
BF3·OEt2-promoted tandem Meinwald rearrangement and nucleophilic substitution of oxiranecarbonitriles†
Chuangchuang
Xu
 and
Jiaxi
Xu
*
and
Jiaxi
Xu
*
State Key Laboratory of Chemical Resource Engineering, Department of Organic Chemistry, College of Chemistry, Beijing University of Chemical Technology, Beijing 100029, People's Republic of China. E-mail: jxxu@mail.buct.edu.cn
First published on 26th November 2019
Abstract
Tandem Meinwald rearrangement and nucleophilic substitution of oxiranenitriles was realized. Arylacetic acid derivatives were readily synthesized from 3-aryloxirane-2-carbonitriles with amines, alcohols, or water in the presence of boron trifluoride under microwave irradiation, and the designed synthetic strategy includes introducing a cyano leaving group into arylepoxides and capturing the in situ generated toxic cyanide with boron trifluoride, making the reaction efficient, safe, and environmentally benign. The reaction occurs through an acid-promoted Meinwald rearrangement, producing arylacetyl cyanides, followed by an addition–elimination process with nitrogen or oxygen-containing nucleophilic amines, alcohols or water. The current method provides a new application of the tandem Meinwald rearrangement.
Introduction
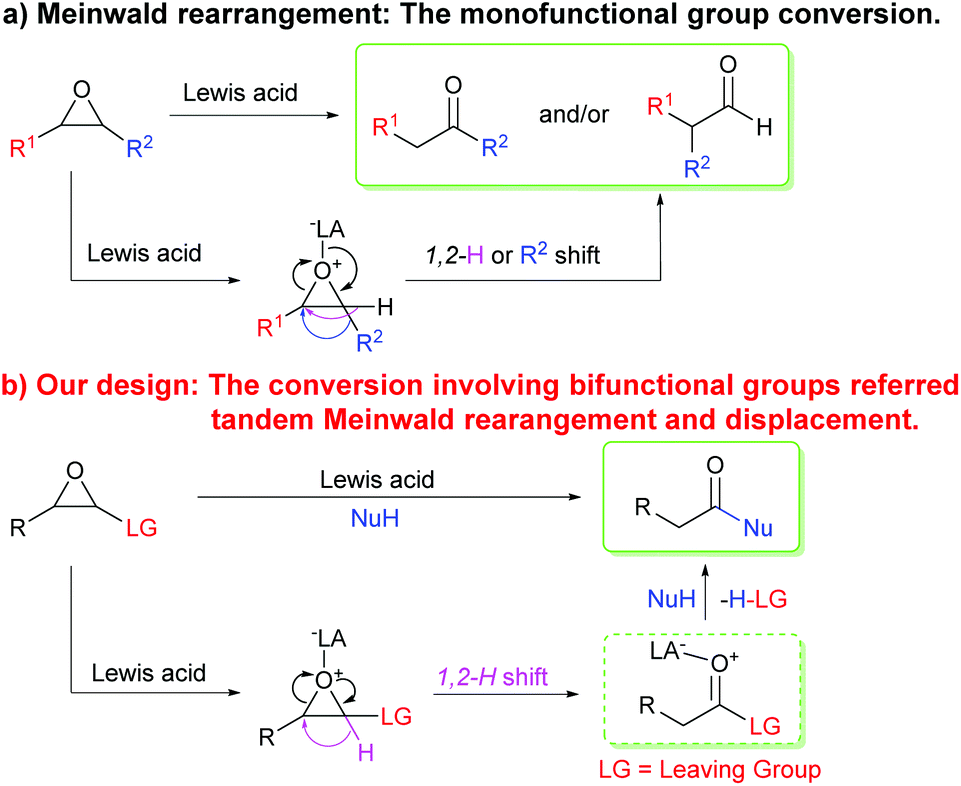
Transformations and reactivity of functional groups are the bases for organic reactions and syntheses in organic chemistry. Studies on the transformations and reactivity of monofunctional groups have made significant progress over the past centuries,1 and many classical organic named reactions have emerged gradually.2 Compared to monofunctional compounds, bifunctional and multifunctional compounds have two or more functional groups that can serve different functions in conversions and reactions. The synergetic functions of bifunctional and multifunctional groups endow them with more complicated and rich reactivity and can be applied in designing novel organic reactions. As a result, introducing a suitable functional group into monofunctional compounds is a challenging strategy to realize tandem transformations in one step and considerable focus on the strategy is required to develop new reactions or tandem reactions.Meinwald rearrangement, a rearrangement reaction of epoxides into aldehydes or ketones by ring-opening and 1,2-shift of hydride or alkyl groups, is a classical monofunctional group conversion reaction that has been widely used in organic and drug syntheses, and even in total syntheses of natural products (Scheme 1a).3 The rearrangement reaction generally occurs in the presence of protonic acids,4 Lewis acids,5 or nucleophiles,6 in which the rearrangement products such as aldehydes or ketones are identified by the formation of the most stable carbenium intermediates, and the migratory aptitude of the substituents attached to the epoxide moiety, Lewis acids or nucleophiles, and solvents. Since the reaction was first reported by Meinwald in 1963,7 it has been studied in-depth and a series of modified Meinwald rearrangement reactions have been developed.4–6 Additionally, the Meinwald rearrangement in tandem processes has also attracted considerable attention for its new application in synthetic organic chemistry.8–18
 | ||
| Scheme 1 Lewis acid-promoted conversions of epoxides into carbonyl derivatives. | ||
On the basis of the Meinwald rearrangement mechanism (Scheme 1a), we envisioned that if a leaving group is introduced into epoxides, they undergo a Meinwald rearrangement (1,2-hydride shift) to produce highly reactive acyl derivatives with a leaving group. The acyl derivatives further react with nucleophiles to give more stable carboxylic acid derivatives (Scheme 1b). In addition, compared to other epoxides possessing a leaving group, oxirane-2-carbonitriles (cyanoepoxides) can be easily prepared from aldehydes and chloroacetonitrile by the Darzens reaction19 and cyanide is a good leaving group.20 Thus, oxirane-2-carbonitriles were eventually selected to explore above idea. A microwave-assisted BF3·OEt2-promoted method to synthesize arylacetic acid derivatives from 3-aryloxirane-2-carbonitriles with nitrogen and oxygen nucleophiles via a tandem Meinwald rearrangement and nucleophilic substitution was developed. Inexpensive and readily available boron trifluoride etherate first acts as a Lewis acid to promote the Meinwald rearrangement and then functions as a scavenger to capture the in situ generated toxic cyanide,21 making the reaction safe and environmentally benign. The reaction features the advantages of microwave irradiation acceleration, short reaction time, metal-free synthesis, readily accessible starting materials, and a wide substrate scope. Such a strategy would not only provide a new application of the Meinwald rearrangement, but also be of importance in the synthesis of arylacetic acids, arylacetates, arylacetamides, and a series of arylacetic acid derivatives that widely exist in nature and drugs.22
Results and discussion
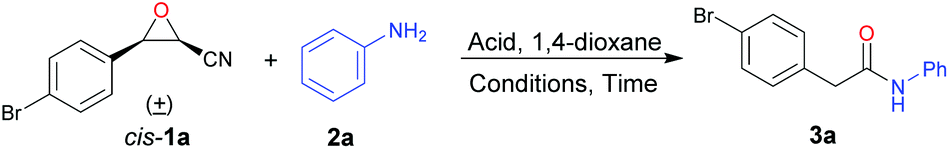
