation Project Glyco3Dysplay, for generous financial support. Open Access funding provided by the Max Planck Society.References
- A. Varki, R. Cummings, J. Esko, H. Freeze, P. Stanley, G. Hart and P. H. Seeberger, Essentials of glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 3rd edn, 2017 Search PubMed.
- B. R. Thakur, R. K. Singh, A. K. Handa and M. Rao, Crit. Rev. Food Sci. Nutr., 1997, 37, 47–73 CrossRef CAS PubMed.
- D.-J. Scheffers and M. G. Pinho, Microbiol. Mol. Biol. Rev., 2005, 69, 585–607 CrossRef CAS PubMed.
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- Y. Van Kooyk and G. A. Rabinovich, Nat. Immunol., 2008, 9, 593 CrossRef CAS PubMed.
- M. Krzesłowska, Acta Physiol. Plant., 2011, 33, 35–51 CrossRef.
- S. Gim, Y. Zhu, P. H. Seeberger and M. Delbianco, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, e1558 Search PubMed.
- M. George and T. E. Abraham, J. Controlled Release, 2006, 114, 1–14 CrossRef CAS PubMed.
- M. Witvrouw and E. De Clercq, Gen. Pharmacol., 1997, 29, 497–511 CrossRef CAS PubMed.
- R. De Philippis and M. Vincenzini, FEMS Microbiol. Rev., 1998, 22, 151–175 CrossRef CAS.
- S. Zhang, Z. Zhang and B. Vardhanabhuti, Food Funct., 2014, 5, 1829–1838 RSC.
- I. Alshanski, J. Blaszkiewicz, E. Mervinetsky, J. Rademann, S. Yitzchaik and M. Hurevich, Chem. – Eur. J., 2019, 25, 12083–12090 CrossRef CAS PubMed.
- L. Schefer, J. Adamcik and R. Mezzenga, Angew. Chem., Int. Ed., 2014, 53, 5376–5379 CrossRef CAS PubMed.
- R. Mezzenga, J.-M. Jung and J. Adamcik, Langmuir, 2010, 26, 10401–10405 CrossRef CAS PubMed.
- V. Castelletto, I. W. Hamley, J. Adamcik, R. Mezzenga and J. Gummel, Soft Matter, 2012, 8, 217–226 RSC.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- P. H. Seeberger, Acc. Chem. Res., 2015, 48, 1450–1463 CrossRef CAS PubMed.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- M. T. Walvoort, H. van den Elst, O. J. Plante, L. Kröck, P. H. Seeberger, H. S. Overkleeft, G. A. van der Marel and J. D. Codée, Angew. Chem., Int. Ed., 2012, 51, 4393–4396 CrossRef CAS PubMed.
- S. Eller, M. Collot, J. Yin, H. S. Hahm and P. H. Seeberger, Angew. Chem., Int. Ed., 2013, 52, 5858–5861 CrossRef CAS PubMed.
- H. S. Hahm, F. Broecker, F. Kawasaki, M. Mietzsch, R. Heilbronn, M. Fukuda and P. H. Seeberger, Chem, 2017, 2, 114–124 CAS.
- C.-F. Liang, H. S. Hahm and P. H. Seeberger, in Glycosaminoglycans, Springer, 2015, pp. 3–10 Search PubMed.
- M. W. Weishaupt, S. Matthies, M. Hurevich, C. L. Pereira, H. S. Hahm and P. H. Seeberger, Beilstein J. Org. Chem., 2016, 12, 1440–1446 CrossRef CAS PubMed.
- A. Pardo-Vargas, M. Delbianco and P. H. Seeberger, Curr. Opin. Chem. Biol., 2018, 46, 48–55 CrossRef CAS PubMed.
- M. Guberman, M. Bräutigam and P. H. Seeberger, Chem. Sci., 2019, 10, 5634–5640 RSC.
- R. S. Loka, E. T. Sletten, U. Barash, I. Vlodavsky and H. M. Nguyen, ACS Appl. Mater. Interfaces, 2018, 11, 244–254 CrossRef PubMed.
- B. Thomas, M. C. Raj, J. Joy, A. Moores, G. L. Drisko and C. M. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- L. T. Mika, E. Csefalvay and A. Nemeth, Chem. Rev., 2017, 118, 505–613 CrossRef PubMed.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 13127–13132 CrossRef CAS PubMed.
- D. Senf, C. Ruprecht, G. H. de Kruijff, S. O. Simonetti, F. Schuhmacher, P. H. Seeberger and F. Pfrengle, Chem. – Eur. J., 2017, 23, 3197–3205 CrossRef CAS PubMed.
- G. Veeneman, S. Van Leeuwen and J. Van Boom, Tetrahedron Lett., 1990, 31, 1331–1334 CrossRef CAS.
- A. M. Vibhute, V. Muvvala and K. M. Sureshan, Angew. Chem., Int. Ed., 2016, 55, 7782–7785 CrossRef CAS PubMed.
- S. Traboni, E. Bedini, M. Giordano and A. Iadonisi, Adv. Synth. Catal., 2015, 357, 3562–3572 CrossRef CAS.
- T. W. Rising, T. D. Claridge, N. Davies, D. P. Gamblin, J. W. Moir and A. J. Fairbanks, Carbohydr. Res., 2006, 341, 1574–1596 CrossRef CAS PubMed.
- T. B. Grindley, Adv. Carbohydr. Chem. Biochem., 1998, 53, 17 CrossRef CAS.
- M. Ohlin, R. Johnsson and U. Ellervik, Carbohydr. Res., 2011, 346, 1358–1370 CrossRef CAS PubMed.
- K. Le Mai Hoang, A. Pardo-Vargas, Y. Zhu, Y. Yu, M. Loria, M. Delbianco and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 9079–9086 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00137f |
| This journal is © The Royal Society of Chemistry 2020 |