To realize our designed tandem reaction, initially, cis-3-(4-bromophenyl)oxirane-2-carbonitrile (cis-1a) was employed as the model substrate and aniline (2a) was selected as the nucleophile to optimize the reaction conditions. Firstly, cyanoepoxide cis-1a (0.5 mmol), aniline (2a) (0.5 mmol), and BF3·OEt2 (0.65 mmol) refluxed for 9 h in different commercial solvents were examined (see Table S1 in the ESI†). We found that the ring-opening product of β-amino-α-hydroxynitrile was detected when the reactions were conducted in alcohols. It could further transfer to the corresponding indole under more acidic conditions as mentioned in our previous report.19 Moreover, in aprotic solvents, the reaction could work well only in THF, MeCN, and 1,4-dioxane, with the best performance in 1,4-dioxane, giving 2-(4-bromophenyl)-N-phenylacetamide (3a) in 42% yield (Table 1, entry 1). Next, different acids were tried (Table 1, entries 2–6), and it was found that FeCl3 and TsOH could also promote the reaction, but gave 3a in lower yields (Table 1, entries 4 and 6). Besides, equivalents of 2a and acid were investigated, respectively (Table 1, entries 7–16), and it was found that 1.1 equivalents of 2a and 1.3 equivalents of BF3·OEt2 were the best choices, giving 3a in 57% yield (Table 1, entry 7). Furthermore, reactions conducted under microwave irradiation in a sealed vessel were tested and it was found that when cyanoepoxide cis-1a (0.5 mmol), aniline (2a) (0.55 mmol), and BF3·OEt2 (0.65 mmol) in 5 mL of commercial 1,4-dioxane were stirred at 110 °C for 1 hour under microwave irradiation in a sealed vessel 3a was obtained in 50% yield (Table 1, entry 17). Moreover, when the reaction was performed in anhydrous 1,4-dioxane, a higher yield of 59% was obtained (Table 1, entry 18), indicating that the reaction proceeded better in anhydrous solvent. The reaction temperature was investigated and it was found that the yield of 3a increased along with increasing temperature (Table 1, entries 19–22). When the reaction was conducted at 180 °C, 3a was obtained in 72% yield (Table 1, entry 22). In addition, investigation of the reaction time revealed that 30 min was a good choice, with 3a being obtained in 72% yield (Table 1, entries 23–27). To further improve the yield, the reaction temperature was increased to 190 °C under the permissible pressure conditions of the microwave reactor, affording 3a in a higher yield of 77% (Table 1, entry 28). To avoid system pressure exceeding the preset pressure of the microwave reactor, the reaction temperature was not further increased. Next, increasing or decreasing the loading amounts of aniline and BF3·OEt2 did not improve the yield (Table 1, entries 29–32). Furthermore, a high boiling-point ether solvent dipropyleneglycol dimethyl ether was also tested, giving the desired product 3a in 48% yield (Table 1, entry 33). A mixed solvent of dipropyleneglycol dimethyl ether and 1,4-dioxane was attempted, affording 3a in 42% yield (Table 1, entry 34). The reaction was also carried out in anhydrous cyclopentyl methyl ether, affording 3a in 24% yield with 30% yield of an indole derivative as the byproduct (Table 1, entry 35). B(C6F5)3 was tested as a Lewis acid promoter, giving product 3a in 45% yield (Table 1, entry 36). Overall, the best reaction conditions for the nitrogen-containing nucleophile: cis-1 (0.5 mmol), 2 (0.55 mmol), and BF3·OEt2 (0.65 mmol) in 5 mL of anhydrous 1,4-dioxane were stirred at 190 °C for 30 min under microwave irradiation in a sealed vessel.|
|
|||||
|---|---|---|---|---|---|
| Entry | 2a/equiv. | Acid/equiv. | Conditions | Time | Yield/% |
a Reactions were conducted on a 0.5 mmol scale of cis-1a in 5 mL of anhydrous 1,4-dioxane. All yields are yields of the isolated products.
b Commercial solvent was used.
c A high boiling-point solvent of dipropyleneglycol dimethyl ether was used.
d A mixture solvent of dipropyleneglycol dimethyl ether and 1,4-dioxane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v:v) was used.
e Anhydrous cyclopentyl methyl ether was used as the solvent.
f Indole derivative was obtained in 30% yield as the byproduct.
g B(C6F5)3 was used as a Lewis acid instead of BF3·OEt2. :3, v:v) was used.
e Anhydrous cyclopentyl methyl ether was used as the solvent.
f Indole derivative was obtained in 30% yield as the byproduct.
g B(C6F5)3 was used as a Lewis acid instead of BF3·OEt2.
|
|||||
| 1b | 1 | BF3·OEt2/1.3 | Refluxed | 9 h | 42 |
| 2b | 1 | AlCl3/1.3 | Refluxed | 9 h | Trace |
| 3b | 1 | ZnCl2/1.3 | Refluxed | 9 h | Trace |
| 4b | 1 | FeCl3/1.3 | Refluxed | 9 h | 34 |
| 5b | 1 | FeCl2/1.3 | Refluxed | 9 h | Trace |
| 6b | 1 | TsOH·H2O/1.3 | Refluxed | 9 h | 15 |
| 7 | 1.1 | BF 3 ·OEt 2 /1.3 | Refluxed | 9 h | 57 |
| 8b | 1.2 | BF3·OEt2/1.3 | Refluxed | 9 h | 43 |
| 9b | 1.5 | BF3·OEt2/1.3 | Refluxed | 9 h | 39 |
| 10b | 1.1 | BF3·OEt2/0.5 | Refluxed | 9 h | 23 |
| 11b | 1.1 | BF3·OEt2/0.8 | Refluxed | 9 h | 31 |
| 12b | 1.1 | BF3·OEt2/1.0 | Refluxed | 9 h | 36 |
| 13b | 1.1 | BF3·OEt2/1.1 | Refluxed | 9 h | 48 |
| 14b | 1.1 | BF3·OEt2/1.2 | Refluxed | 9 h | 46 |
| 15b | 1.1 | BF3·OEt2/1.4 | Refluxed | 9 h | 41 |
| 16b | 1.1 | BF3·OEt2/2.0 | Refluxed | 9 h | 34 |
| 17b | 1.1 | BF3·OEt2/1.3 | MW/110 °C | 1 h | 50 |
| 18 | 1.1 | BF3·OEt2/1.3 | MW/110 °C | 1 h | 59 |
| 19 | 1.1 | BF3·OEt2/1.3 | MW/120 °C | 1 h | 62 |
| 20 | 1.1 | BF3·OEt2/1.3 | MW/140 °C | 1 h | 64 |
| 21 | 1.1 | BF3·OEt2/1.3 | MW/160 °C | 1 h | 66 |
| 22 | 1.1 | BF3·OEt2/1.3 | MW/180 °C | 60 min | 72 |
| 23 | 1.1 | BF 3 ·OEt 2 /1.3 | MW/180 °C | 30 min | 72 |
| 24 | 1.1 | BF3·OEt2/1.3 | MW/180 °C | 10 min | 69 |
| 25 | 1.1 | BF3·OEt2/1.3 | MW/180 °C | 20 min | 70 |
| 26 | 1.1 | BF3·OEt2/1.3 | MW/180 °C | 40 min | 68 |
| 27 | 1.1 | BF3·OEt2/1.3 | MW/180 °C | 80 min | 69 |
| 28 | 1.1 | BF 3 ·OEt 2 /1.3 | MW/190 °C | 30 min | 77 |
| 29 | 1.0 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 71 |
| 30 | 1.2 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 68 |
| 31 | 1.1 | BF3·OEt2/1.2 | MW/110 °C | 30 min | 70 |
| 32 | 1.1 | BF3·OEt2/1.4 | MW/110 °C | 30 min | 62 |
| 33c | 1.1 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 48 |
| 34d | 1.1 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 42 |
| 35e | 1.1 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 24f |
| 36g | 1.1 | BF3·OEt2/1.3 | MW/190 °C | 30 min | 45 |
To test the influence of different diastereomers of cyanoepoxide 1a, trans-1a was subjected to the optimal conditions, affording product 3a in 52% yield, lower than that of its cis-diastereomer cis-1a (Scheme 2).
 | ||
| Scheme 2 Tandem reaction of trans-1a. | ||
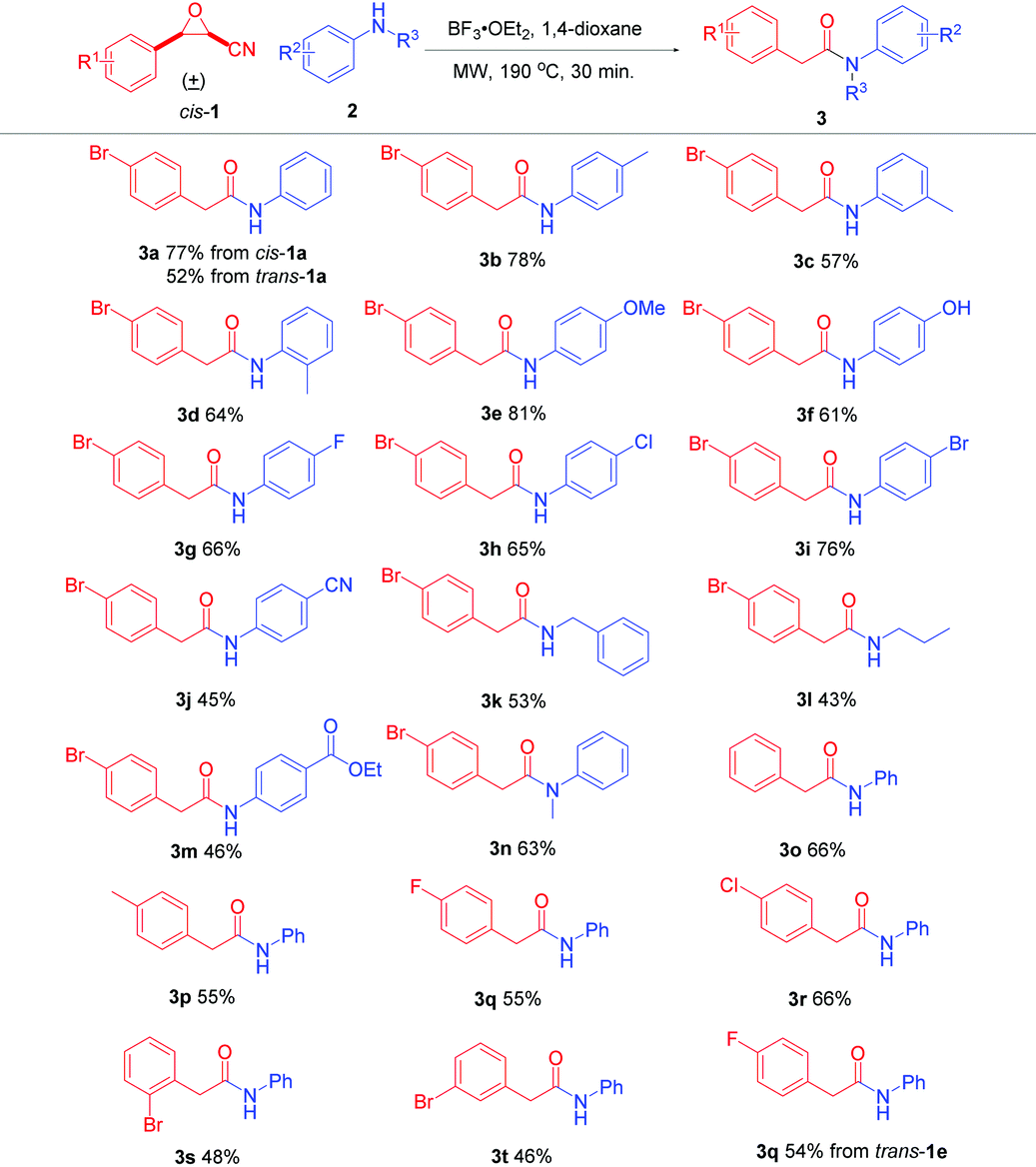
With the optimal conditions in hand, cis-cyanoepoxides cis-1 were applied. The scope and generality of the reaction with different nitrogen-containing nucleophiles 2 were investigated and the results are summarized in Table 2. For different anilines 2, both electron-withdrawing and electron-donating substituted anilines were well suited for the reaction and gave the corresponding products in acceptable to good yields (3b–3j). p,m,o-Toluidines gave the corresponding products 3b, 3c, and 3d in 78%, 57%, and 64% yields, respectively. The lower yields of o,m-toluidines can be attributed to the slight steric hindrance (o-isomer) and the electron-withdrawing property (m-isomer), respectively. 4-Methoxyaniline gave 3e in 81% yield, but 4-hydroxyaniline generated 3f in 61% yield, which may be due to the stronger interaction between the hydroxyl group and BF3, making the oxonium in the BF3-coordinated hydroxyl group an electron-withdrawing group. Additionally, 4-fluoro, 4-chloro, 4-bromo, and 4-cyano substituted anilines gave 3g–3j in 66%, 65%, 76%, and 45% yields, respectively. For aliphatic amines, phenylmethanamine and propan-1-amine were also evaluated to give the corresponding products 3k and 3l in 53% and 43% yields, respectively. The lower yields compared to anilines are possibly attributed to the stronger basicity of aliphatic amines, making them slightly strong interaction with Lewis acid BF3, decreasing their nucleophilicity. Furthermore, functionalized aniline ethyl 4-aminobenzoate gave the desired product 3m in 46% yield. Besides, N-methylaniline was used to generate 3n in 63% yield. Finally, the reactions of different 3-aryloxirane-2-carbonitriles with anilines were conducted. cis-3-Phenyloxirane-2-carbonitrile (cis-1b) gave 3o in 66% yield. cis-3-Aryloxirane-2-carbonitrile with an electron-donating group on the phenyl ring, such as cis-3-(4-methyl)oxirane-2-carbonitrile (cis-1c), yielded product 3p in 55% yield. cis-3-(4-Fluorophenyl)oxirane-2-carbonitrile (cis-1d) produced 3q in 55% yield. The electron-withdrawing substrate cis-3-(4-chlorophenyl)oxirane-2-carbonitrile (cis-1e) gave product 3r in 66% yield. Besides, cis-3-(2-bromophenyl)- and cis-3-(3-bromophenyl)oxirane-2-carbonitriles (cis-1f and cis-1g) gave products 3s and 3t in 48% and 46% yields, respectively.
| a Reaction conditions: cis-1 (0.5 mmol), 2 (0.55 mmol), and BF3·OEt2 (0.65 mmol) in 5 mL of anhydrous 1,4-dioxane were stirred at 190 °C for 30 min under microwave irradiation in a sealed vessel. Yields of the isolated products are indicated. |
|---|
|
To further study the influence of different diastereomers of cyanoepoxides on the yield of the tandem reaction, trans-3-(4-fluorophenyl)oxirane-2-carbonitrile (trans-1d) was also examined under the optimal conditions, giving product 3q in 54% yield, almost the same yield as that of its cis-diastereomer cis-1e (Table 2). The results indicate that both cis- and trans-cyanoepoxides 1 can undergo the Meinwald rearrangement and subsequent displacement under our reaction conditions.
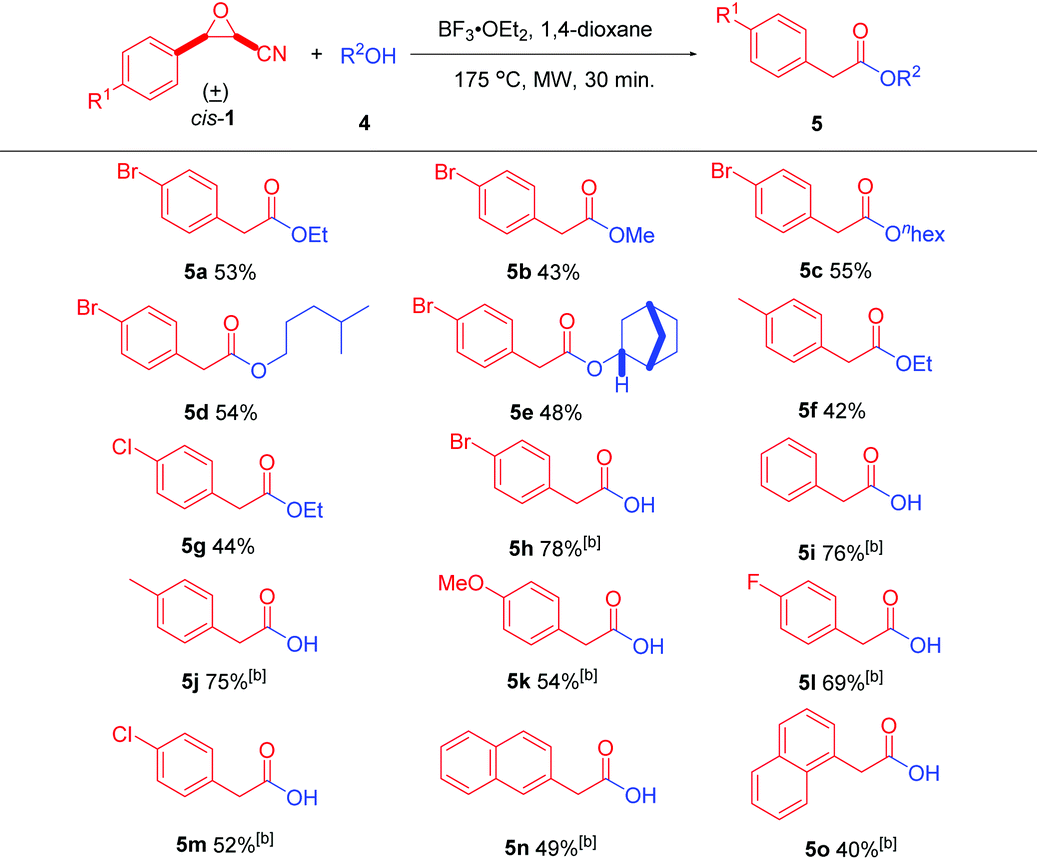
In addition to nitrogen-containing nucleophiles, oxygen-containing nucleophiles are also suitable for this reaction to generate the corresponding arylacetic acids and their esters after optimization of the reaction conditions. The optimized reaction conditions for alcohols are that cis-cyanoepoxide cis-1 (0.5 mmol), alcohol 4 (0.9 mmol), and BF3·OEt2 (0.5 mmol) in 5 mL of anhydrous 1,4-dioxane were stirred at 175 °C for 30 min under microwave irradiation in a sealed vessel (for details, see Table S2 in the ESI†). The best reaction conditions to prepare arylacetic acids are just changing the nucleophile to H2O and loading amount of BF3·OEt2 to 1.2 equivalents (for details, see Table S3 in the ESI†).
With the optimal reaction conditions, the generality of the reaction with different oxygen-containing nucleophiles 4 was evaluated (Table 3). The reactions of cis-3-(4-bromophenyl)oxirane-2-carbonitrile (cis-1a) with several alcohols were first investigated. Anhydrous ethanol and methanol gave 5a (53% yield) and 5b (43% yield), respectively. Besides, long-chain anhydrous alcohols n-hexanol and 4-methylpentanol produced 5c and 5d in 55% and 54% yields, respectively. Additionally, (+)-endo-norborneol also gave product 5e in 48% yield. Furthermore, different 3-aryloxirane-2-carbonitriles were reacted with anhydrous ethanol. cis-3-(4-Methylphenyl)- and 3-(4-chlorophenyl)oxirane-2-carbonitriles (cis-1b and cis-1c) gave products 5f and 5g in 42%, and 44% yields, respectively.
| a Reaction conditions: cis-1 (0.5 mmol), 4 (0.9 mmol), and BF3·OEt2 (0.5 mmol) in 5 mL of anhydrous 1,4-dioxane were stirred at 175 °C for 30 min under microwave irradiation in a sealed vessel. Yields of the isolated products are indicated. b cis-1 (0.5 mmol), 4 (0.6 mmol), and BF3·OEt2 (0.6 mmol) were used. |
|---|
|
Finally, the reactions of different cis-3-aryloxirane-2-carbonitriles with water were investigated. cis-3-Aryloxirane-2-carbonitriles with both electron-withdrawing and electron-donating groups on the aryl ring were well suited for the reaction, giving the corresponding arylacetic acids 5h–5m in moderate to good yields ranging from 52% to 78%. For further application, cis-3-(naphthalene-2-yl) and 3-(naphthalene-1-yl)oxirane-2-carbonitriles (cis-1h and cis-1i) were used and gave products 5n and 5o in 49% and 40% yields, respectively. 2-(Naphthalen-1-yl)acetic acid (5o) is a synthetic plant hormone in the auxin family and is an ingredient in many commercial plant rooting horticultural products. It can be used for the vegetative propagation of plants by stem and leaf cutting. It is also used for plant tissue culture.23
Overall, the designed reaction possesses a wide substrate scope and produces arylacetic acids, arylacetates, arylacetamides, and a series of arylacetic acid derivatives.
Yields of most products are in a moderate range. In the case of nitrogen nucleophiles, side reactions for the formation of indole derivatives occur sometimes.19 Additionally, in the cases of oxygen nucleophiles, relatively low yields were obtained probably because of the stronger interaction between BF3 and the oxygen atom, further forming [B(OR)2F2]− or [B(OH)2F2]− and resulting in lower concentration of oxygen nucleophiles in the reaction mixture.
To gain insight into the reaction mechanism, control experiments were conducted and are summarized in Scheme 3. The reaction of cis-1a under the standard conditions without any nucleophiles was investigated and 2-(4-bromophenyl)acetyl cyanide (6a) was observed in both HRMS and 1H NMR spectra, indicating that cyanoepoxide cis-1a truly underwent the Meinwald rearrangement, producing the corresponding arylacetyl cyanide 6a under our reaction conditions. However, a mixture of 5h and 6a was isolated after work-up (Scheme 3a and c). Besides, when the reaction mixture was quenched with water before purification, only 24% of 5h was isolated and no product 6a was detected, revealing that 2-(4-bromophenyl)acetyl cyanide (6a) is unstable and a water sensitive compound. Previously, Mohan and coworkers reported that 3-phenyloxirane-2-carbonitrile (1b) could not generate the Meinwald rearrangement product when it was refluxed in DCM under the catalysis of 0.1 mol% of Bi(OTf)3.24 Here, cis-3-phenyloxirane-2-carbonitrile (cis-1b) was examined under the catalysis of 0.1 mol% of Bi(OTf)3 in anhydrous 1,4-dioxane at 190 °C for 30 min under microwave irradiation in a sealed vessel. The corresponding 2-phenylacetyl cyanide (6b) was detected by GC-MS. Product 6b shows a different retention time and mass spectral fragmentation from cyanoepoxide 1b. Moreover, cis-3-phenyloxirane-2-carbonitrile (cis-1b) was reacted with 1.1 equivalents of water under the catalysis of 0.1 mol% of Bi(OTf)3 in anhydrous 1,4-dioxane at 190 °C for 30 min under microwave irradiation in a sealed vessel, eventually giving 5i in 43% yield. The results further prove that cyanoepoxides can undergo the Meinwald rearrangement, producing the corresponding acyl cyanides at a higher temperature (Scheme 3d and e).
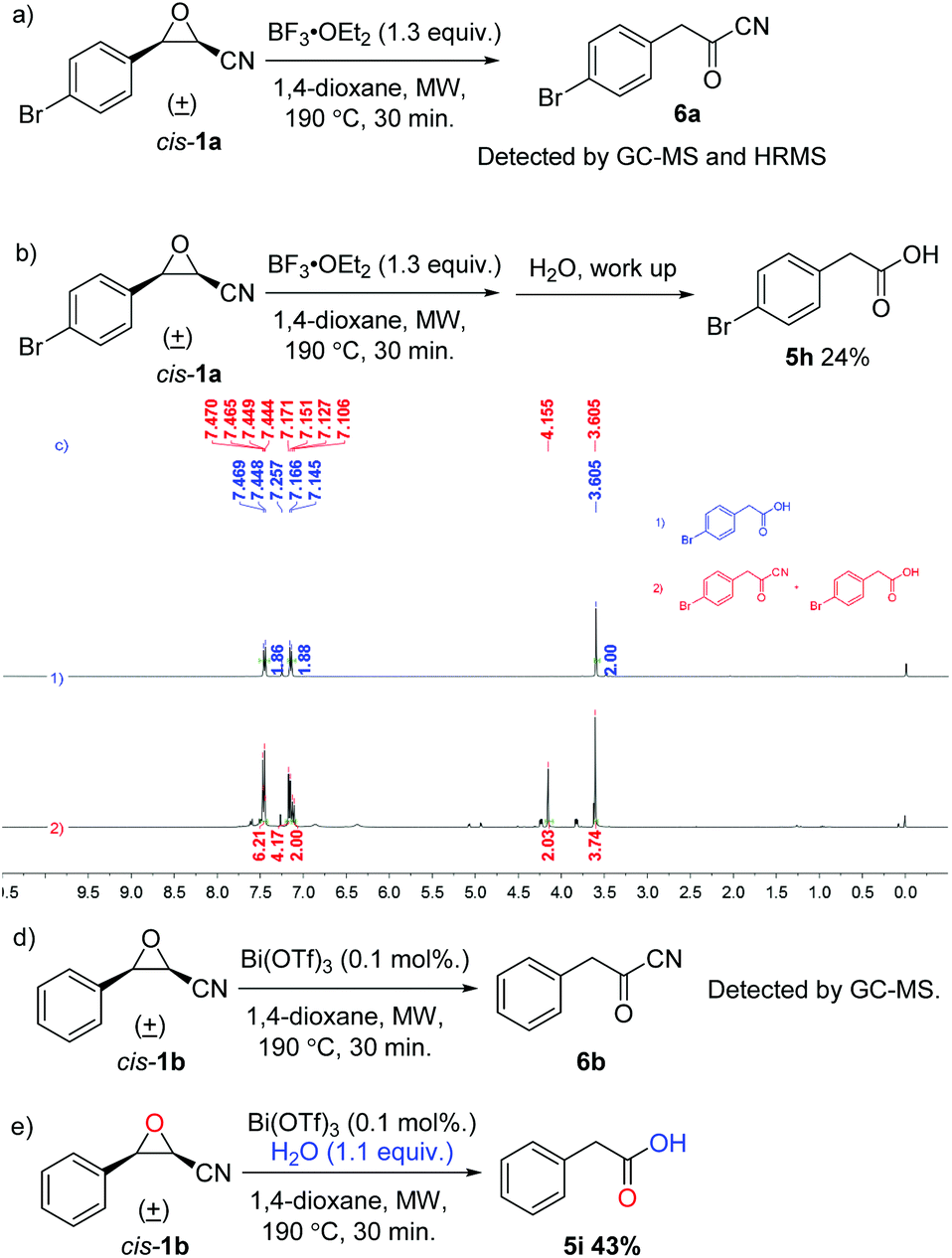 | ||
| Scheme 3 Control experiments. | ||
Based on the reported mechanism of the Meinwald rearrangement4–6 and our experimental results, the following plausible mechanism is proposed (Scheme 4). The reactions with nitrogen- and oxygen-nucleophiles exhibit different mechanisms. For the reaction with nitrogen nucleophiles 2, BF3·OEt2 first coordinates with epoxides 1 to afford intermediates I, followed by ring-opening of the epoxide ring to give benzylic carbocation intermediates II. The predominant formation of the benzylic carbocations makes the ring-opening regiospecific.25 The intermediates II undergo a 1,2-hydride shift to generate arylacetyl cyanides III. They possess high reactivity and easily undergo an addition–elimination process with nitrogen nucleophiles 2 to give substitution products arylacetic amide derivatives 3 through tetrahedral intermediates IV, which eliminate a cyanotrifluoroborate anion because the coordination between BF3 and cyanide is stronger than that between BF3 and Et2O.
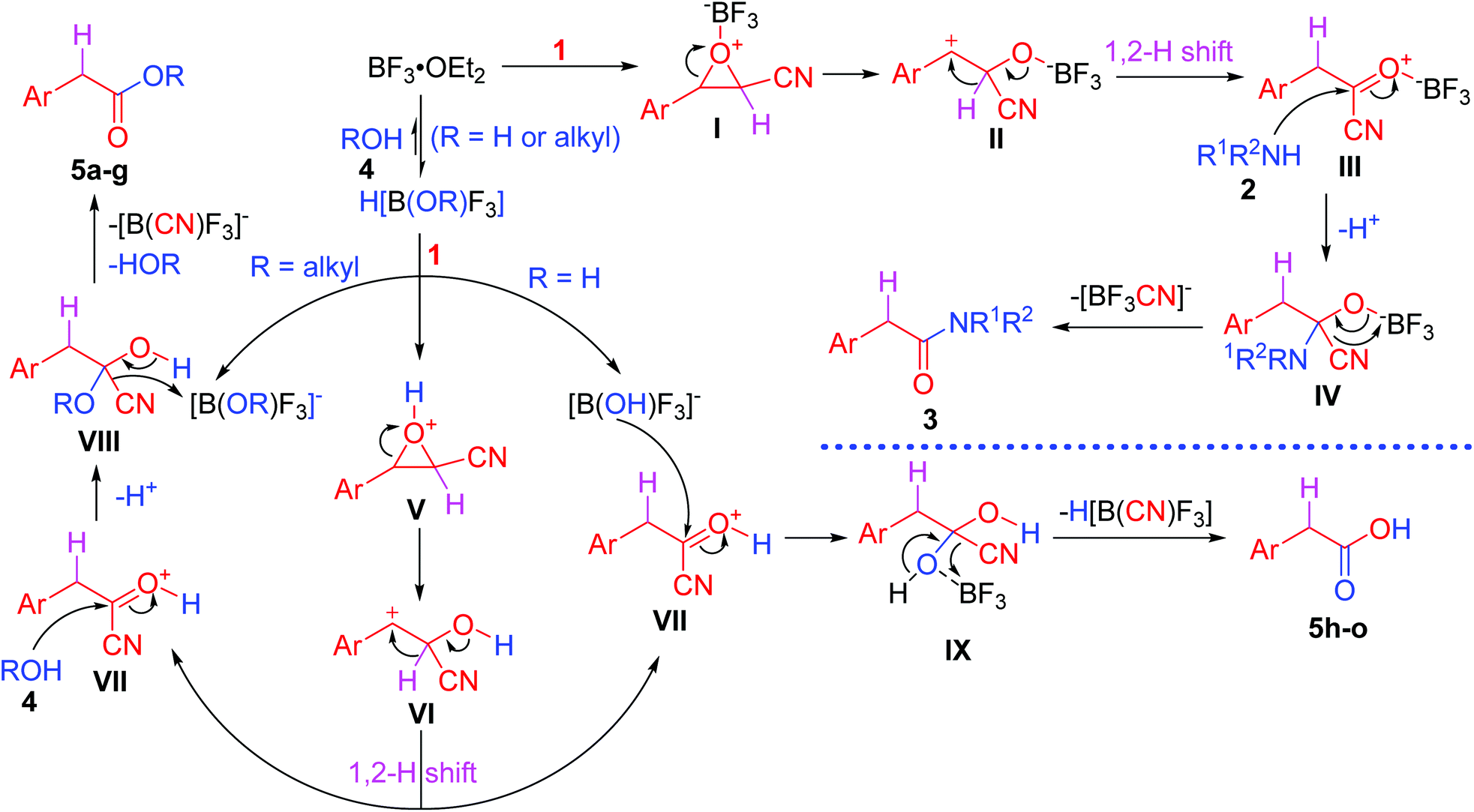 | ||
| Scheme 4 Proposed mechanism for the tandem reactions. | ||
In the case of oxygen nucleophiles 4, oxygen nucleophiles 4 (alcohols and water) first react with BF3 to generate super acids H[B(OR)F3],26 which can dissociate into protons and [B(OR)F3]− ([B(OH)F3]− when water is used as an oxygen nucleophile). Protons can catalyze epoxides to undergo a Meinwald rearrangement to form protonated arylacetyl cyanides VII through intermediates V and VI. When alcohols are used as oxygen nucleophiles, they nucleophilically attack intermediates VII to generate tetrahedral intermediates VIII, which eliminate cyanide. Cyanide and [B(OR)F3]− generate [B(CN)F3]− and alcohols ROH after proton transfer from protonated products 5 to the RO group in [B(OR)F3]−. However, when water is used as an oxygen nucleophile, because equal amounts of BF3 and water are added, no free water exists in the reaction system. It should be noted that [B(OH)F3]− nucleophilically attacks intermediates VII to form tetrahedral intermediates IX, which eliminates H[B(CN)F3] to afford arylacetic acids 5h–o. Although the amount of alcohols is more than that of BF3 when alcohols are used as nucleophiles, a similar process of [B(OR)F3]− instead of [B(OH)F3]− as an oxygen nucleophile nucleophilically attacking intermediates VII is not exclusive in the synthesis of arylacetates 5a–g.
Cyanotrifluoroborate [−B(CN)F3], even dicyanodifluoroborate [−B(CN)2F2], tricyanofluoroborate [−B(CN)3F], and tetracyanoborate [−B(CN)4] were prepared previously from the reaction of boron trifluoride etherate or potassium tetrafluoroborate and potassium cyanide in acetonitrile21a or trimethylsilyl cyanide and potassium tetrafluoridoborate,21b respectively. This is the reason why more than one equivalent of BF3 is required in our reaction. In all reactions, the amount of BF3 is no less than that of cyanoepoxides to capture the in situ generated toxic cyanide, keeping no free cyanide in the reaction mixture. [−B(CN)4] is stable in water.21b revealing that cyanotrifluoroborate cannot release CN− when treated with aqueous solution during workup, making the procedure safe and environmentally benign.
In our previous work, 3-aryloxirane-2-carbonitriles 1 reacted with anilines 2 catalyzed by a catalytic amount of BF3·OEt2 or AlCl3 in the presence of trifluoroethanol, producing indoles.19 However, in this work, 3-aryloxirane-2-carbonitriles 1 reacted with anilines 2 catalyzed by a stoichiometric amount of BF3·OEt2 in 1,4-dioxane under higher temperature conditions, giving the Meinwald rearrangement products followed by nucleophilic addition and elimination, affording arylacetic derivatives. By comparing the differences of these two reaction conditions, we think that 1,4-dioxane as the solvent and a higher reaction temperature make it easier for cyanoepoxides to undergo the Meinwald rearrangement process, producing arylacetyl cyanides as key intermediates followed by the substitution of nucleophilic amines, alcohols, and water to afford the final products arylacetic acids and their derivatives. But refluxing in alcohols under lower temperature conditions makes the ring-opening process easy, followed by intramolecular aromatic electrophilic substitution, generating the indole products. This is the reason why in a previous report cyanoepoxides could not generate the corresponding products acyl cyanides.24
Conclusions
In conclusion, tandem Meinwald rearrangement and nucleophilic substitution of oxiranenitriles are realized successfully under the catalysis of BF3·OEt2 at a high reaction temperature. The reaction has been applied in the synthesis of arylacetic acid derivatives from 3-aryloxirane-2-carbonitriles. The reaction first undergoes an acid-promoted Meinwald rearrangement of 3-aryloxirane-2-carbonitriles, producing arylacetyl cyanides, followed by an addition–elimination process with nitrogen or oxygen-containing nucleophiles, in which the cyanide group functions as a leaving group. Although cyanide is generated in the reaction, it coordinates strongly with boron trifluoride to form a non-toxic tightly tetrahedral cyanotrifluoridoborate anion, making the method safe and environmentally benign. Boron trifluoride plays two important roles in the reaction, first as a Lewis acid to promote the Meinwald rearrangement and second as a scavenger to capture the in situ generated toxic cyanide. The tandem reaction features the advantages of microwave irradiation acceleration, short reaction time, metal-free synthesis, readily accessible starting materials, and a wide substrate scope.Experimental
General information
Melting points were measured on Yanaco MP-500 melting point apparatus and were uncorrected. 1H, 13C, and 19F NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer. Chemical shifts are reported in ppm and referenced to tetramethylsilane (TMS) or residual solvent peaks as internal standards (for CDCl3, tetramethylsilane 0 ppm for 1H and CDCl3 77.00 ppm for 13C; for DMSO-d6, 2.50 ppm for 1H and 39.50 ppm for 13C). IR spectra (KBr pellets, v (cm−1)) were recorded on a Nicolet 5700 FT-IR spectrometer. The high-resolution mass spectra were obtained by ESI ionization using an Agilent LC/MSD TOF mass spectrometer. Microwave reactions were performed with a CEM Discover microwave reactor. Specific rotations were measured on an Anton Paar MCP500 polarimeter. Column chromatography was carried out on silica gel (200–300 mesh) with a mixture of petroleum ether (PE, 60 °C–90 °C) and ethyl acetate (EA) as the eluent. All reactions were followed by thin-layer chromatography (TLC) where practical, using silica gel 60 F254 fluorescent treated silica gel plates, which were visualized under UV light (254 nm).Commercial-grade reagents and solvents were used as received without further purification unless otherwise noted, and anhydrous solvents were purified using the standard processes. The reaction pressure was about 200 psi when reactions were conducted in 1,4-dioxane at 190 °C under microwave irradiation in a sealed vessel. All 3-aryloxirane-2-carbonitriles 1 used in this work were synthesized in our previous work.19,27
Caution: After workup, the waste silica gel and water should be treated with aqueous sodium hypochloride solution for decomposition of the possibly present cyanide residue.
General procedure for the synthesis of cyanoepoxides 1.19,27
A solution of chloroacetonitrile (905 mg, 12 mmol) and KOH (675 mg, 12 mmol) in 75 mL of THF was added to an aldehyde (10 mmol) in one jet. The reaction mixture was stirred at room temperature for 10 h. After the resulting solution was concentrated in vacuo and extracted with ethyl acetate–water, the organic phase was washed with 30 mL of water and then mixed with 2 × 30 mL of saturated NaHSO3. The mixture was stirred at room temperature each time for 6 h until the disappearance of the residual aldehyde as detected by GC-MS. The organic phase was separated and washed with 3 × 30 mL of saturated NaHCO3 and 30 mL of brine. The resulting organic phase was dried over anhydrous Na2SO4. After being filtered and concentrated in vacuo the residue was purified by flash column chromatography on silica gel (PE/EA 30:1, v/v) to afford the corresponding cis- and trans-oxirane-2-carbonitriles 1. Without treatment with saturated NaHSO3, pure cis-oxirane-2-carbonitriles cis-1 were obtained. However, trans-oxirane-2-carbonitriles trans-1 were obtained with the corresponding aldehydes due to very similar polarities (for details, see the ESI†).
General procedure for the synthesis of arylacetamides 3
A solution of 3-aryloxirane-2-carbonitrile 1 (0.5 mmol), aniline (0.55 mmol), and BF3·OEt2 (0.65 mmol) in 5 mL of anhydrous 1,4-dioxane was stirred at 190 °C for 30 min under microwave irradiation in a sealed vessel. After being cooled to room temperature, the solution system was transferred into a 25 mL flask and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with petroleum ether and ethyl acetate (8:1, v/v) as eluents to afford the corresponding arylacetamides 3.
General procedure for the synthesis of arylacetates 5a–5g
A solution of 3-aryloxirane-2-carbonitrile 1 (0.5 mmol), anhydrous alcohol (0.9 mmol) purified with the standard process, and BF3·OEt2 (0.5 mmol) in 5 mL of anhydrous 1,4-dioxane was stirred at 175 °C for 30 min under microwave irradiation in a sealed vessel. After being cooled to room temperature, the solution system was transferred into a 25 mL flask and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with petroleum ether and ethyl acetate (30:1, v/v) as eluents to afford the corresponding arylacetates 5a–5g.
General procedure for the synthesis of arylacetic acids 5h–5o
A solution of 3-aryloxirane-2-carbonitrile 1 (0.5 mmol), H2O (0.6 mmol), and BF3·OEt2 (0.6 mmol) in 5 mL of anhydrous 1,4-dioxane was stirred at 175 °C for 30 min under microwave irradiation in a sealed vessel. After being cooled to room temperature, the solution system was transferred into a 25 mL flask and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel with petroleum ether and ethyl acetate (10:1, v/v) as eluents to afford the corresponding arylacetic acids 5h–5o.
General procedure for the Meinwald rearrangement of cyanoepoxides 1
A solution of 3-aryloxirane-2-carbonitrile 1 (0.5 mmol) and BF3·OEt2 (0.65 mmol) or Bi(OTf)3 (0.4 mg, 0.0005 mmol) in 5 mL of anhydrous 1,4-dioxane was stirred at 190 °C for 30 min under microwave irradiation in a sealed vessel. After being cooled to room temperature, the solution system was transferred into a 25 mL flask and concentrated in vacuo. The residue was subjected to GC-MD, NMR, and HRMS analyses, and further purified by flash column chromatography on silica gel with petroleum ether and ethyl acetate (8:1, v/v) as eluents to afford a mixture of the corresponding arylacetyl cyanide 6 and arylacetic acid 5.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21572017 and 21772010) and the Fundamental Research Funds for the Central Universities (XK1802-6).References
- (a) V. R. C. Larock, Comprehensive Organic Transformations, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 1990 Search PubMed; (b) B. M. Trost, Comprehensive Organic Synthesis, ed. I. Fleming, Pergamon, Oxford, 1991 Search PubMed.
- J. J. Li, Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, Springer International: University of San Francisco, San Francisco, United States, 5th edn, 2014 Search PubMed.
- (a) Z. Wang, in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., 2010, ch. 422, pp. 1880–1882 CrossRef; (b) R. Kumar, J. Halder and S. Nanda, Tetrahedron, 2017, 73, 809–818 CrossRef CAS; (c) X. Liu, J. Liu, J. Zhao, S. Li and C. C. Li, Org. Lett., 2017, 19, 2742–2745 CrossRef CAS PubMed.
- (a) K. Maruoka, T. Ooi and H. Yamamoto, J. Am. Chem. Soc., 1989, 111, 6431–6432 CrossRef CAS; (b) S. Niwayama, S. Kobayashi and M. Ohno, Tetrahedron Lett., 1988, 29, 6313–6316 CrossRef CAS.
- (a) N. Jeedimalla, C. Jacquet, D. Bahneva, J. J. Youte Tendoung and S. P. Roche, J. Org. Chem., 2018, 83, 12357–12373 CrossRef CAS PubMed; (b) J. M. Fraile, J. A. Mayoral and L. Salvatella, J. Org. Chem., 2014, 79, 5993–5999 CrossRef CAS PubMed; (c) D. J. Vyas, E. Larionov, C. Besnard, L. Guenee and C. Mazet, J. Am. Chem. Soc., 2013, 135, 6177–6183 CrossRef CAS PubMed; (d) M. J. González, J. González, C. Pérez-Calleja, L. A. López and R. Vicente, Catal. Sci. Technol., 2013, 3, 932–925 RSC; (e) M. W. C. Robinson, K. S. Pillinger, I. Mabbett, D. A. Timms and A. E. Graham, Tetrahedron, 2010, 66, 8377–8382 CrossRef CAS; (f) M. W. Robinson, A. M. Davies, R. Buckle, I. Mabbett, S. H. Taylor and A. E. Graham, Org. Biomol. Chem., 2009, 7, 2559–2564 RSC; (g) M. W. C. Robinson, K. S. Pillinger and A. E. Graham, Tetrahedron Lett., 2006, 47, 5919–5921 CrossRef CAS; (h) K. Maruoka, N. Murase, R. Bureau, T. Ooi and H. Yamamoto, Tetrahedron, 1994, 50, 3663–3672 CrossRef CAS; (i) X. M. Deng, X. L. Sun and Y. Tang, J. Org. Chem., 2005, 70, 6537–6540 CrossRef CAS PubMed; (j) J. Izquierdo, S. Rodriguez and F. V. Gonzalez, Org. Lett., 2011, 13, 3856–3859 CrossRef CAS PubMed.
- (a) S. Q. Li, Y. Shi, P. F. Li and J. X. Xu, J. Org. Chem., 2019, 84, 4443–4450 CrossRef CAS PubMed; (b) Y. Tian, E. Jürgens and D. Kunz, Chem. Commun., 2018, 54, 11340–11343 RSC; (c) E. Jurgens, B. Wucher, F. Rominger, K. W. Tornroos and D. Kunz, Chem. Commun., 2015, 51, 1897–1900 RSC.
- J. Meinwald, S. S. Labana and M. S. Chadha, J. Am. Chem. Soc., 1963, 85, 582–585 CrossRef CAS.
- V. L. Mamedova and G. Z. Khikmatova, Chem. Heterocycl. Compd., 2017, 53, 976–978 CrossRef CAS.
- H. Zhu, W. Jin, J. He, Y. Zhang and G. Zhu, Adv. Synth. Catal., 2016, 358, 3730–3735 CrossRef CAS.
- S. Wang, C. Zhao, T. Liu, L. Yu, F. Yang and J. Tang, Tetrahedron, 2016, 72, 7025–7031 CrossRef CAS.
- M. R. Tiddens, R. J. M. Klein Gebbink and M. Otte, Org. Lett., 2016, 18, 3714–3717 CrossRef CAS PubMed.
- V. A. Mamedov, V. L. Mamedova, G. Z. Khikmatova, E. V. Mironova, D. B. Krivolapov, O. B. Bazanova, D. V. Chachkov, S. A. Katsyuba, I. d. K. Rizvanov and S. K. Latypov, RSC Adv., 2016, 6, 27885–27895 RSC.
- A. K. Pandey, A. Ghosh and P. Banerjee, Eur. J. Org. Chem., 2015, 2517–2523 CrossRef.
- J. R. Lamb, Y. Jung and G. W. Coates, Org. Chem. Front., 2015, 2, 346–349 RSC.
- W. Zhu, J. Ren and Z. Wang, Eur. J. Org. Chem., 2014, 3561–3564 CrossRef CAS.
- J. Totobenazara, H. Haroun, J. Remond, K. Adil, F. Denes, J. Lebreton, C. Gaulon-Nourry and P. Gosselin, Org. Biomol. Chem., 2012, 10, 502–505 RSC.
- R. Taylor and J. Donald, Synlett, 2009, 59–62 Search PubMed.
- V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677–12682 CrossRef CAS PubMed.
- C. C. Xu and J. X. Xu, J. Org. Chem., 2018, 83, 14733–14742 CrossRef CAS PubMed.
- (a) A. Lapworth, J. Chem. Soc., Trans., 1903, 83, 995–1005 RSC; (b) A. Lapworth, J. Chem. Soc., Trans., 1904, 85, 1206–1214 RSC; (c) L. Y. Zhang, C. Zhang, T. Wang, Y. L. Shi, M. T. Ban and D. M. Cui, J. Org. Chem., 2019, 84, 536–543 CrossRef CAS PubMed; (d) K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050–16053 CrossRef CAS PubMed.
- (a) E. Bernhardt, M. Finze and H. Willner, Z. Anorg. Allg. Chem., 2003, 629, 1229–1234 CrossRef CAS; (b) K. Bläsing, S. Ellinger, J. Harloff, A. Schulz, K. Sievert, C. Täschler, A. Villinger and C. Zur Täschler, Eur. J. Inorg. Chem., 2016, 1175–1183 CrossRef.
- (a) A. L. Blobaum and L. J. Marnett, J. Biol. Chem., 2007, 282, 16379–13690 CrossRef CAS PubMed; (b) A. J. Souers, J. Gao, M. Brune, E. Bush, D. Wodka, A. Vasudevan, A. S. Judd, M. Mulhern, S. Brodjian, B. Dayton, R. Shapiro, L. E. Hernandez, K. C. Marsh, H. L. Sham, C. A. Collins and P. R. Kym, J. Med. Chem., 2005, 48, 1318–1321 CrossRef CAS PubMed; (c) Y. Wang, W. Cai, Y. Cheng, T. Yang, Q. Liu, G. Zhang, Q. Meng, F. Han, Y. Huang, L. Zhou, Z. Xiang, Y. G. Zhao, Y. Xu, Z. Cheng, S. Lu, Q. Wu, J. N. Xiang, J. D. Elliott, S. Leung, F. Ren and X. Lin, ACS Med. Chem. Lett., 2015, 6, 787–792 CrossRef CAS PubMed.
- (a) N. B. Shibin, R. Sreekanth, U. K. Aravind, K. M. Afsal Mohammed, N. V. Chandrashekhar, J. Joseph, S. K. Sarkar, D. B. Naik and C. T. Aravindakumar, J. Phys. Org. Chem., 2014, 27, 478–483 CrossRef CAS; (b) A. Navalón, R. Blanc and J. L. Vilchez, Microchim. Acta, 1997, 126, 33–38 CrossRef.
- K. A. Bhatia, K. J. Eash, N. M. Leonard, M. C. Oswald and R. S. Mohan, Tetrahedron Lett., 2001, 42, 8129–8132 CrossRef CAS.
- (a) C. Zhou and J. X. Xu, Prog. Chem., 2011, 23, 165–180 CAS; (b) X. Y. Li, Z. Y. Yang and J. X. Xu, Curr. Org. Synth., 2013, 10, 169–177 CAS; (c) C. Zhou and J. X. Xu, Prog. Chem., 2012, 24, 338–347 CAS; (d) L. G. Ma and J. X. Xu, Prog. Chem., 2004, 16, 220–235 CAS.
- (a) G. F. Hennion, H. D. Hinton and J. A. Nieuwland, J. Am. Chem. Soc., 1933, 55, 2857–2860 CrossRef CAS; (b) G. K. S. Prakash, C. Panja, A. Shakhmin, E. Shah, T. Mathew and G. A. Olah, J. Org. Chem., 2009, 74, 8659–8668 CrossRef CAS PubMed.
- C. C. Xu, Y. Lu, K. N. Xu and J. X. Xu, Synthesis, 2019 DOI:10.1055/s-0039-1690243.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1, S2, and S3 on the optimization of the reaction conditions; full spectroscopic data for all isolated compounds 1, 3, 5 and 6a; HRMS of 6a; and GC-Mass of 1a, 1b and 6b. See DOI: 10.1039/c9ob02428j |
| This journal is © The Royal Society of Chemistry 2020 |